
Abstract
Williams-Beuren syndrome (WBS) and supravalvular aortic stenosis (SVAS) are genetic syndromes marked by the propensity to develop severe vascular stenoses. Vascular lesions in both syndromes are caused by haploinsufficiency of the elastin gene. We used these distinct genetic syndromes as models to evaluate the feasibility of using engineered zinc-finger protein transcription factors (ZFPs) to achieve compensatory expression of haploinsufficient genes by inducing augmented expression from the remaining wild-type allele. For complex genes with multiple splice variants, this approach could have distinct advantages over cDNA-based gene replacement strategies. Targeting the elastin gene, we show that transcriptional activation by engineered ZFPs can induce compensatory expression from the wild-type allele in the setting of classic WBS and SVAS genetic mutations, increase elastin expression in wild-type cells, induce expression of the major elastin splice variants, and recapitulate their natural stoichiometry. Further, we establish that transcriptional activation of the mutant allele in SVAS does not overcome nonsense-mediated decay, and thus ZFP-mediated transcriptional activation is not likely to induce production of a mutant protein, a crucial consideration. Finally, we show in bioengineered blood vessels that ZFP-mediated induction of elastin expression is capable of stimulating functional elastogenesis. Haploinsufficiency is a common mechanism of genetic disease. These findings have significant implications for WBS and SVAS, and establish that haploinsufficiency can be overcome by targeted transcriptional activation without inducing protein expression from the mutant allele.
Introduction
In humans, the ELN gene is located on chromosome 7 in a region (7q11.23) designated the WBS locus. WBS is caused by heterozygous non-allelic homologous recombination at this locus, resulting in deletion of the entire ELN gene and 25–27 additional genes (Pober et al., 2008a). The intact ELN allele is noncompensatory in WBS, and expression is actually often lower than explainable on the basis of pure haploinsufficiency alone (e.g., 30–40% of normal levels). There are at least 50 identified heterozygous ELN mutations that can result in SVAS. These mutations generally cause frame-shifts that create premature termination codons (PTC), targeting the mutant transcripts for destruction by the nonsense-mediated decay pathway (Micale et al., 2010; Urban et al., 2000). As in WBS, the intact ELN allele is noncompensatory, thus resulting in elastin haploinsufficiency.
Replacement therapy for haploinsufficiency syndromes can be preclusively difficult, either by protein replacement or gene therapy. For genes encoding intracellular proteins or complex extracellular structural proteins like elastin, replacement by providing the mature protein product is not generally possible. For all complex genes, such as those encoding multiple potential splice variants, cDNA-based gene replacement therapy is also highly problematic. The ELN gene is structurally and functionally complex. The coding region contains at least 34 exons in the human ELN gene, 17 of which have been shown to undergo alternative splicing, resulting in a substantial number and variety of elastin splice variants (Heim et al., 1991; Mecham, 2008; Parks et al., 1992). Although it has been reported that exons 26A and 32 are splice deleted in human skin fibroblasts (Fazio et al., 1988; Holzenberger et al., 1993), exon 22 participates in intramolecular and intermolecular cross-linking (Dyksterhuis and Weiss, 2010; Wise et al., 2005), and the c-terminus is important for binding microfibrils (Kozel et al., 2003), relatively little is known about the individual functions of each exon and splice variant. This not only limits understanding of elastin biology, but this complexity is a crucial issue in the development of therapeutic strategies to promote elastogenesis and to rescue ELN haploinsufficiency.
One potential alternative therapeutic approach in WBS, SVAS, and other haploinsufficiency syndromes is to increase expression of the intact wild-type allele to full compensatory levels. Theoretically, this approach should promote expression of all naturally occurring splice variants of a targeted gene. Further, by activating the endogenous expression program for that specific gene, this approach might also promote normal posttranslational processing and assembly of complex proteins, like elastin. Elastogenesis, the formation and maturation of new elastin fibers, involves ELN expression combined with an array of complex coordinated events (Czirok et al., 2006; Wagenseil and Mecham, 2007). In mammals, ELN encodes a 60–70 kDa protein called tropoelastin, comprised of alternating hydrophobic and lysine-rich cross-linking domains (Mecham, 2008). After synthesis, soluble tropoelastin is secreted into the extracellular space and undergoes cross-linking by lysyl oxidase (LOX) and lysyl oxidase–like 1 (LOXL1) to form insoluble mature elastin fibers that also contain microfibril proteins, such as fibrillin-1 and fibulin-5 (Kielty et al., 2002). While elastogenesis involves multiple genes and additional proteins, it is clear that ELN expression is a major driver of this process.
Augmenting transcription from the intact wild-type allele could conceptually achieve compensatory expression of ELN. One way to achieve this would be to exploit endogenous ELN regulatory mechanisms. Unfortunately, how ELN gene expression is regulated is poorly understood, though of high developmental and clinical relevance. ELN expression begins around mid-gestation, peaks near birth and the early neonatal period (P7-P14 in mice), then drops sharply to low levels that persist into adulthood. It is also tightly tissue and cell-type restricted. The ELN 5′ flanking region is G+C rich (66%), does not have a canonical TATA box, and though containing a large number of potential binding sites for various transcription factors, it remains unknown how temporal and cell type–specific expression is maintained (Oleggini et al., 2007; Rosenbloom et al., 1991). While factors that can regulate ELN expression have been identified, such as TGFβ1 and IGF1 (activation), and bFGF (negative regulation), these factors have broad biological effects that preclude exploiting them to therapeutically modulate ELN expression (Carreras et al., 2002; Oleggini et al., 2007).
Herein we explore the feasibility of using engineered zinc-finger protein (ZFP) transcription factors to achieve compensatory expression from the normal ELN allele in WBS and SVAS. We (and others) have previously shown that ZFPs can be engineered to direct highly specific gene expression in vivo and in vitro, and that activation of an endogenous gene using this approach can lead to normal stoichiometric expression of that gene's natural splice variants—a distinct advantage for a complex gene like ELN (Klug, 2010a; Mandell and Barbas, 2006; Rebar et al., 2002; Tan et al., 2003; Yu et al., 2006). The ZFP approach exploits cumulative knowledge of how specific amino acids within the alpha-helix of a zinc-finger protein can direct binding to targeted DNA sequences with high specificity (Christensen et al., 2011; Gonzalez et al., 2010; Pabo et al., 2001). Each finger binds a specific 3-bp DNA sequence. Building multi-finger ZFPs allows longer sequences to be targeted. By combining the DNA-binding fingers with an appropriate functional domain, highly specific transcription factors can be constructed. A number of papers and excellent reviews describe the principles involved in designing engineered ZFPs (Christensen et al., 2011; Klug 2010b; Miller et al., 2007; Pabo et al., 2001; Rebar et al., 2002).
We describe here the successful construction of an ELN-targeted ZFP that achieves compensatory expression of elastin from the intact allele in both WBS and SVAS, and explore how transcriptional activation of the mutant allele in SVAS affects the surveillance mechanism of nonsense-mediated decay. We further show the utility of this approach by demonstrating ZFP-induced ELN expression and elastogenesis in bioengineered blood vessels. We contend that this approach to force compensatory expression from the remaining intact allele in haploinsufficiency syndromes may have significant clinical applicability.
Materials and Methods
Generation of ZFP transcription factors for human ELN gene
A library of n>106 3-finger ZFPs was screened for specific binding to a 10 bp target sequence (GGGAAAGAGa, 292 bp upstream translation initiation site) on human elastin promoter using a bacterial one-hybrid system (B1H) as described (Meng et al., 2005; Meng and Wolfe, 2006). The resulting human elastin gene targeted zinc finger protein (hELN-ZFP) clones were then inserted to pcDNA3.1 (Clontech) containing cytomegalovirus (CMV) promoter, the nuclear translocation signal from simian virus 40 (SV40) large T antigen, the herpes simplex virus herpes simplex virus virion protein 16 (VP16) transactivation domain spanning amino acids 413–490, and a FLAG peptide (Rebar et al., 2002). The plasmid without ZFP was referred to as VP16 alone and used as control. The uppercase letters in the binding sites denote bases directly contacted by the ZFP, and the extra base shown in lowercase denotes an extra overlapping target site.
Lipofectamine transfection and electroporation
Human dermal fibroblast (HDF) cells were electroporated with ELN-ZFP plasmids using Amaxa nucleofector kit for primary fibroblasts (Lonza). Human embryonic kidney 293 cells (HEK293) were transiently transfected with ELN-ZFP plasmids using lipofectamine 2000 (Invitrogen). Messenger RNA (mRNA) or protein were harvested 48 h later for analysis.
Retroviral or lentiviral contructs containing ELN-ZFPs
Retroviral constructs containing ELN-ZFP were made and used in transduction as described (Zhang et al., 2010). Briefly, ELN-ZFP or VP16 were inserted into LZRSpBMNZ retroviral vectors through the multiple cloning sites. The correct orientation and the DNA sequence of the insertion were verified by sequencing. The retroviral constructs were transfected into the Phoenix-Ampho packaging cell line by using Lipofectamine 2000. Puromycin-resistant Phoenix-Ampho packaging cells were selected and used to condition the medium. Retrovirus-conditioned medium was passed through 0.45 μm filters (Millipore), supplemented with 8 μg/mL polybrene (Sigma-Aldrich), and then added on target cells for 15 hr. Lentiviral constructs were generated by subcloning ELN-ZFP or VP16 into pLVX-Puro (Clontech). Forty-eight hours after 293 T cell were transfected with lentiviral plasmids, fresh lentivirus-containing media were collected and used to infect target cells at 5U MOI (multiplicity of infection of 5). mRNA or protein were harvested 72 hours later after infection.
Cell culture
HDF cells, isolated from human foreskin or adult skin, were grown in low glucose Dulbecco's modified Eagle's medium (DMEM; JRH) with 10% heat-inactivated fetal bovine serum (FBS; Lonza), 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen). Additional HDF cells were purchased from Coriell cell repositories. WBS dermal fibroblast and age-matched HDF were purchased from NICHD Brain and Tissue Bank for Developmental Disorders (University of Maryland). Human vascular smooth muscle cells (VSMC) and SVAS pulmonary VSMC were explanted by outgrowth as described (Zhang et al., 2010) and cultured in 20% FBS in DMEM with supplements and antibiotics.
Cell proliferation assay and scratch assay
Transduced and nontransduced cells of the same number were seeded on plates in complete media. Cell numbers at selected time points were determined by hemocytometer readings. Scratch wounds were created by a p-200 pipet tip on confluent cell monolayers as described (Liang et al., 2007). Wounds were rinsed with phosphate buffered saline (PBS) and cultured in 0.25% FBS media. Wound closure areas 15 hours later were stained by 0.2% crystal violate and quantified with ImageJ software (National Institutes of Health [NIH]).
qPCR
Primers for quantitative polymerase chain reaction (qPCR) were designed by PerlPrimer and experimentally validated by qPCR melting curve and product agarose gel electrophoresis. Primer sequences include human ELN (non-splicing exon 18 unless specified otherwise) forward aggggttgtgtcaccagaag, reverse cagctccaaccccgtaagta; ELN exon 3 forward cctggggccattcctggtgg, reverse cctccgggaactggcttaa; ELN exon 10 forward ctgccaggtgtatacccagg, reverse ccacctacacctggagcctt; ELN exon 13 forward ggctatggactgccctacac, reverse gggccaacccctgtccctgt; ELN exon 23 forward gttagttcctggtgtcggcg, reverse tgcaactccaccagggccaa; ELN exon 25 forward agctgcagctgggcttggtg, reverse gccagggctccaggtactgcc; human COL1A1 forward aagggacacagaggtttcag, reverse gtagcaccatcatttccacg; human COL3A1 forward ctcctaatggtcaaggacct, reverse gagaatagttctgaggaccag. Primers for vascular endothelial growth factor A (VEGFA), hypoxia-inducible factor 1α (HIF-1α), and 18S ribosomal RNA (18S) were designed as described (Huang et al., 2004). Cycle threshold (Ct) values of the target mRNA were normalized by subtracting 18S Ct value to achieve ΔCt.
Western blot
To measure tropoelastin expression in the media, cells were grown in 0.5% FBS DMEM media for the final 24 hr, and media proteins were precipitated by 10% trichloroacetic acid (TCA) and resolved on 10% SDS-PAGE. To measure elastin protein associated with cell membrane, equal number of cells were lysed in 6 M urea-containing sample-loading buffer and loaded onto 10% SDS-PAGE. Western membranes were probed with a polyclonal antibody recognizing human elastin (a gift from Dr. Robert Mecham, Washington University, St. Louis, MO). Ponceau red staining or probing with monoclone antibody against β-actin (Sigma) were used to normalize protein loading among the lanes. ImageJ (NIH) software was used to quantitate the intensity of bands.
Emetine treatment
After lentiviral transduction, SVAS pulmonary VSMC were treated with 100 μg/mL emetine. mRNA were collected at indicated time points and qPCR analyzed by Taqman probes for ELN and COL1A1 (Applied biosystem). Specific primer pairs used to generate PCR products covering the mutation site in SVAS were ctgccaggtgtatacccagg (forward) and ccacctacacctggagcctt (reverse).
Culture and analyses of tissue-engineered vessels
Engineered arteries having dimensions of 1 mm in diameter and 3 cm in length were grown as previously described (Niklason et al., 1999). Briefly, human VSMC (0.8×106 cells/ml) were seeded onto a polyglycolic acid (PGA) scaffold (Albany International) around a silicone tube for support. Vessels were cultured in a bioreactor for 6 weeks in low-glucose DMEM with 10% FBS, 10% human serum, 2.6 mg/ml hydroxyethyl piperazineethanesulfonic acid (HEPES), 100 U/ml penicillin, 50 μg/ml proline, 20μg/ml alanine, 50 μg/ml glycine, 3 ng/ml CuSO4, 10 ng/ml basic fibroblastic growth factor (bFGF), and 10 ng/ml platelet-derived growth factor (PDGF-bb), as previously described (Dahl et al., 2007). A pulsatile pump forced PBS through the silicone tubing at a rate of 90 beats per minute to simulate an adult pulse rate.
Light and transmission electron microscopy
Vessel samples were also fixed in 10% formalin, embedded in paraffin, cut into 5 μm sections, and stained with hematoxylin and eosin (H&E) and Masson's trichrome stain, which stains collagen blue. For transmission electron microscopy (TEM), tissues were fixed in 2.5% glutaraldehyde, 1% formaldehyde in sodium cacodylate buffer, and embedded in Epon. Images were taken at Yale transmission electron microscope (TEM) center.
Desmosine and hydroxyproline analysis
Desmosine and hydroxyproline, demonstrating cross-linked elastin and collagen levels respectively, were measured as described (Starcher and Conrad, 1995).
Microarray analysis
Total RNA from ELN-ZFP3–transduced or VP16–transduced HDF cells in triplicates were isolated as described and hybridized to GeneChip Human Gene 1.0 ST Array that covers 28,869 genes (Affymetrix) at Yale Center for Genome Analysis.
Statistical analysis
Comparisons between two groups were by unpaired student t test. Statistical analyses were performed using Prism 4 software (GraphPad). P values were two-tailed and values <0.05 were considered to indicate statistical significance. For microarray analysis, normalized expression signals were calculated from Affymetrix CEL files using RMA (Irizarry et al., 2003). Statistical differences were calculated using the Limma package of lineal model analysis from the Bioconductor project (Smyth, 2005). The p values were obtained from moderated t statistics using empirical Bayesian methods. P values were adjusted for multiple testing with Benjamini and Hochberg's (Klipper-Aurbach et al., 1995).
Results
Design and in vitro testing of ELN-targeted ZFPs
A 9-bp target sequence in the promoter of the ELN gene was selected by screening the 5′ regulatory region for known transcription factor–binding sites and indentifying a sequence without significant overlap with these sites, but with features that have been previously shown to facilitate effective ZFP targeting. The 9-bp sequence chosen, GGGAAAGAG, is located ∼200-bp upstream of the major translational start site. Three three-finger ELN-ZFP DNA binding domains were selected by a bacterial one-hybrid screening method (Meng et al., 2005). Individual cDNAs encoding these DNA-binding domains were cloned in front of the VP16 transactivation domain to generate CMV-driven expression cassettes encoding these ELN-ZFP transcription activators, as shown in Figure 1. Their ability to activate the endogenous ELN gene was first tested by transient transfection into HEK293 cells, followed by qRT-PCR analysis of elastin mRNA. In comparison to the vehicle (lipofectamine), a control plasmid containing VP16 alone, and a plasmid encoding a previously validated VEGF-ZFP (Rebar et al., 2002), all three ELN-ZFPs directed a >6-fold induction of elastin mRNA, with ELN-ZFP3 having the highest activation (more than 58-fold) (Fig. 2A). ELN-ZFPs had little effect on VEGF-A and HIF-1α mRNA in HEK293 cells, whereas the VEGF targeted ZFP-activated VEGF expression, as expected. Next, we electroporated HDF with ELN-ZFP plasmids. As seen (Fig. 2B and C), ELN-ZFP1 and ELN-ZFP3 increased elastin mRNA 12- or 32-fold respectively, and ELN-ZFP treatment dramatically increased the levels of secreted tropoelastin protein from these cells (Fig. 2C).
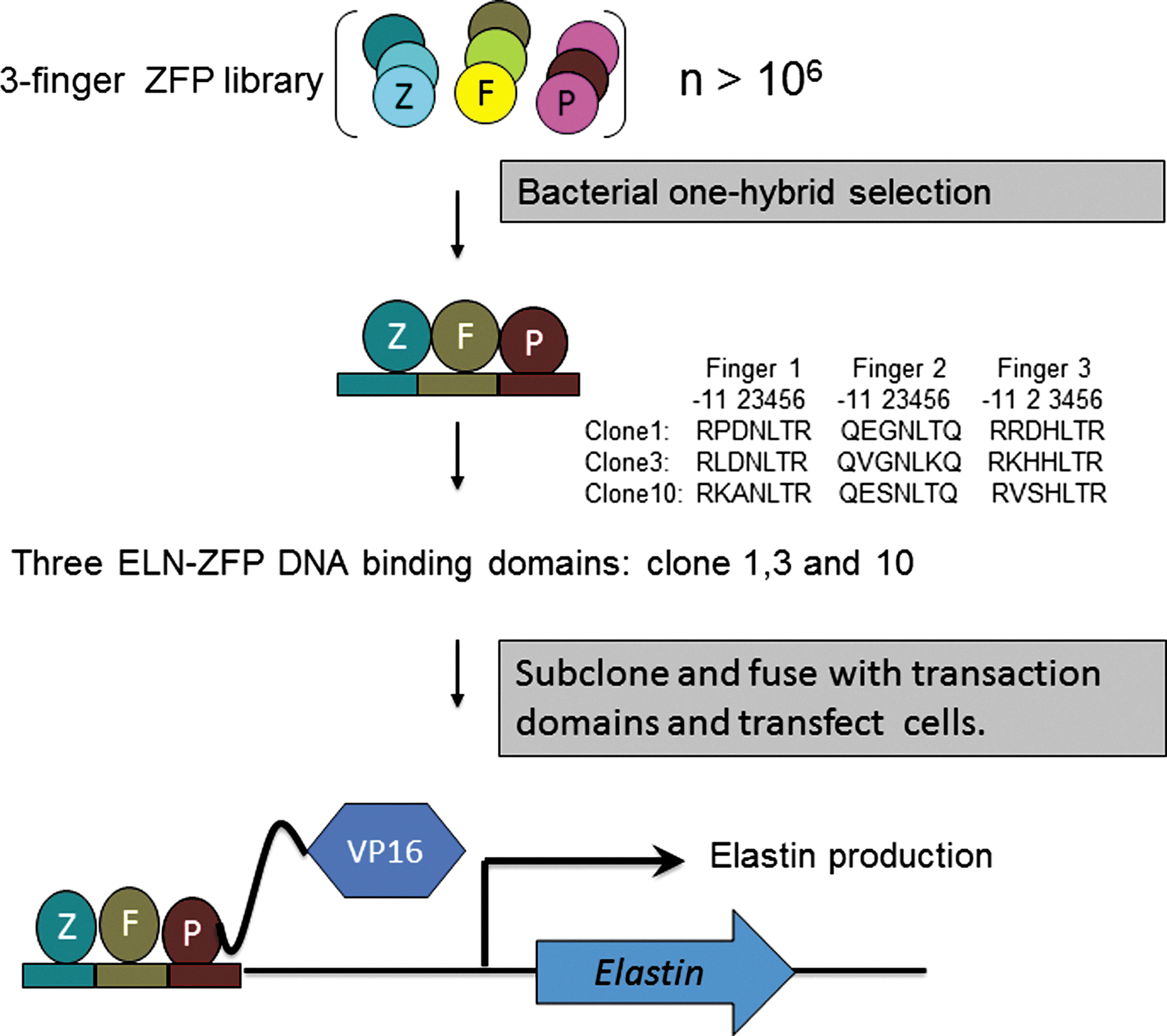
ELN-ZFP design. Three-finger zinc-finger proteins (ZFPs) targeting a 9-bp target sequence in the ELN promoter were selected from a random three-finger library using a bacterial one-hybrid screening approach. Each finger binds a specific 3-bp sequence on the sense DNA strand, based on the sequence of specific amino acids within the alpha helix of that finger. The sequences of the DNA-binding amino acids within each finger of the three most highly represented clones are shown. cDNA expression cassettes encoding the ELN-ZFP transcription factors were created by cloning cDNA encoding the ZFP DNA-binding fingers in front of sequences encoding the VP-16 transactivation domain, as depicted. The ELN-ZFP expression cassettes were driven by a CMV promoter (not shown). ELN, loss of elastin gene; ZFP, zinc-finger protein, CMV, cytomegalovirus. Color images available online at
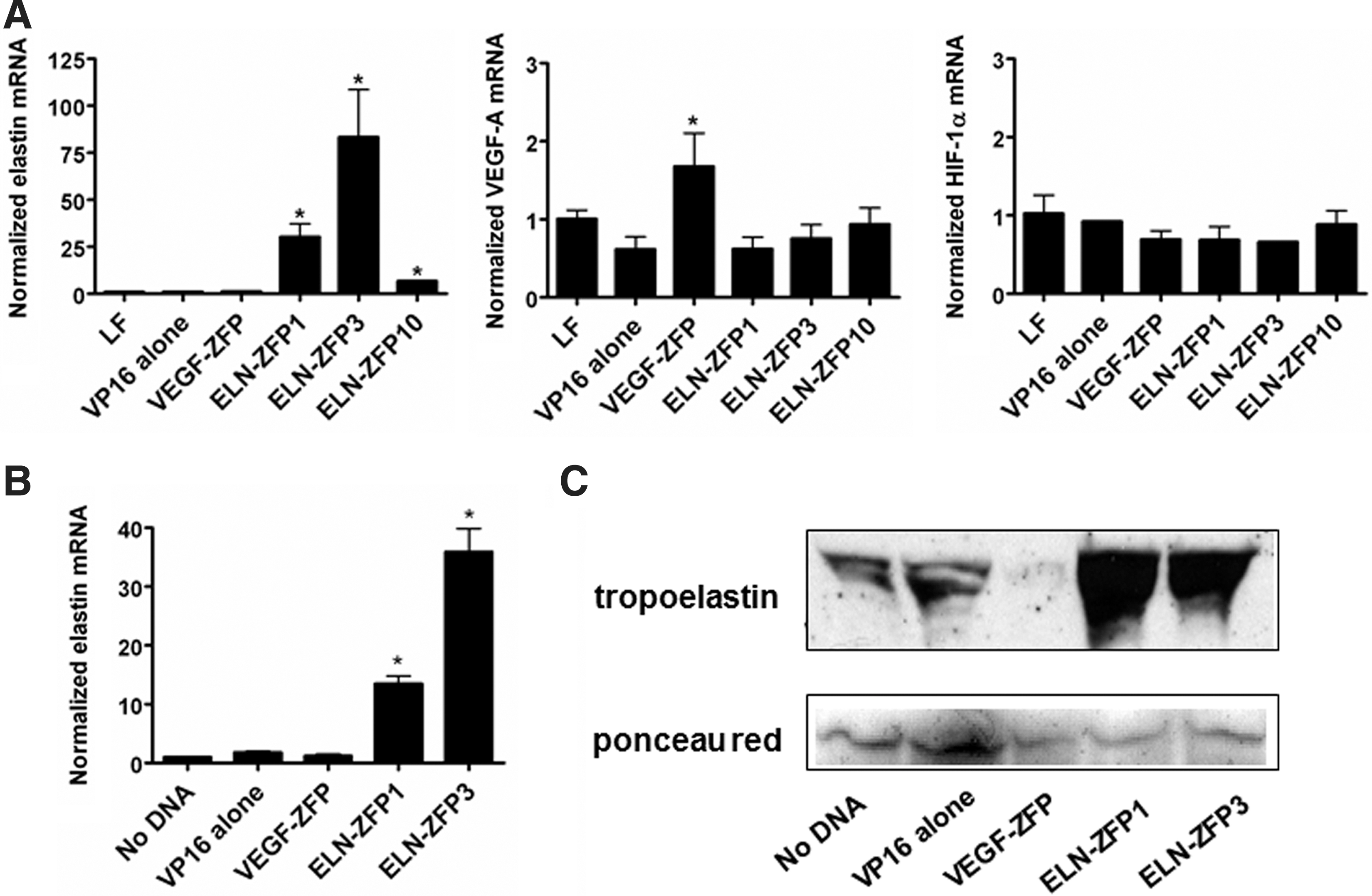
Validation of ELN-ZFP clones.
VSMC express ELN and play a crucial role in elastogenesis within the blood vessel wall. To evaluate the effects of our ZFP constructs more efficiently in these cells, we generated lentiviral constructs encoding ELN-ZFP constructs one and three. As shown (Fig. 3A), transduction with ELN-ZFP3 lentivirus increased elastin mRNA >8-fold over basal expression levels in VSMC, as compared to transduction with a viral vector encoding VP16 alone (no DNA-binding fingers) or vehicle alone (no virus). There were no effects on the expression of collagens COL1A1 and COL3A1, demonstrating that gene activation by the ELN-ZFPs was not a generalized effect involving all matrix components. ELN-ZFP3 also induced marked increases in both cell-associated elastin protein and soluble tropoelastin protein (Fig. 3B). ELN-ZFP1 was less effective in these cells, a finding that may reflect the ∼10-fold lower expression of this ZFP in these cells when compared to ELN-ZFP3 (based on quantitative reverse transcriptase polymerase chain reaction [q-RT-PCR] analysis of VP16 expression as a marker of ELN-ZFP expression levels). The reason for this reduced expression of ELN-ZFP1 is unclear, but for experiments evaluating the effects of ELN-ZFP on splice variant expression and nonsense mediated decay (see below) we used ELN-ZFP3, the construct that demonstrated the highest efficacy in our initial testing.
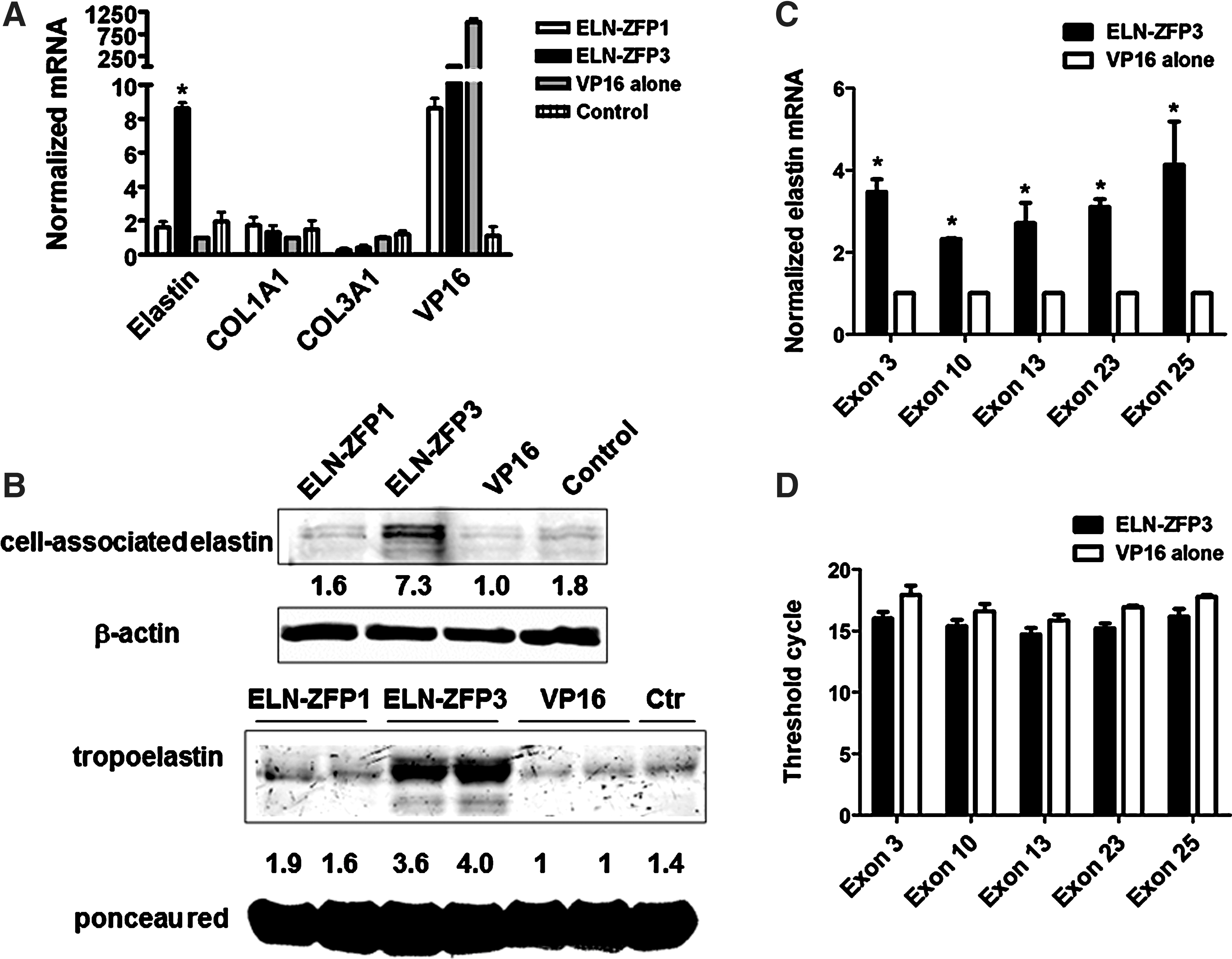
ELN-ZFP–mediated induction of ELN expression in vascular smooth muscle cells (VSMC).
One of our expectations was that endogenous activation of the ELN gene would lead to expression of all the major splice variants and not to a selected population of these variants. To test this we used exon-specific primers to determine by q-RT-PCR the expression levels of key ELN exons that are spliced out in specific ELN variants. As seen in Figure 3C and D, elastin mRNAs containing the known exons 3, 10, 13, 23, and 25 were significantly increased in ELN-ZFP3 transduced cells, and the stoichiometric relationship between them maintained.
Rescue of ELN haploinsufficiency in WBS and SVAS cells
Next, we examined the ability of ELN-ZFP3 to induce compensatory expression from the intact wild-type allele in dermal fibroblast cells isolated from a patient with WBS. Using comparative genomic hybridization (CM-CGH) testing through the Yale Genetics Core, we verified that these cells have a typical heterozygous ∼1.5 Mb microdeletion at 7q11.23 encoding ∼25 genes, including ELN (Pober, 2010). These elastin-haploinsufficient cells express only 26–36% of elastin mRNA compared to age-matched normal fibroblast cells from the same bank. ELN-ZFP3 increased elastin mRNA close to 7-fold and cell-associated elastin protein by 4.9-fold, levels close to that seen in normal cells (Fig. 4A and B). ELN-ZFP1 increased elastin mRNA significantly, but to a much lesser extent in these cells. Both ELN-ZFPs appeared to increase COL3A1 mRNA levels to a small extent, although not COL1A1 in these WBS cells. Strong augmentation of ELN mRNA by ELN-ZFP3 was also seen in dermal fibroblasts from another WBS patient in comparison to levels in matched normal cells (12.2±2.2 vs 1.5±0.7, p<0.05, graph not shown).
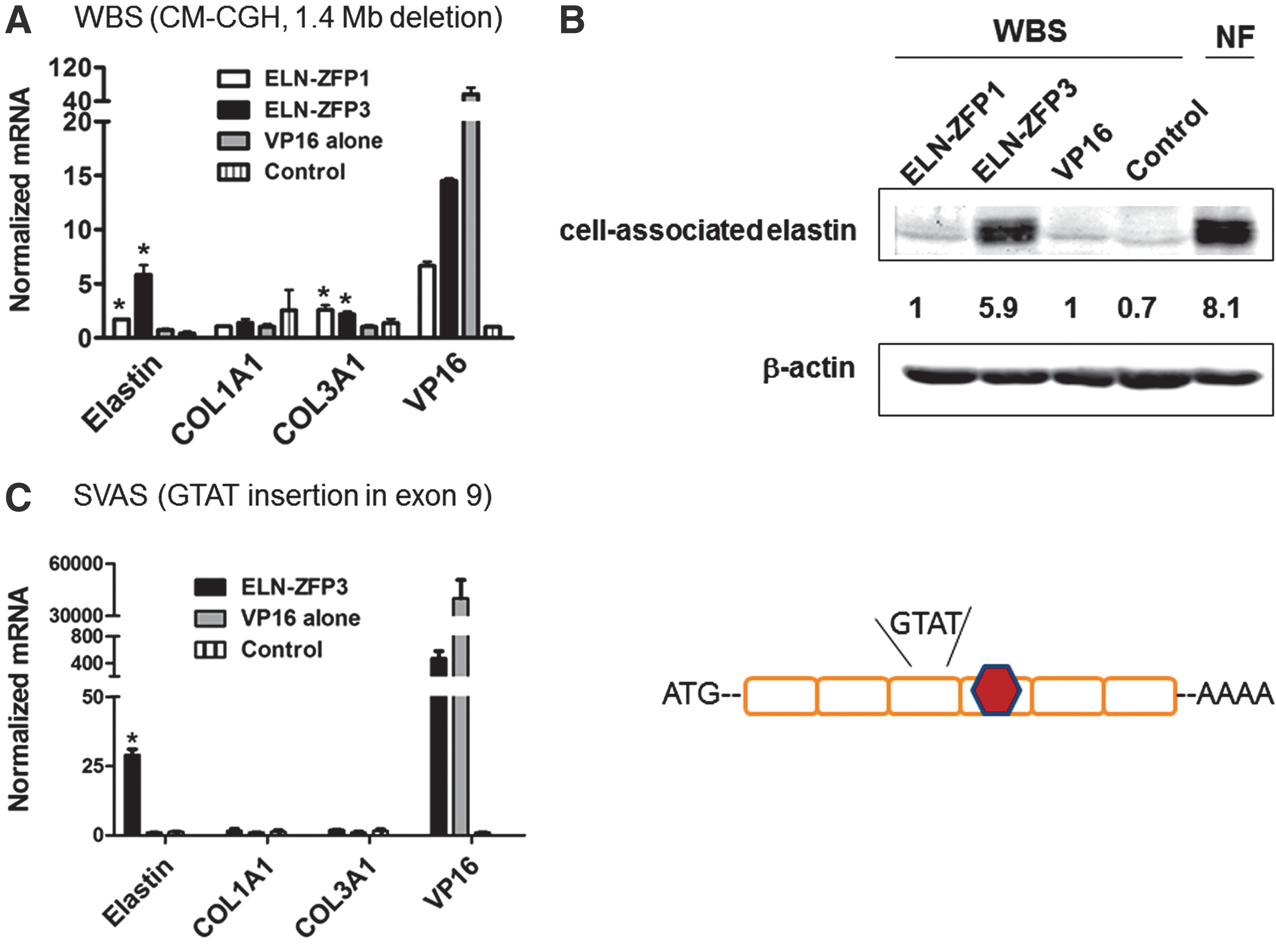
ELN-ZFP3 induces compensatory ELN expression from the wild-type allele in haploinsufficient WBS and SVAS cells.
We also examined the ability of ELN-ZFP to increase ELN expression in pulmonary VSM cells isolated from a patient with SVAS. In this particular patient, ELN haploinsufficiency is caused by a haploid 4-bp insertion (GTAT) in exon 9 of the ELN gene, causing a frameshift and a consequent premature termination codon (PTC) in exon 10. This PTC targets the mutant transcript to nonsense-mediated decay (NMD), thus leading to the ELN haploinsufficiency. NMD of the mutant transcripts is the major mechanism causing ELN haploinsufficiency in most cases of SVAS. Transduction of these SVAS cells with lentivirus encoding ELN-ZFP3 induced an ∼5-fold increase in elastin mRNA from these cells as compared to cells transduced with a control virus, while inducing no significant changes in collagen synthesis (Fig. 4C).
Effects of ELN-ZFP on nonsense mediated decay of mutant ELN transcripts
A major mechanism preventing the expression of potentially deleterious mutant proteins in many genetic syndromes, including the majority of patients with SVAS, is nonsense-mediated decay (NMD). In NMD, mutant genes are transcribed, but gene mutations that cause a reading frameshift and a PTC result in rapid clearance of the mutant transcripts—before translation into mutant proteins. In haploid genetic syndromes in which whole genes are deleted, such as WBS, mutant transcripts are not made and thus NMD is not required. Conversely, for many ELN mutations that result in SVAS, and in many other genetic syndromes, NMD is essential to prevent mutant protein production.
The DNA sequence targeted by the ELN-ZFPs is present in the regulatory region of the mutant as well as the wild-type allele, thus, the ELN-ZFPs could conceivably increase expression of the mutant ELN transcripts (Tassabehji et al., 1997; Urban et al., 2000). This raises the question whether ELN-ZFP–mediated increases in transcription of mutant ELN mRNA could overwhelm clearance by NMD and lead to production of a mutant ELN protein. This possibility is a crucial consideration in assessing the feasibility of any strategy to treat haploinsufficiency by augmenting transcription when there is a mutant allele present. To investigate this, we evaluated the effects of ELN-ZFP3 on levels of the mutant ELN transcript in SVAS cells, in the presence and absence of the translation inhibitor emitine. NMD occurs when a PTC is encountered during translation. Thus, blocking translation also blocks NMD (Noensie and Dietz, 2001). SVAS cells were transduced with either ELN-ZFP3 or control lentivirus constructs and then treated with emitine (100 μg/mL) 72 hours later. At various time points after blocking translation with emitine, levels of total elastin mRNA and of the mutant ELN transcript were evaluated using a combination of q-RT-PCR and acrylamide gel electrophoresis.
Compared to VP16 alone, ELN-ZFP3 transduced SVAS cells had a >4.5 fold increase in elastin mRNA prior to treatment with emitine. Interestingly, 7 hr post-emetine treatment, there was a further 63% increase in total elastin mRNA (Fig. 5A). Both ELN-ZFP3 and emetine treatments had negligible effect on COL1A1 mRNA in SVAS, although there appeared to be a slight increase (40%) in COL1A1 mRNA in ELN-ZFP3 transduced cells 7hr. post-emetine treatment. Standard agarose gel electrophoresis of PCR products using a specific primer pair around the mutation site after 30 cycles of amplification showed that both ELN-ZFP3 and emetine increased elastin mRNA in SVAS cells, although the mutant and wild-type alleles could not be resolved with this approach (Fig. 5B, top). After longer amplification (35 cycles) and electrophoresis on 12% acrylamide, the wild-type and mutant ELN mRNAs were well separated, allowing assessment of the effects of ELN-ZFP3 and emetine (Fig. 5B, middle panel). At baseline (72 hr post-transduction and pre-emetine), there was a barely perceptible mutant elastin mRNA band present, with no significant differences apparent between the ELN-ZFP and control virus transduced cells. Progressively, at incremental time points after addition of emetine, there was an increase in mutant ELN mRNA levels in both the ELN-ZFP and control-treated cells, although the increase was markedly greater in cells treated with the ELN-ZFP. These data demonstrate that the ELN-ZFPs do indeed increase transcription from the mutant ELN allele, but that this does not overcome the surveillance mechanism of NMD and is thus unlikely to result in the expression of a mutant elastin protein.
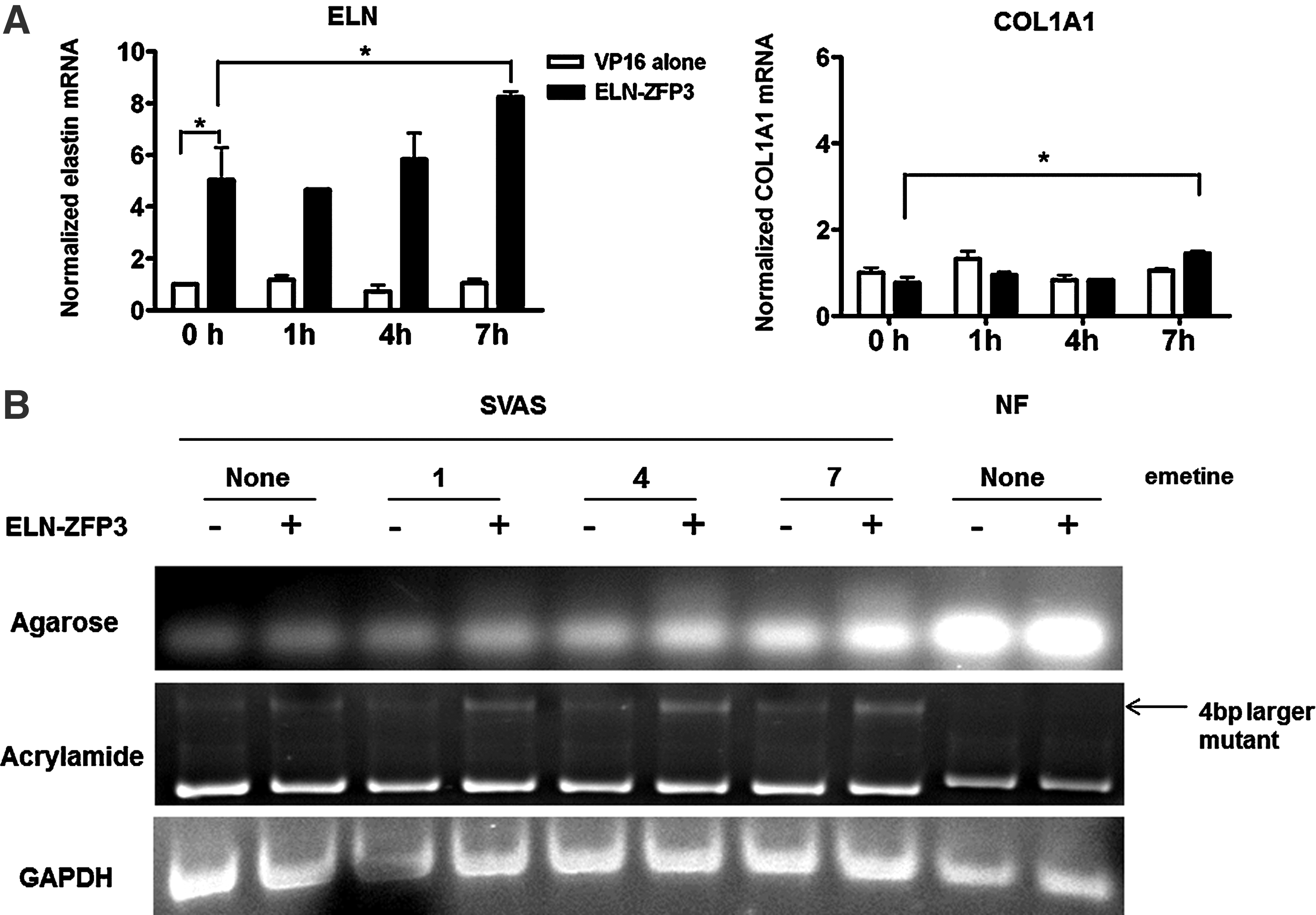
ELN-ZFP–treatment does not bypass nonsense-mediated decay (NMD). A 4-bp insertion in one ELN allele causes destruction of the mutant transcript by NMD, resulting in elastin haploinsufficiency and consequent SVAS. To determine if transcriptional activation of the mutant allele with an ELN-ZFP could bypass NMD, leading to an accumulation of mutant transcript, cells were transduced with ELN-ZFP3 and then 72 hours later treated with the translation and NMD inhibitor emitine. qRT-PCR of RNA collected at sequential time points
ELN-ZFP induction of elastogenesis in bioengineered blood vessels
To determine whether transcriptional activation of ELN could enhance elastogenesis we examined the effects of ELN-ZFP3 in bioengineered blood vessels being developed for clinical use (Niklason et al., 1999; Solan et al., 2009). These vessels are grown in bioreactors by seeding cells onto a PGA scaffold and exposing to pulsatile flow for extended periods (Niklason et al., 1999). Although they exhibit many crucial properties of human arteries, they do not express sufficient amounts of elastin and do not develop elastic lamellae. We transduced early passage human VSMC with lentivirus encoding either ELN-ZFP3 or the VP16 control, seeded ∼1×106 cells onto each PGA scaffold, and grew vessels under identical conditions in a bioreactor for 8 weeks.
After this 8-week period, immunofluorescence analysis demonstrated that ELN-ZFP3 treated vessels contained more of the matrix proteins fibronectin and fibrillin-1, and more elastin than controls (Fig. 6). This increase in elastin was corroborated at the ultrastructural level with transmission electron microscopy. Quantitatively, ELN-ZFP3–treated vessels had ∼twice the amount of desmosine, a marker of cross-linked elastin content, vs. controls (Fig. 6K). Thus mature elastin appeared to have been generated in these vessels. There was no concomitant increase in the amount of hydroxyproline, a marker of collagen content, and no difference in cell number in ELN-ZFP3 treated vessels vs. controls (Fig. 6L and M).

ELN-ZFP–mediated elastogenesis in bioengineered blood vessels. Low passage human vascular smooth muscle cells (VSMC) were transduced with lentivirus encoding ELN- ZFP3. Bioengineered blood vessels were grown in a bioreactor for 8 weeks after seeding these cells onto a polyglycolic acid (PGA) scaffolding. As shown by immunofluorescence, vessels grown from cells transduced with ELN-ZFP3
ELN-ZFP3 does not affect cellular proliferation or migration in vitro
Since we did not see a difference in cellular proliferation between ELN-ZFP3–treated vs. VP16-treated bioengineered vessels (Fig. 6M), we also examined the effects of ELN-ZFP3 on normal fibroblasts (NF) or WBS fibroblasts on the proliferative and migratory characteristics of these cells in culture (Fig. 7). Neither proliferation (measured by cell counts and by a WST-1 assay) or cellular migration (determined by scratch assay and corroborated by ECIS cell impedance experiments) was significantly altered by ELN-ZFP3 transduction, as compared to VP16 transduced and nontransduced cells (Fig. 7).
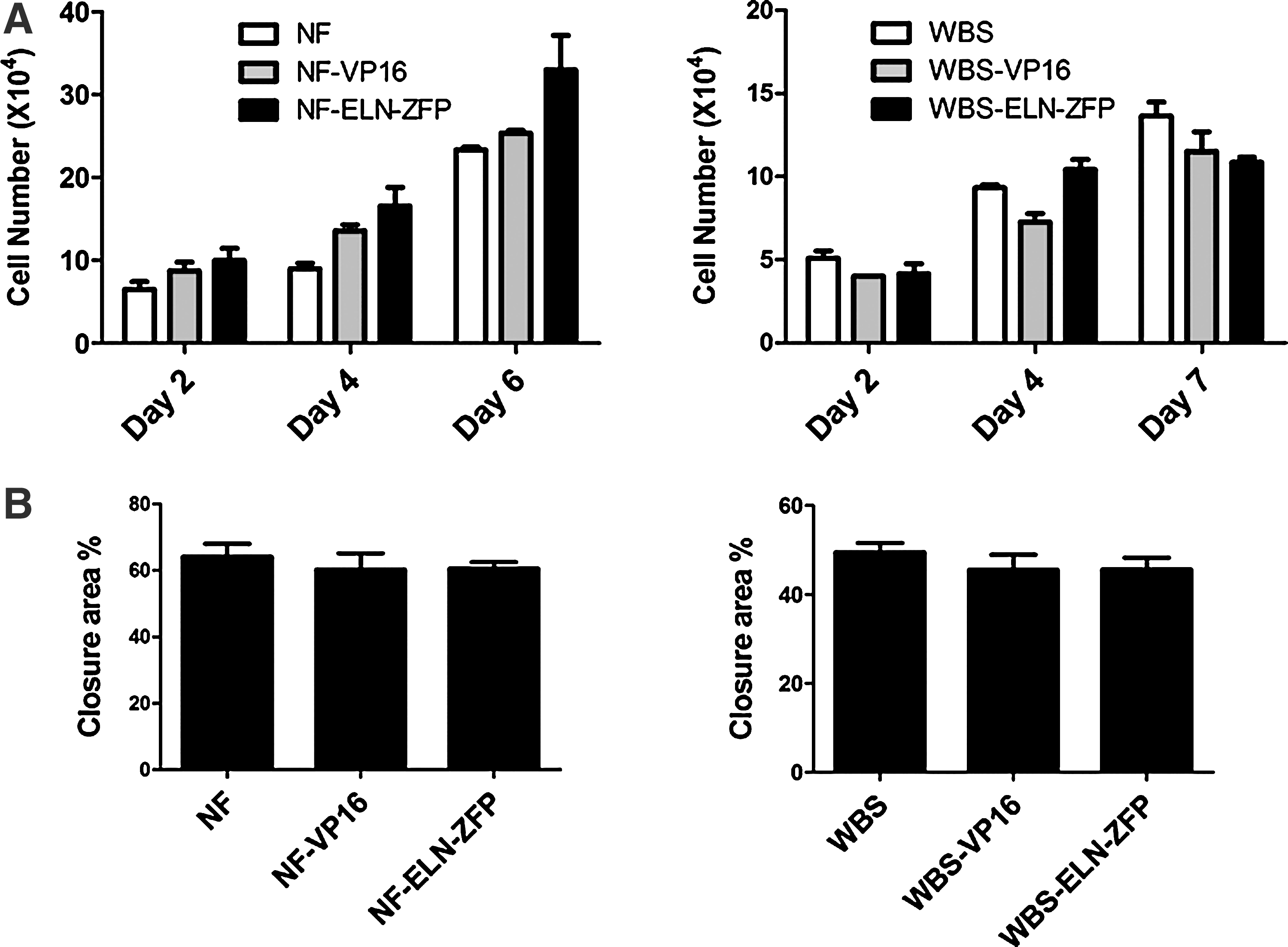
ELN-ZFP does not affect cellular proliferation or migration in vitro. Normal fibroblast cells (NF) or WBS fibroblast were transduced with lentivirus encoding ELN-ZFP3 or VP16.
ELN-ZFP3 is highly specific
Having established that ELN-ZFP3 can drive significant ELN activation in both wild-type and ELN haploinsufficient cells, we next evaluated its functional specificity. Independent microarray experiments were performed to compare the genome-wide expression changes after ELN-ZFP3 transduction in human dermal fibroblasts. We found that among the 30K genes (764K probes) on the Affymetrix chip, only four (ELN, SERPINA3, PRSS35, and PTPRN) had significant (p<0.05) changes in gene expression after ELN-ZFP3 treatment, and only one (SERPINA3) showed a fold change greater than ELN (5.14-fold vs 2.07-fold; Table I). Interestingly, the human SERPINA3 gene also contains the same 10 bp DNA sequence targeted by ELN-ZFP3 (between exons 1 and 2; chr14:94133491-94133500; GRCh37 assembly of the human genome).
Global gene expresssion in ELN-ZFP3–tranduced human dermal fibroblasts (HDF) were compared to those in VP16-transduced HDF. Out of close to 30K probes on the GeneChip Human gene array, only four gene expressions had significant (p<0.05) changes.
Discussion
Here we show that targeted transcriptional activation of the intact wild-type allele with engineered ZFPs is a viable strategy to induce compensatory expression of ELN and rescue haploinsufficiency of this gene in two distinct genetic syndromes, WBS and SVAS. In SVAS, we provide evidence that concomitant transcriptional upregulation of the mutant allele does not bypass surveillance by nonsense mediated decay, and is thus not likely to lead to augmented expression of a mutant elastin protein. These are important preliminary steps in the potential development of targeted transcriptional strategies to treat haploinsufficiency syndromes. It remains unclear why heterozygous loss of specific genes leads to haploinsufficiency, while heterozygous loss of other genes is compensated by the remaining allele. It has also remained unclear whether the pathology of haploinsufficiency is primarily due to an absolute reduction in amount of the encoded protein, or is due to an altered stoichiometry of that protein relative to other proteins with which it interacts. This has raised concerns that attempted therapeutic overexpression of a haploinsufficient gene might lead to toxicities based on an inability to correct this stoichiometric imbalance. Work on haploinsufficiency in yeast supports the contention that haploinsufficiency is primarily a consequence of reduced protein production, but it is likely that stoichiometric balance is also important for specific haploinsufficient proteins (Deutschbauer et al., 2005). Targeted transcriptional activation of the wild-type endogenous allele with engineered ZFPs engenders several potential advantages in this context. First, the levels of induced expression are potentially titratable, either by using weaker or stronger transcriptional activation domains, altering the regulatory sequence targeted, or by altering specific properties of the ZFPs such as half-life or target sequence binding affinity. Second, if stoichiometry is indeed important, it is reasonable to assume that splice variant stoichiometry is also important, and endogenous gene activation by the engineered ZFP approach can recapitulate normal splice variant stoichiometry (Rebar et al., 2002).
In the case of ELN haploinsufficiency, the argument for targeted endogenous gene activation as a therapeutic approach is particularly strong, given the complexity of this gene and the number of potential splice variants. In a mouse model in which the entire human ELN gene was inserted, expression from this transgene partially reversed the cardiovascular phenotype associated with heterozygous deletion of the mouse Eln gene and rescued the lethality of the homozygous mouse Eln deletion (Hirano et al., 2007), supporting our ELN compensation strategy. Although these and other published findings strongly suggest that rescuing elastin haploinsufficiency can prevent WBS vascular pathology, further preclinical work in elastin haploinsufficient animal models (Hirano et al., 2007; Li et al., 1998a) will be required prior to making a leap to clinical testing of our ZFP approach or other approaches to increase elastin expression.
Indeed, several issues remain to be addressed before any attempted clinical application of engineered ELN-ZFPs, including how to deliver them to essential cells and tissues at specific developmental stages. A majority of SVAS and WBS patients develop vascular lesions, often requiring surgical intervention (Pober, 2010; Pober et al., 2008b). Despite the propensity to develop focal aortic stenosis just above the aortic valve, severe vascular stenosis can occur in many different arterial territories and can be recurrent. Thus, an effective therapeutic strategy would arguably necessitate an approach that achieves compensation for ELN haploinsufficiency throughout the susceptible vasculature, likely requiring systemic delivery. Expressing ZFPs from cDNA cassettes is effective, but in vivo systemic delivery of these cassettes to the vasculature is a significant hurdle, even with advanced generation viral vectors.
To prevent the development or recurrence of vascular lesions at specific locations, catheter-based local ELN-ZFP gene delivery to specific arterial segments could be accomplished, potentially with existing technology. However, a broader delivery strategy that achieves rescue of elastin haploinsufficiency throughout the susceptible vasculature would likely be significantly more effective, and is thus more appealing. One potential approach would be to deliver the ZFP-encoding cassettes during the prenatal or early postnatal period using a gene delivery method that achieved genomic integration and thus ELN-ZFP expression in progeny cells (e.g., lentivirus transduction). The goal would be to transduce enough cells such that after developmental proliferation there would be a sufficient number to rescue the elastin haploinsufficiency and to substantively decrease the propensity to develop vascular lesions. The potential risks of genomic integration would, however, have to be carefully considered. Another possible approach would be to stably transduce pluripotent cells from an affected fetus or early postnatal child (e.g., cord blood–derived stem cells) with the ELN-ZFP construct, and then deliver these cells back with hopes that they would integrate, differentiate, and express normal amounts of elastin. Although both of these strategies present challenging technical hurdles, the potential to modify genetic diseases with early intervention using these approaches is attractive and could be applicable to other genes in the WBS locus and to a significant number of additional genetic syndromes marked by haploinsufficiency.
Although the prenatal and early postnatal approaches described above could theoretically have a significant positive impact, a major consideration for any such approach is the ability to replicate the concise temporal and cell-specific expression patterns that normally occur during the fetal or early postnatal periods. Arguably, this is particularly important for elastogenesis, which is a complex process with tight spatial and temporal control. Although ELN-ZFP expression could be restricted to specific cell types by using tissue-specific promoters, replicating specific temporal patterns of ELN expression will be difficult. For point mutations more readily amenable to gene correction approaches (e.g., gene correction using ZFP endonucleases) (Urnov et al., 2010), correction of mutations in pluripotent cells in the fetal or early postnatal periods would have significant theoretical advantages, including intact temporal and cell-type specific gene expression control. For deletion syndromes, including those in which whole genes are lost (e.g. ELN deletion in WBS), gene correction would require whole gene replacement, and thus transcriptional activation (e.g., our ZFP approach) would be a more viable option. Despite the substantial complexities and technical hurdles, we maintain that transcriptional upregulation of the wild-type allele in WBS and other genetic syndromes that cause haploinsufficiency remains appealing. Advancements in technology, for example, delivering ZFPs as cell permeable proteins, may help to address specific hurdles to clinical implementation.
Effective treatment of adults, and children who are beyond the major developmental periods of elastogenesis, is dependent upon whether increasing ELN expression, by an ELN-ZFP or any means, will result in the effective production of new incorporated elastin, and whether this will have significant functional effects on the vasculature. On the positive side, we and others have shown that antagomirs to microRNA miR29 can increase elastin expression (Zhang et al., 2011), and one study demonstrated that this approach alters aortic aneurysm formation in a mouse model (Boon et al., 2011). This suggests that functionally significant elastin production can occur in an established vasculature. Nonetheless, whether induction of ELN expression with ZFPs in a child or adult can prevent or modify WBS or SVAS vascular lesions remains unclear. Underscoring the biological complexity and thus clinical challenge is the broad phenotypic variability in patients with WBS or SVAS. Although it is now well established that ELN loss or mutation is the primary driver of the vascular lesions in these genetic syndromes, there are likely multiple modifiers that contribute to the phenotype variation. A recent study using quantitative trait locus analysis in mice identified several potential genetic modifiers of the ELN deficiency vascular phenotype, including genes involved in reactive oxygen species generation (Kozel et al., 2011). We and others have identified several additional factors that can modify ELN expression at transcriptional and post-transcriptional levels, including specific miRNA species, growth factors, and control of the cell cycle (Kuang and Goldstein, 2003; Sen et al., 2011; Shi et al., 2012; Zhang et al., 2011). The ELN promoter also contains a CpG island, and it is possible that methylation can epigenetically modify ELN expression.
Another necessary consideration is that the precise mechanism whereby elastin haploinsufficiency leads to vascular stenosis remains unclear. Nonstenotic areas of aorta in WBS show medial thickening, and areas of supravalvular aortic stenosis often demonstrate areas of increased smooth muscle cell proliferation along with fragmented elastic lamellae. Ex vivo examination of cultured arterial segments from elastin-deficient mice revealed increased rates of smooth muscle cell proliferation (Li et al., 1998a; Li et al., 1998b), and in vitro studies have shown that adding exogenous soluble elastin to the media can reduce the proliferation rate of cells from individuals with WBS and SVAS (Urban et al., 2002). These data have led to a widely held hypothesis that elastin insufficiency in the vessel wall leads to an increase in smooth muscle proliferation, thus leading to stenosis. However, the pathology of SVAS lesions can be variable, and this variability in vascular pathology, along with the identification of potential genetic modifiers, suggests that the mechanisms underlying vascular lesion formation involve more than this inverse relationship between elastin levels and smooth muscle proliferation. In our studies, despite marked induction of elastin mRNA and protein, we did not see a reduction in cellular proliferation in normal fibroblasts, or those from a WBS patient when transduced with a lentivirus encoding ELN-ZFP. Whether this is a function of the specific cells we used, the fact that we relied upon endogenous tropoelastin production rather than exogenous application, or perhaps relates somehow to the specific pattern of ELN expression induced by the ELN-ZFP (e.g., specific splice variants) is unclear and will require further study in additional cell lines and conditions.
To be effective in treating heterozygous genetic haploinsufficiency syndromes, it is important that ZFP-mediated transcriptional activation not induce the expression of a potentially deleterious mutant protein. Our findings suggest that transcriptional activation of the mutant ELN allele by ELN-ZFP in SVAS does not overwhelm the capacity of the NMD mechanism to target destruction of the mutant transcripts. In WBS, with complete deletion of one ELN allele and consequently no mutant allele, ZFP-mediated transcriptional overriding of NMD is not a concern. For most SVAS mutations, however, NMD is the major mechanism preventing production of a mutant protein. Thus the possibility that transcriptional activation of the mutant allele can overcome NMD and result in production of a potentially pathological mutant protein is a significant and legitimate concern. Although our results demonstrate that this does not occur with the current batch of ELN-ZFPs in the specific SVAS cells we tested, the possibility that transcriptional activation could bypass NMD in cells with other SVAS mutations, or in different cell types, has not been ruled out. Further, the effects of transcriptional activation on NMD surveillance may be different for mutations in other genes, and this will have to be investigated further before concluding that NMD is generally unaffected by the ZFP approach. Whether strategies to increase mRNA half-life, such as our miR29 antagomir approach, will bypass NMD and lead to mutant protein production is also an unanswered question, and strategies to treat haploinsufficiency in this manner will also require clarification of this issue.
One prominent advantage of engineered ZFPs is that they are designed or selected for high affinity binding to specific target DNA sequences. For this study, we used a 3-finger ZFP targeted to a specific 9-bp DNA sequence, but the modularity of ZFP design allows us to make even longer ZFPs (Wright et al., 2006). The most effective ELN-targeted ZFP we constructed, ELN-ZFP3, significantly increased the expression of only four genes two-fold or greater out of ∼30,000 genes on an Affymetrix array, including ELN. One of the other 3, SERPINA3, contains our targeted sequence in the intron between exons 1 and 2. Another of these 3, PTPRN, contains a 9-bp sequence in its promoter that differs from our target sequence by one base. It is possible that binding of our ZFP to these sites directs the increased expression of these genes. The other method of ELN upregulation we have investigated, stabilization of elastin mRNA by an anti-miR29 antagomir approach, has potential advantages regarding the deliverability in a clinical context. Conversely, current antagomirs generally alter the expression of a significant number of genes and thus lack the specificity of the ZFP approach. Likely, technological advances will improve the clinical potential of each of these approaches. Further, given that ZFPs increase mRNA synthesis while the antagomirs stabilize mRNA transcripts, these approaches may prove to be therapeutically synergistic.
The ability to induce elastogenesis has broad clinical implications. The integrity of elastin is of crucial biological importance (Kielty, 2006), and degradation/loss of elastin is a feature of emphysema (Pierce et al., 1995; Snider, 2000), decreased skin elasticity with aging (Sherratt, 2009; Uitto, 2008), vascular aneurysms and other vascular pathologies (Hellenthal et al., 2009; Lee and Oh, 2010; Sawabe, 2010), and numerous additional clinical entities (Sherratt, 2009; Word et al., 2009). As elastogenesis is a complex process, involving both ELN expression and crucial post-translational processing, it was important to determine whether transcriptional upregulation of ELN could lead to the deposition of mature elastin in the extracellular matrix. The clinical use of bioengineered vessels as arterial conduits (e.g., coronary bypass surgery) is limited because of their paucity of elastin and consequent weak mechanical properties (Kielty et al., 2007). Our data establishes that increased deposition of cross-linked elastin in these vessels can be achieved by ZFP-induced ELN transcription. While an important first step toward the goal of achieving stronger bioengineered vessels, a significant amount of additional studies and optimization steps will be necessary before concluding that the ELN-ZFP methodology should be incorporated into the production of bioengineered vessels. Currently, we did not observe mature elastin lamella in these vessels and transduction efficiency in these experiments was suboptimal, but with optimization in transduction efficiency, and with combined efforts to improve other steps involved in elastogenesis, such as increasing the activity of cross-linking enzymes (LOX) and the expression of microfibril proteins, more natural patterns of mature elastin deposition may be achieved.
In summary, this study establishes that compensation for elastin haploinsufficiency in WBS and SVAS can be achieved by augmenting expression from the wild-type allele using engineered transcription factors. Further, this approach can be very highly specific and is unlikely to induce mutant protein production by overwhelming NMD surveillance. Finally, induction of ELN expression in this manner appears capable of stimulating elastogenesis. These findings have significant implications specific to WBS and SVAS and generally suggest that treating haploinsufficiency states by inducing expression of the wild-type endogenous allele may eventually prove to be a viable clinical strategy.
Footnotes
Acknowledgments
This work was funded by a joint award from Marshall and Johanna Kiev and the Williams Foundation and by National Institutes of Health RO1 HL075616-02 (FJG).
We thank Robert P. Mecham for kindly providing human ELN antibody; Scot A. Wolfe for consultation in bacterial one-hybrid screening; George Tellides for insightful review of the work; and Marshall, Johanna, and Chloe Kiev, without whom this work would not have been initiated.
Author Disclosure Statement
There are no conflicts of interest to report.