
Abstract
Genes encoding transcription activator-like effector (TALE) proteins may be engineered to target specific DNA sequences. TALEs fused with a transcription activator can be used to specifically induce the expression of a gene. This could lead to completely new therapies for several diseases. We have applied this potential therapeutic approach to Friedreich ataxia (FRDA), as an example. FRDA is due to reduced expression of frataxin because of elongation of a trinucleotide (GAA) repeat in intron 1. Our aim was to develop a potential treatment for FRDA by increasing the expression of the frataxin gene. We engineered 12 TALE genes (TALEFrat) encoding TALEFrat proteins, each specifically targeting different 14-bp DNA sequences within the proximal region of the human frataxin promoter. When the genes encoding these TALEFrat proteins were fused with a transcription activator, that is, four VP16 peptides (i.e., VP64), the resulting TALEFrat-VP64 proteins induced the expression of an mCherry reporter gene fused to a mini-cytomegalovirus promoter able to be activated by the insertion of the frataxin proximal promoter upstream to the minipromoter. These TALEFrat-VP64 proteins also increased, by 2- to 3-fold, frataxin gene expression (detected by qRT-PCR) in the cells. We conclude that TALEFrat proteins targeting the frataxin promoter may be used to increase the expression of frataxin mRNA and potentially could alleviate the symptoms of Friedreich ataxia. TALE methodology opens a new field of research, which could be used to develop TALE proteins to treat other diseases by inducing the expression of specific genes.
Introduction
As an example of this therapeutic approach, we have applied this strategy to Friedreich ataxia (FRDA). FRDA is an autosomal recessive neurodegenerative and cardiac disease, caused by a trinucleotide (GAA) repeat expansion in the first intron of the frataxin gene located on chromosome 9 (Campuzano et al., 1996). The mutation leads to reduced expression of the frataxin gene without changing the protein. The pathological mechanisms have been reviewed by Pandolfo (2006, 2012). Frataxin is essential for proper functioning of mitochondria (Campuzano et al., 1997). It is involved in the removal of iron and when frataxin is reduced, the iron builds up and causes free radical damage. Neurons and cardiomyocytes are particularly sensitive to these deleterious effects (Wallis et al., 1989; Rotig et al., 1997; Becker and Richardson, 2001). In the classic form, FRDA symptoms appear in or before the second decade of life. It is characterized by ataxia, areflexia, loss of vibratory sense and proprioception, and dysarthria (Harding, 1981; Pandolfo, 1999; Lynch et al., 2002; Cooper and Schapira, 2003; Babady et al., 2007). Moreover, patients with FRDA often have systemic involvement, with cardiomyopathy, diabetes mellitus, and scoliosis. Early death can result from cardiomyopathy or associated arrhythmias (Harding, 1981; Singh et al., 2001).
A potential treatment of Friedreich ataxia is thus to increase the expression of frataxin, using TALE proteins coupled with a transcription activator, for example, four VP16 sequences (VP64) and specifically targeting the human frataxin promoter. In the present paper, we show that several plasmids encoding such TALEFrat-VP64 proteins effectively increase the expression of a reporter gene placed under the control of the frataxin promoter and that some of them were able to increase transcription of the frataxin gene in human cells.
Materials and Methods
Construction of TALEFrat genes targeting the frataxin promoter
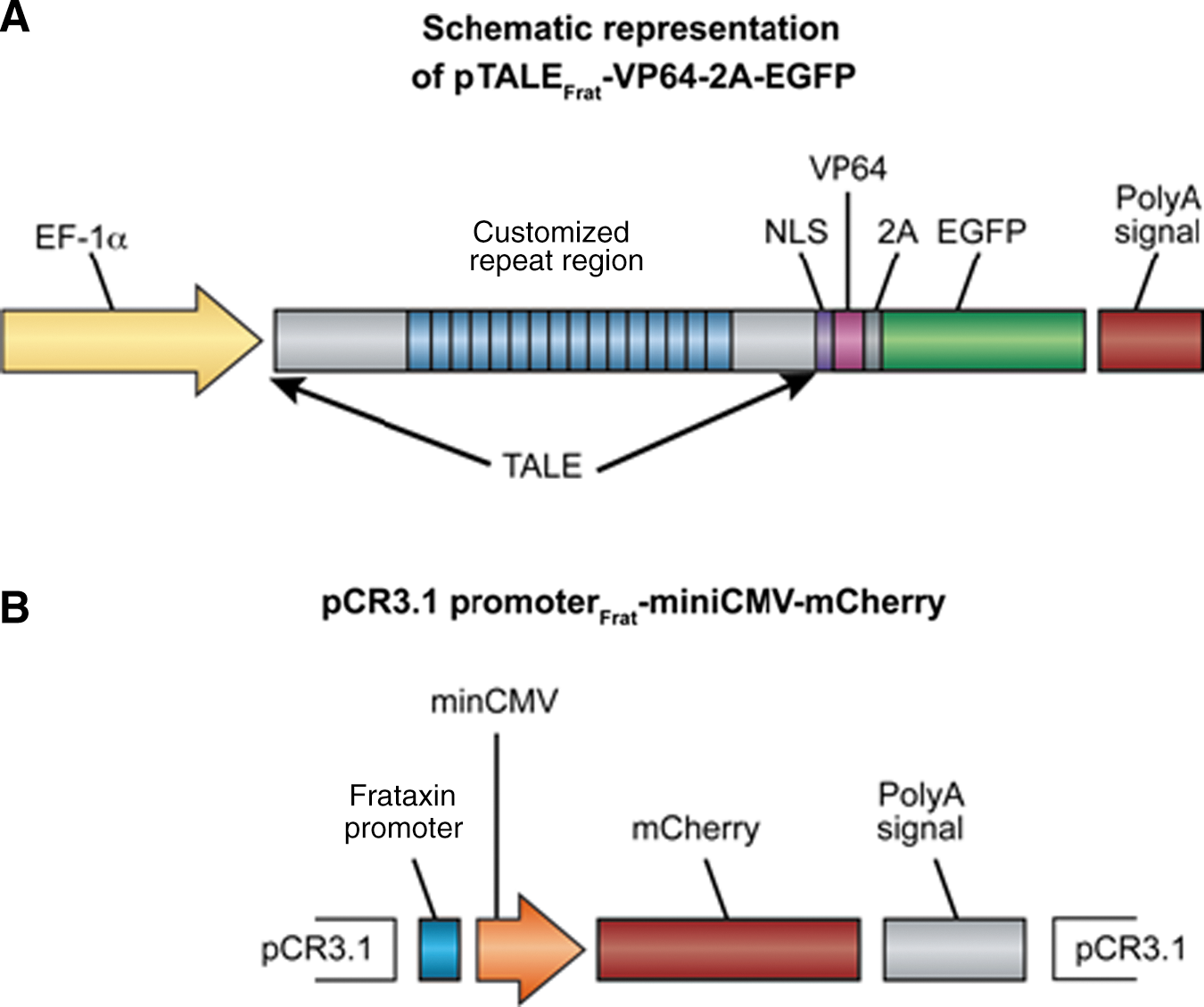
We have engineered 12 TALEFrat genes (Table 1) targeting the proximal region of the human frataxin promoter (Fig. 1), using a technique described by Zhang and colleagues (2011) and using reagents distributed by Addgene (Cambridge, MA). The final TALE expression vectors were the same as those originally used by Zhang and colleagues (2011), containing a nuclear localization signal (NLS), a transcriptional effector domain (VP64), and a 2A sequence followed by an enhanced green fluorescent protein (EGFP)-encoding gene (Paul et al., 1998; Furler et al., 2001). Thus the final vectors (pTALEFrat-VP64-2A-EGFP) produced two separate proteins (TALEFrat-VP64 and EGFP) from a single mRNA (Fig. 2A).
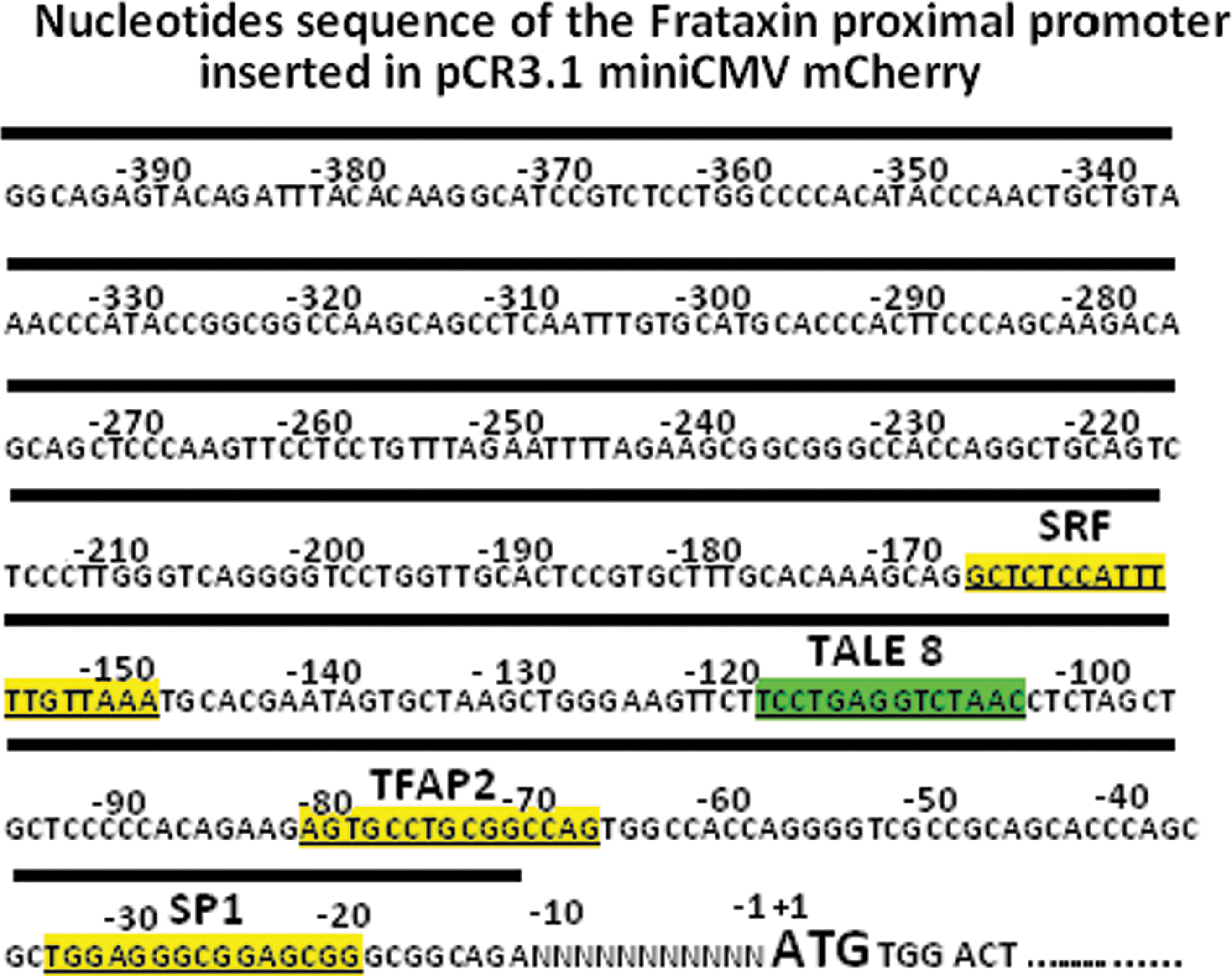
Sequence of the proximal region of the frataxin promoter. The sequence was obtained from GenBank NM_000144. The binding sites of TALEFrat #8 and of three transcription factors, that is, serum response factor (SRF), transcription factor-activating enhancer-binding protein-2 (TFAP2), and specificity protein-1 (SP1), are indicated. Binding sites of the transcription factors were identified by K. Li and colleagues (2010). The sequence under the dark band has been introduced into the pCR3.1-promoterFrat-miniCMV-mCherry reporter plasmid. Color images available online at
Schematic representation of
Summarized here are the results with various TALEFrat-VP64 proteins, the positions of the sequences targeted (according to NCBI reference sequence NM_000144.4), and the corresponding repeat-variable diresidue (RVD). 293FT cells in 24-well plates were cotransfected with 1 of the 12 p. TALEFrat-2A-EGFP plasmids and with pCR3.1-promoterFrat-miniCMV-mCherry. Red and green fluorescence levels were determined by flow cytometric analysis for the various p. TALEFrat-VP64 proteins as described by Zhang and colleagues (2011). Mean red fluorescence intensity is indicated in column 5 and mean green fluorescence intensity is shown in column 6 for cells located in quadrant 2, which express both red and green fluorescence. In column 7, the ratio of column 5 to column 6 data indicates how strongly TALEFrat-VP64 induced expression of the reporter gene. It is interesting to note that TALEFrat proteins 6 to 8, which induced the strongest mCherry expression, are targeting sequences found next to each other (indicated in boldface).
Construction of reporter vector
We have modified the TALE-VP64 transcription activator system reporter vector named TALE mini-CMV promoter mCherry provided by Addgene by replacing the original pAAV vector by pCR3.1 (Invitrogen Canada, Burlington, ON, Canada). The new reporter vector was named pCR3.1-promoterFrat-miniCMV-mCherry (Fig. 2B). It contains the proximal region of the frataxin promoter (385 bp) (Fig. 1) and was constructed as follows. The miniCMV-mCherry sequence from the plasmid pAAV-minCMV-mCherry (Addgene) was cloned into the pCR3.1 vector from which the original long cytomegalovirus (CMV) promoter had been removed. The proximal region of the frataxin promoter was amplified by PCR with Taq DNA polymerase (New England BioLabs, Ipswich, MA) from the genomic DNA of normal human cells with primers containing an XbaI site at the 5′ end (5′-AGTCTAGAGGCAGAGTACAGATTTACACAAGGCA) and a BamHI site at the 3′ end (5′-GGATCCTCTGCCGCCCGCTCCGCCCTCCAGCGCTG-3′), producing a 380-bp fragment. This promoter amplicon was then digested with XbaI and BamHI and cloned between the XbaI and BamHI sites of the pCR3.1-miniCMV-mCherry vector, that is, before the miniCMV-mCherry nucleotide sequence.
Quantitative real-time PCR for frataxin mRNA
qRT-PCR was done according to a technique previously described by Luu-The and colleagues (2005). Briefly, total RNA was extracted with an RNeasy mini kit (Qiagen, Valencia, CA). RNA quality was assessed with an Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, CA). First-strand cDNA synthesis was done with 5 μg of isolated RNA. Frataxin mRNA was amplified with primers designed from GenBank sequence NM_000144 (Fig. 1). Four reference RNAs were also amplified, that is, ribosomal RNA (18S), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), hypoxanthine phosphoribosyltransferase (HPRT), and glucose-6-phosphate dehydrogenase (G6PD) (see Table 2 for primer sequences). All experiments for qRT-PCR analysis were performed with total RNA extracted from cells cultured in 6-well plates.
S/AS, sense/antisense.
Results
Expression of TALEFrat-VP64 proteins
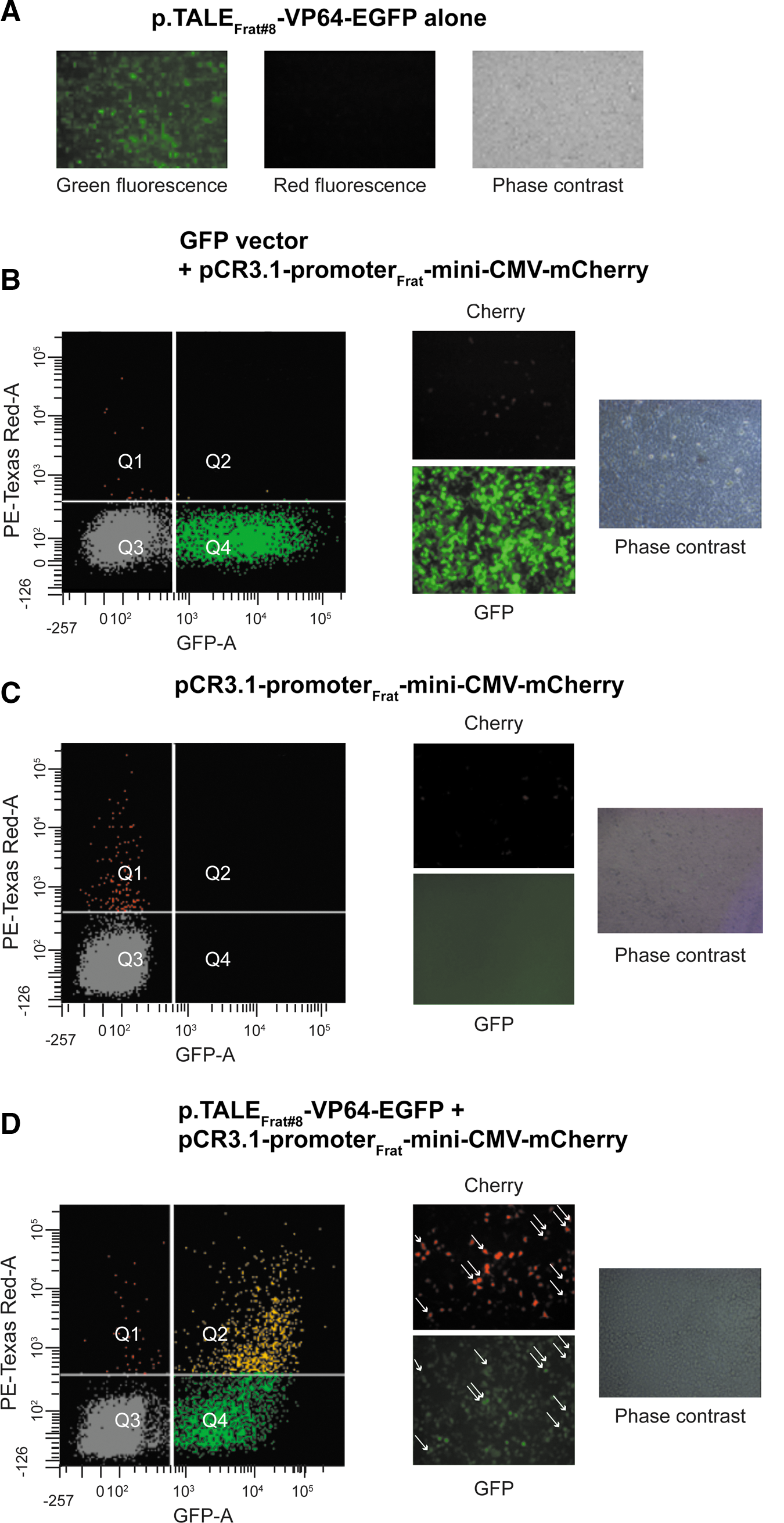
Table 1 summarizes the nucleotide sequences, in the proximal region of the human frataxin promoter, that have been targeted by TALEFrat proteins and correspond to 14 repeat-variable di-residues (RVD) (Cermak et al., 2011). When a p.TALEFrat-VP64-2A-EGFP expression plasmid was transfected alone into human cells (293FT cells), only green fluorescence was detected (Fig. 3A). The presence of green fluorescence confirmed the expression of EGFP protein, thus indirectly confirming the expression of TALEFrat-VP64 protein.
Activation of the pCR3.1-promoterFrat-miniCMV-mCherry reporter in 293FT cells by pTALEFrat #8. Analysis was done by flow cytometry
TALEFrat-VP64 proteins induce expression of a reporter gene under the control of the frataxin promoter
The pCR3.1-promoterFrat-miniCMV-mCherry reporter plasmid was initially transfected at 30 ng/ml with 2 μl of Lipofectamine 2000 into human cells in 24-well plates (i.e., 293FT cells) with a GFP vector (500 ng) (Fig. 3B) or alone (Fig 3C). In both cases, few cells expressed red fluorescence. However, when this reporter plasmid was cotransfected into human cells with one of the p.TALEFrat-VP64-2A-EGFP expression plasmids (500 ng), green fluorescence was detected, confirming expression of the TALEFrat-VP64 protein. Moreover, a much higher number of cells also expressing mCherry (red fluorescence) were also detected (Fig. 3D). This indicated that TALEFrat-VP64 was attaching to the frataxin promoter sequence, inducing expression of the mCherry gene. Table 1 summarizes the results. The intensity of red fluorescence was normalized by dividing it by the green fluorescence intensity to compensate for variations in the efficacy of transfection. TALEFrat #6, #7, and #8 induced the strongest expression of mCherry. The best TALEFrat proteins were targeting the frataxin proximal promoter sequences close to each other between the binding sequences of the serum response factor (SRF) and transcription factor-activating enhancer-binding protein-2 (TFAP2) transcriptional factors identified by K. Li and colleagues (2010a).
TALEFrat-VP64 proteins increase expression of frataxin mRNA
Several plasmids encoding TALEFrat-VP64 proteins were further investigated by qRT-PCR for induction of frataxin gene expression. 293FT cells were transfected with a plasmid encoding one of the p.TALEFrat-VP64-2A-EGFP expression plasmids. The transfection was done in a 6-well plate with 4 μg of plasmid, using 10 μl of Lipofectamine 2000. mRNAs were extracted 48 to 55 hr later. Frataxin, HPRT, and GAPDH mRNAs, and ribosomal RNA, were quantified by qRT-PCR. Frataxin mRNA was amplified by qRT-PCR and normalized relative to three different housekeeping mRNAs (HPRT1, G6PD, and GAPDH) and the ribosomal RNA (18S) in the same sample. The results were then further normalized with those obtained from 293FT cells not transfected with a plasmid (Table 3). TALEFrat #8 gave a consistent increase relative to all reference RNAs, ranging from 1.7- to 3.1-fold.
18S, 18S ribosonal RNA; G6PD, glucose-6-phosphate dehydrogenase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HPRT1, hypoxanthine phosphoribosyl transfeiase-1; TALEFrat, transcription activato-like effector targeting the frataxin prometer.
qRT-PCR results were normalized relative to different housekeeping genes in 293FT cells not transfected with a TALEFrat plasmid. TALEFrat #8 (boldface) produced the best increase in frataxin mRNA relative to the four housekeeping genes, the results varying between 1.7- and 3.1-fold increases.
Additional experiments were made with the three TALEFrat plasmids that gave the best results in the induction of mCherry. In these experiments, 293FT cells were either not transfected, transfected with a control plasmid encoding only EGFP (pCR3.1-EGFP), or transfected with one of the p.TALEFrat-VP64-2A-EGFP expression plasmids (#6, #7, and #8). Frataxin mRNA results were normalized with those of GAPDH mRNA, which was the most consistent under the various types of conditions. Two independent experiments were made for TALEFrat #6 and #7, whereas three experiments were made for TALEFrat #8. To compare the results from one experiment with those of another, the results were normalized either with nontransfected cells or with cells transfected with the EGFP plasmid (Fig. 4). Only TALEFrat #8 produced a consistent increase in frataxin mRNA relative to nontransfected cells (i.e., 1.7- to 3.1-fold increases; Fig. 4, left) and relative to EGFP-transfected cells (1.9- to 2.1-fold increases; Fig. 4, right). Thus TALEFrat #8 significantly increased expression of frataxin mRNA by roughly 2- to 3-fold above control values.
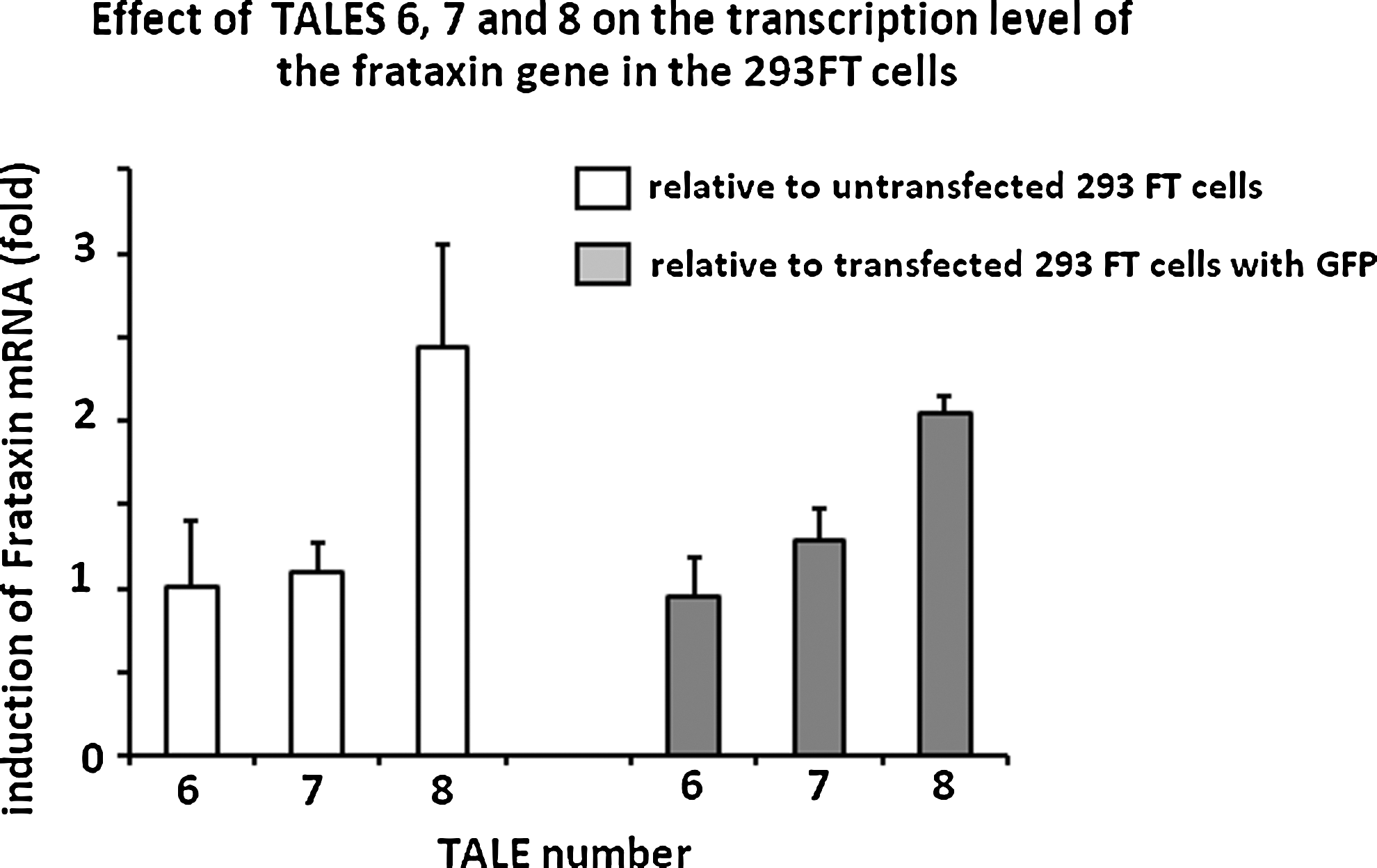
Induction of frataxin mRNA by p.TALEFrat-VP64-EGFP in 293FT cells. Frataxin mRNA was quantified by qRT-PCR. Results were normalized with the GAPDH housekeeping gene. Control cells were either not transfected or transfected with pCR3.1-EGFP plasmid. Frataxin mRNA was normalized with both types of control cells. Two or three independent experiments were done for each condition. TALEFrat #8 consistently increased frataxin mRNA up to 2- to 3-fold.
Discussion
Friedreich ataxia is due to reduced expression of frataxin following expansion of a trinucleotide (GAA) repeat in intron 1. Frataxin is a mitochondrial protein, and its reduction leads to a mitochondrial malfunction leading in turn to oxidative stress and accumulation of iron in the mitochondria. These changes, particularly the increased oxidative stress, lead to cell death, including that of neurons and cardiomyocytes. This progressive cell death results in several neurological and cardiac symptoms. Thus all the symptoms of Friedreich ataxia are due to the reduction of frataxin.
A new method to increase frataxin expression is to engineer genes encoding TALE proteins targeting specific sequences in the frataxin promoter coupled with a transcription activator. To identify the target sequence within the human frataxin promoter to which a TALE-VP64 could bind to produce the largest increase in frataxin transcription, we have targeted all possible sequences within the proximal promoter that start with a thymidine. We have, however, not targeted regions of the promoter to which transcription activators (SRF, TFAP2, and specificity protein-1 [SP1]) bind. Our results clearly demonstrate that several different TALEFrat-VP64 proteins effectively increased expression of a reporter gene under the control of the human frataxin promoter. Moreover, TALEFrat #8 increased expression of the frataxin gene by 2- to 3-fold in human cells. It is important to note that such an increase is significant. Indeed, depending on the length of the trinucleotide repeat, patients with Friedreich ataxia produce 5–30% of the normal frataxin level. However, heterozygous carriers of this disease produce roughly 50% of the normal frataxin level and are asymptomatic. Thus, for the majority of patients an increase of 2- to 3-fold in the production of frataxin would represent a level of production comparable to that of carriers.
Our results suggest that TALEFrat-VP64 protein #8 should be able to increase expression of the frataxin gene in patient cells. Increased expression of frataxin may reduce or completely prevent the symptoms of Friedreich ataxia. However, for therapeutic applications TALEFrat-VP64 will have to be expressed in most of the cells of patients with FRDA, especially the neurons and cardiomyocytes, to prevent the development of nervous system and cardiac symptoms. There are two potential methods by which to obtain such intracellular expression. The first is to deliver the TALEFrat-VP64 gene systemically, using an adeno-associated viral (AAV) vector. However, immune response against AAV-infected cells has been reported (Herzog, 2007; Hauck et al., 2009; Ohshima et al., 2009) and large-scale production of AAV vector under Good Manufacturing Practice conditions is still problematic. A second solution would be to couple the TALEFrat-VP64 with a cell-penetrating peptide (CPP) such as Tat or Pep-1 (Becker-Hapak et al., 2001; Morris et al., 2001; Vives et al., 2003; Tilstra et al., 2007). These fusion proteins could be delivered systemically; they would enter the cells and, it is hoped, drive the expression of frataxin. The fusion proteins would, however, have to be readministered on a regular basis.
The therapeutic approach that we are proposing for FRDA could also be used to treat any disease in which the mutation leads to a reduction of mRNA without changing the sequence encoding a protein. In addition, any hereditary and nonhereditary diseases, in which the increased expression of a compensatory gene would produce beneficial effects, could be a target for such a therapeutic approach. For example, the expression of utrophin could be induced with a TALE targeting its promoter to compensate for the absence of dystrophin in the muscle fibers of patients with Duchenne muscular dystrophy (Tinsley et al., 1996).
Footnotes
Acknowledgment
This work was supported by a grant from the Canadian Association of Familial Ataxia (CAFA).
Author Disclosure Statement
A patent has been applied for, for the treatment of FRDA with TALEs.