
Abstract
We have previously shown that injury-induced neointima formation was rescued by adenoviral-Nogo-B gene delivery. Integrase-competent lentiviral vectors (ICLV) are efficient at gene delivery to vascular cells but present a risk of insertional mutagenesis. Conversely, integrase-deficient lentiviral vectors (IDLV) offer additional benefits through reduced mutagenesis risk, but this has not been evaluated in the context of vascular gene transfer. Here, we have investigated the performance and genetic safety of both counterparts in primary human vascular smooth muscle cells (VSMC) and compared gene transfer efficiency and assessed the genotoxic potential of ICLVs and IDLVs based on their integration frequency and insertional profile in the human genome. Expression of enhanced green fluorescent protein (eGFP) mediated by IDLVs (IDLV-eGFP) demonstrated efficient transgene expression in VSMCs. IDLV gene transfer of Nogo-B mediated efficient overexpression of Nogo-B in VSMCs, leading to phenotypic effects on VSMC migration and proliferation, similar to its ICLV version and unlike its eGFP control and uninfected VSMCs. Large-scale integration site analyses in VSMCs indicated that IDLV-mediated gene transfer gave rise to a very low frequency of genomic integration compared to ICLVs, revealing a close-to-random genomic distribution in VSMCs. This study demonstrates for the first time the potential of IDLVs for safe and efficient vascular gene transfer.
Introduction
Nogo (RTN4)-spliced variants A, B, and C are members of the reticulon superfamily of transmembrane proteins associated primarily with the endoplasmic reticulon (ER) (Oertle and Schwab, 2003; Teng and Tang, 2008). Nogo-B has many biological roles; for example, it acts as a pro-apoptotic protein/tumor suppressor (Tagami et al., 2000; Li et al., 2001; Oertle et al., 2003; Watari and Yutsudo 2003; Tambe et al., 2004; Kuang et al., 2006); plays a role in the acute inflammation response (Di Lorenzo et al., 2010) and in pulmonary hypertension (Munoz and Zorzano 2011; Sutendra et al., 2011); is a regulator in vascular (Acevedo et al., 2004), renal (Marin et al., 2010) and hepatic (Zhang et al., 2011) injuries; and plays roles in inflammation of the asthmatic lung (Wright et al., 2010; Xu et al., 2011) and inflammation and tissue repair in response to ischemia (Yu et al., 2009). Previous studies have documented high levels of Nogo-B in vascular endothelial cells (VECs), VSMCs (Acevedo et al., 2004) and monocytes/macrophages (Paszkowiak et al., 2007; Rodriguez-Feo et al., 2007; Yu et al., 2009).
Endogenous Nogo-B expression is important for vascular maintenance and remodeling (Acevedo et al., 2004), as well as cardiac function (Bullard et al., 2008). Indeed, mice deficient in Nogo-A/B demonstrated accelerated rates of NIF after acute vascular injury, which were attenuated/rescued by adenoviral gene transfer of Nogo-B (Acevedo et al., 2004). Nogo-B expression is down-regulated in human arterial atherosclerotic plaques (Rodriguez-Feo et al., 2007; Lee et al., 2009), stenotic lesions (Rodriguez-Feo et al., 2007), and lesions of aortic aneurysms (Pan et al., 2007), and therefore can be regarded as a biomarker for injury-induced NIF (Paszkowiak et al., 2007). Hence, Nogo-B is a therapeutic candidate gene in the vascular gene therapy setting for the prevention of the NIF following acute vascular injury.
Nogo-B acts as a positive and negative regulator of vascular cells by mediating the chemo-attraction of VECs (Acevedo et al., 2004; Miao et al., 2006; Zhao et al., 2010) and antagonizing the migration (Acevedo et al., 2004; Kritz et al., 2008) and proliferation (Kritz et al., 2008) of VSMCs, respectively. We have previously shown that NIF was rescued in a porcine vein graft model and a wire injured mouse model by adenoviral gene transfer of Nogo-B (Kritz et al., 2008).
In comparison to Ad5-based systems, there are many advantageous characteristics that suggest lentiviral vectors (LV) to be favorable gene delivery vectors for vascular gene transfer. LVs are highly efficient at transducing vascular cells (Dishart et al., 2003; Cefai et al., 2005; Qian et al., 2006; Yang et al., 2010) and have been considered as a useful vector for delivering therapeutic interventions to reduce NIF following acute vascular injury in vivo (Qian et al., 2006; Yang et al., 2010). LVs are integration-competent (ICLV) and integrate a transgene copy into the chromosome of the target cell, which allows for efficient, stable, and long-term transgene expression (Matrai et al., 2010).
In contrast, integration-deficient lentiviral vectors (IDLV) are very attractive gene transfer tools since they maintain the efficiency of gene transfer yet have a reduced frequency of integration (Yanez-Munoz et al., 2006; Wanisch and Yanez-Munoz, 2009). IDLVs mediate transient gene expression in proliferating cells (Nightingale et al., 2006; Philippe et al., 2006; Apolonia et al., 2007; Matrai et al., 2011) and provide stable and efficient transgene expression in nondividing cells (Philippe et al., 2006; Yanez-Munoz et al., 2006; Apolonia et al., 2007; Rahim et al., 2009; Matrai et al., 2011), with minimal integration frequencies (Yanez-Munoz et al., 2006; Apolonia et al., 2007; Wanisch and Yanez-Munoz 2009; Matrai et al., 2011) associated with a reduced risk of insertional mutagenesis or oncogenesis (Hacein-Bey-Abina et al., 2003a; Hacein-Bey-Abina et al., 2003b; Hacein-Bey-Abina et al., 2008; Howe et al., 2008; Kane et al., 2010).
Additionally, IDLVs offer an alternative yet promising option that would be highly advantageous in vascular cell gene transfer (George et al., 2006; White et al., 2007) associated with a higher safety profile. Large-scale studies denoting the integration pattern and genotoxicity of LVs have shown a substantially reduced frequency of insertional induced alteration of cellular genes compared to gammaretroviral vectors (Montini et al., 2006; Modlich et al., 2009; Montini et al., 2009; Balaggan et al., 2011). However, an inherent genotoxic risk based on vector design is still present (Nowrouzi et al., 2011). In particular, the interaction of the LV integration machinery with cellular factors may be responsible for the proposed cell type- and species-specific integration pattern of LV-based vectors (Lewinski et al., 2006; Bartholomae et al., 2011; Biasco et al., 2011; Biasco et al., 2012).
Integration-site (IS) analyses of the first clinical trial for treatment of adrenoleukodystrophy (ALD) with LV-based vectors has not revealed any insertional-induced side effects (Cartier et al., 2009). A comparative analysis of IS hotspots has demonstrated a mega base-wide LV integration preference without obvious skewing of oncogene targeting (Biffi et al., 2011). To date, large-scale studies of LV integration patterns were performed in cell lines and primary murine and human cells of the hematopoietic system (Bushman et al., 2005; Nowrouzi et al., 2011). Due to an inserted mutation in the LV integrase gene, the integration of IDLV-based vectors is not driven by the viral integrase but by transient DNA-double strand (DSB) sites in the host genome, with residual integrase activity leading to lower frequency of random integration frequency as compared to ICLVs (Yanez-Munoz et al., 2006; Wanisch and Yanez-Munoz 2009; Matrai et al., 2011). However, insertional mutagenesis remains a risk, even with such a rare integrating vector system; therefore, comprehensive genome-wide analysis of integration frequency and pattern of integrating and rare integrating vector systems in the target cell-type remains a requirement. The frequency of IDLV vectors to integrate as well as the integration pattern of ICLV vectors in human VSMCs is current unknown. This study addresses whether IDLV gene transfer of Nogo-B can mediate overexpression in VSMCs to induce phenotypic effects associated with a reduced risk of genotoxicity.
Materials and Methods
Materials
Human saphenous vein segments were obtained from patients, who gave informed consent, undergoing coronary artery bypass grafting. Ethical permission was obtained from the West Glasgow Ethics Committee (reference number: 06/S0703/110). Reagents were purchased from Sigma-Aldrich (Poole, United Kingdom) and tissue culture reagents from Gibco (Invitrogen, Paisley, United Kingdom), unless otherwise stated. All primers and probes (oligonucleotides) were obtained from MWG-Biotech (Ederberg, Germany). Nuclease-free water (not diethyl pyrocarbonate–treated) was purchased from Ambion (Applied-Biosystems/Ambion, Warrington, United Kingdom).
Plasmids
We used the lentiviral construct plasmid encoding the marker gene, enhanced green fluorescent protein (eGFP), (pHR'SIN-cPPT-SFFV-eGFP-WPRE) under the control of the spleen focus forming virus (SFFV) promoter, as published previously (Demaison et al., 2002). Nogo-B complementary DNA (haemagglutinin tagged) (Acevedo et al., 2004) was cloned into the lentiviral construct plasmid containing a multiple cloning site (MCS) (pHR'SIN-cPPT-SFFV-MCS-WPRE) (Ward et al., 2011) using BamHI and XhoI sites. All LVs were pseudotyped with the vesicular stomatitis virus glycoprotein (VSV-g) expressed by the pMD.G2 plasmid, as previously described (Demaison et al., 2002). The second-generation LV packaging plasmid containing either the wild-type integrase gene (pCMV delta R8.74) or integrase-deficient gene (pCMV delta R8.74 D64V) was used, as detailed elsewhere (Yanez-Munoz et al., 2006).
Lentiviral production and titre assay
Self-inactivating (SIN) HIV-1–based LVs were produced using a three-plasmid transient transfection system and concentrated by ultracentrifugation at 90353 × g (23,000 rpm) for 1 hr and 7 min at 4°C using a Beckmann ultracentrifuge, as previously described (Demaison et al., 2002; Buckley et al., 2008). LV stocks were titrated by infecting 293T cells (ATCC-LGC Standards, Middlesex, United Kingdom) with serial dilutions of LVs (Demaison et al., 2002). We quantified LV genomic copy number in DNA extracted from 293T cells, 3 days post-infection, by TaqMan quantitative polymerase chain reaction (PCR), as previously described, according to a published protocol for late reverse transcriptase amplicon quantification (Butler et al., 2001), and standard curves were generated using LV construct plasmid DNA as described (Butler et al., 2001). LV titres were calculated as infectious viral units per ml (iu/ml).
SMC isolation, SMC culturing, and lentiviral transduction
VSMCs were isolated from human saphenous vein segments using the explant technique (Southgate and Newby, 1990) and cultured in smooth muscle cell (SMC) growth media 2 (PromoCell, Heidelberg, Germany) with the addition of the supplement mix, plus 10% (v/v) fetal calf serum (FCS) (for a 15% (v/v) FCS final concentration), 1% (v/v) penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine (15% complete SMC media). Passages 3–8 were used for experiments. Ad5-expressing haemagglutinin (HA)-tagged full-length human Nogo-B (Ad-Nogo-B) was generated as previously described (Acevedo et al., 2004; Kritz et al., 2008) and Ad5 expressing eGFP (Ad-eGFP) was obtained from Crucell (Crucell, The Netherlands). A multiplicity of infection (MOI) per cell of LV or Ad was calculated on the basis of infective titres, iu/ml for LVs and plaque forming units per ml (pfu/ml) for Ad, and an accurate cell count before infection. Equivalent “amounts” were used for ICLVs and IDLVs in relation to their TaqMan quantitative PCR titres. Cells were infected with viral vectors for 18 hr in complete SMC media, subsequently washed twice in phosphate buffered saline (PBS), and maintained in complete SMC media. LV transductions were performed in the presence of 8 μg/ml of polybrene.
Fluorimetry and bicinchoninic acid assay
Expression of eGFP was visualized by fluorescence microscopy prior to cell lysis using 0.2% (v/v) Triton-X-100 PBS. The expression of eGFP was quantified by plate assay (fluorimetry) using a Wallac Victor 2 and recombinant eGFP (Clontech, Basingstoke, United Kingdom) as a standard, as previously described (Dishart et al., 2003). Expression of eGFP was normalized to total mg of protein present (determined by a bicinchoninic acid [BCA] assay [Fisher Scientific, Loughborough, United Kingdom]), and results were expressed as relative fluorescent units (RFU)/mg protein (Dishart et al., 2003).
Immunocytofluorescence
Nogo-B immunocytofluorescence in VSMCs was performed 5 days post-infection as described previously (Acevedo et al., 2004; Rodriguez-Feo et al., 2007; Kritz et al., 2008; Yu et al., 2009). Briefly, VSMCs were fixed in 4% paraformaldehyde (PFA) for 10 min, permeabilized for 10 min in 0.1% (v/v) Triton X-100 in PBS, blocked using 10% (v/v) donkey serum and 1% (w/v) BSA in PBS for 30 min, and incubated with 2 μg/ml of goat anti-human Nogo (N-18) immunoglobulin G (IgG) or isotype-matched goat IgG control in PBS containing 10% donkey serum and 1% (w/v) BSA for 18 hr at 4°C. Nogo-B staining was visualized using 4 μg/ml of Alexa-Fluor 555 donkey anti-goat IgG (Invitrogen) and mounted with Prolong-Gold anti-fade reagent with 4',6-diamidino-2-phenylindole (DAPI; Invitrogen). Micrographs were captured using a Carl Zeiss LSM 510 Meta confocal microscope at ×630 magnification, and images were digitized under constant exposure time, gain, and offset.
Migration assay
VSMC migration was assessed by a scratch-wound–mediated cell migration assay (Liang et al., 2007). Briefly, VSMCs were seeded for 50 to 60% confluence into 8-well culture slides (BD Biosciences, Oxford, United Kingdom) for 16 to 18 hr prior to LV infection. Once VSMCs reached 100% confluence, a scratch was created using a sterile p200 pipette tip. VSMCs were washed twice in PBS to remove cell debris and to smooth the edge of the scratch. VSMCs were maintained in 15% complete SMC media and in the incubator at 37°C. Reference lines close to the edge of the scratch were marked and images of the scratch acquired at 0 hours and 10 hours using bright-field microscopy. After 10 hours post-wounding, VSMCs were fixed in 4% PFA and haematoxylin stained and mounted in DPX. Absolute migration was measured (μm) as farthest distance the VSMCs migrated from the wound edge (average of 10 migration distances of three independent microscope fields for each condition in triplicate) using Image Pro-Plus software (Media Cybernetics, Bethesda, Maryland).
Proliferation and apoptosis assays
For both assays, VSMCs were seeded for 30% confluence after 16 to 18 hr incubation and VSMC quiescence was induced with serum-free media [MEM supplemented with 1% (v/v) penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, and 1mM sodium pyruvate (Sigma-Aldrich)] for 48 hr prior to LV infection with 15% complete SMC media. Proliferation was assessed using a CellTiter 96 Non-Radioactive Cell Proliferation (MTT) assay (Promega, Southampton, United Kingdom) according to manufacturer's instructions and optical density quantified using a Wallac Victor 2 at wavelength of 570 nm (Kritz et al., 2008). Apoptosis was assessed using Caspase-Glo 3/7 assay (Promega) according to the manufacturer's instructions and luminescence quantified using a Wallac Victor 2. Caspase 3/7 activities for all samples were normalized to that of an equal protein amount, and the results were expressed as relative luminescence units (RLU)/mg protein.
Integration site analysis
Linear amplification-mediated (LAM)-PCR insertion site analyses, 454 pyrosequencing and bioinformatical datamining
5’long terminal repeat (LTR)- and 3’LTR-mediated LAM-PCR analyses were performed as previously described (Schmidt et al., 2007; Paruzynski et al., 2010). In brief, linear PCR was performed on 1μg of DNA from ICLV- and IDLV-transduced smooth muscle cells. After magnetic capture of the biotinylated single-stranded linear PCR products second-strand DNA synthesis was performed. The resulting double-stranded fragments were digested with Tsp509I or MseI respectively, and after ligation of a linker cassette the LAM-PCR amplicons were amplified by two exponential PCR steps. The resulting LAM-PCR amplicons were further prepared for 454 pyrosequencing (GS Flx; Roche Diagnostics, Basel) as described previously (Paruzynski et al., 2010). Raw LAM-PCR amplicon sequences were separated according to the introduced barcode, trimmed further, and aligned to the human genome sequence using BLAT (bioinformatics) (assembly February 2009) (Kent, 2002; Paruzynski et al., 2010). The propensity of IS-forming common insertion sites (CIS) was analyzed based on previously published mathematical methods (Suzuki et al., 2002; Abel et al., 2007; Abel et al., 2011) modified based on a new statistical comparison with a generated random data set equivalent to the actual numbers of retrieved IS (Fronza et al., manuscript in preparation). Briefly, to each CIS is associated a probability value that reduce the number of false positive calls resulting from increasing numbers of total IS in the genome.
Q-PCR for vector copy number
Vector copy number estimation was performed as described previously (Paruzynski et al., 2012) with modifications for calculating the relative quantification of vector copies per genome. Briefly, vector copies were amplified with LTR specific primers (SKLTR 3: AGCTTGCCTTGAGTGCTTCA; LV 2 rev: GAGTCCTGCGTCGAGAGAGC) and cell copy numbers by hEPO specific primers (hEpoR-fw: CTGCTGCCAGCTTTGAGTACAC; hEpoR-rev: GAGATGCCAGAGTCAGATACCAC ). Q-PCR was performed on the LightCycler LC480 system using the SYBRGREEN I Mix (Roche) with 30 ng of genomic DNA from ICLV- and IDLV-transduced samples 14 days post transduction. For calculating vector copy numbers relative to genome copies, the LightCycler LC480 Software (Roche) was used, and values from the ICLV- and IDLV- transduced samples were normalized to a reference DNA obtained from a HeLa single-cell clone (D10) harboring one vector copy per genome.
Statistical analysis
All values are expressed as the mean±SEM of triplicates and three independent experiments were performed (representative of each other). Data were tested for, and shown to exhibit, Gaussian distribution by applying the Shapiro-Wilk normality test to the data. Results were statistically analyzed using the student's unpaired t-test or one-way ANOVA for Bonferroni's multiple comparisons. Statistical significance was assigned when p<0.05 (GraphPad Prism v4, La Jolla, California).
Results
IDLV-mediated gene transfer in human VSMCs
We first assessed the transduction efficiency mediated by IDLVs (IDLV-eGFP) versus their integrase-competent counterparts (ICLV-eGFP) at increasing MOIs of 5, 10, 25, and 50 in VSMCs. Expression of eGFP in VSMCs at 3, 7, and 14 days post-infection were visualized by fluorescence microscopy, prior to VSMC lysates, and quantified for eGFP expression by fluorimetry (normalized to protein). IDLV-eGFP facilitated efficient transgene expression in VSMCs, parallel to its integration-competent version (Fig. 1A). As expected, during cell division, transgene expression profiles of IDLVs decreased in a time-dependent manner from 3 days to 14 days post infection at all MOIs and mediates a bit less transgene expression than ICLV counterparts at all MOIs (Fig. 1A). Representative micrographs illustrated efficient eGFP expression in VSMCs mediated by IDLVs 3 and 7 days post infection at an MOI of 50 (Fig. 1 B). Immunocytofluorescence staining for Nogo-B indicated that both ICLV-Nogo-B and IDLV-Nogo-B mediated efficient overexpression of Nogo-B in VSMCs 5 days post-infection at an MOI of 50, compared to their eGFP configurations and endogenous Nogo-B levels observed in uninfected VSMCs (Fig. 1C). Collectively, these data support the notion that IDLVs mediate efficient gene transfer to primary human VSMCs, provide robust induction of transgene expression, and transgene expression was lost over time.
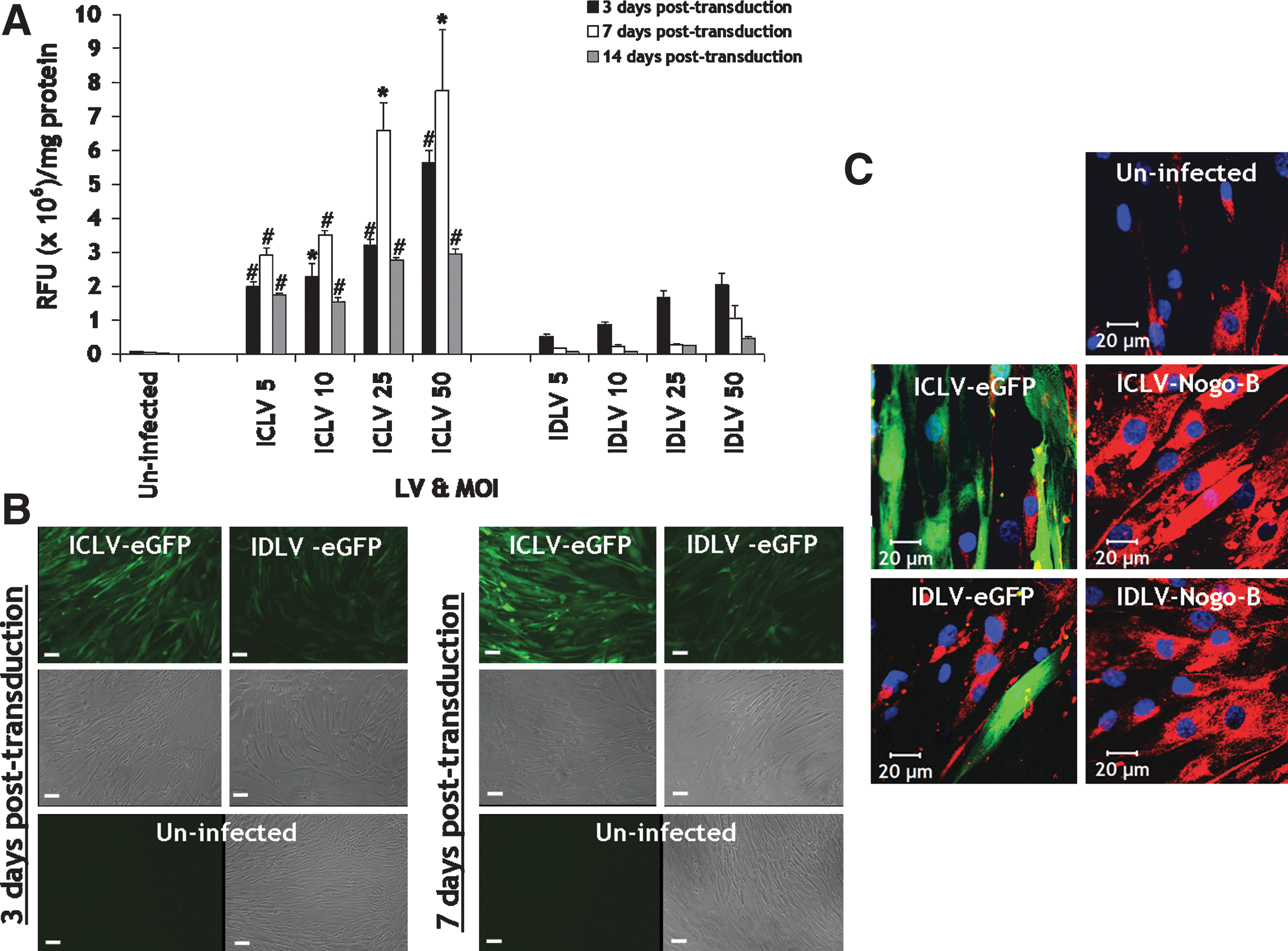
IDLV-mediated gene transfer in VSMCs.
Effect of IDLV-Nogo-B on human VSMC migration
Previous reports have demonstrated that the N-terminus of Nogo-B mediates a dose-dependent inhibition of VSMC migration induced by platelet-derived growth factor-BB (PDGF-BB) (Acevedo et al., 2004). We have previously shown that adenoviral overexpression of Nogo-B reduced PDGF-BB–induced migration rate of VSMCs (Kritz et al., 2008). We therefore examined the effect of Nogo-B overexpression mediated by IDLV-Nogo-B on VSMC phenotypes. Migration was assessed using a scratch-wound–mediated cell migration assay and quantified 3 days post infection. A significant reduction in VSMC migration was observed after infection with IDLV-Nogo-B, compared to IDLV-eGFP, at both MOIs of 25 and 50 and uninfected VSMCs (MOI of 25: 106.24±7.87 μm vs. 174.16±6.49 μm [39% significant reduction in VSMC migration] or 170.15±8.04 μm [38%], p<0.001, respectively, and MOI of 50: 125.98±8.66 μm vs. 178.9±8.78 μm [30%] or 170.15±8.04 μm (26%), p<0.001 or p<0.01, respectively) (Fig. 2A). Micrographs also illustrated IDLV-Nogo-B transduction in VSMCs inhibited migration, compared with IDLV-eGFP and untransduced VSMCs (Fig. 2B). Interestingly, our results also indicated IDLV-Nogo-B was as efficient as ICLV-Nogo-B in mediating a reduction in VSMC migration (Fig. 2A and B). Therefore, IDLV-mediated overexpression of Nogo-B attenuates VSMC migration. To note, the differences between VSMC migration at MOIs of 25 and 50 were not different in this scratch migration assay due to the sensitivity of this assay not depicting differences precisely.
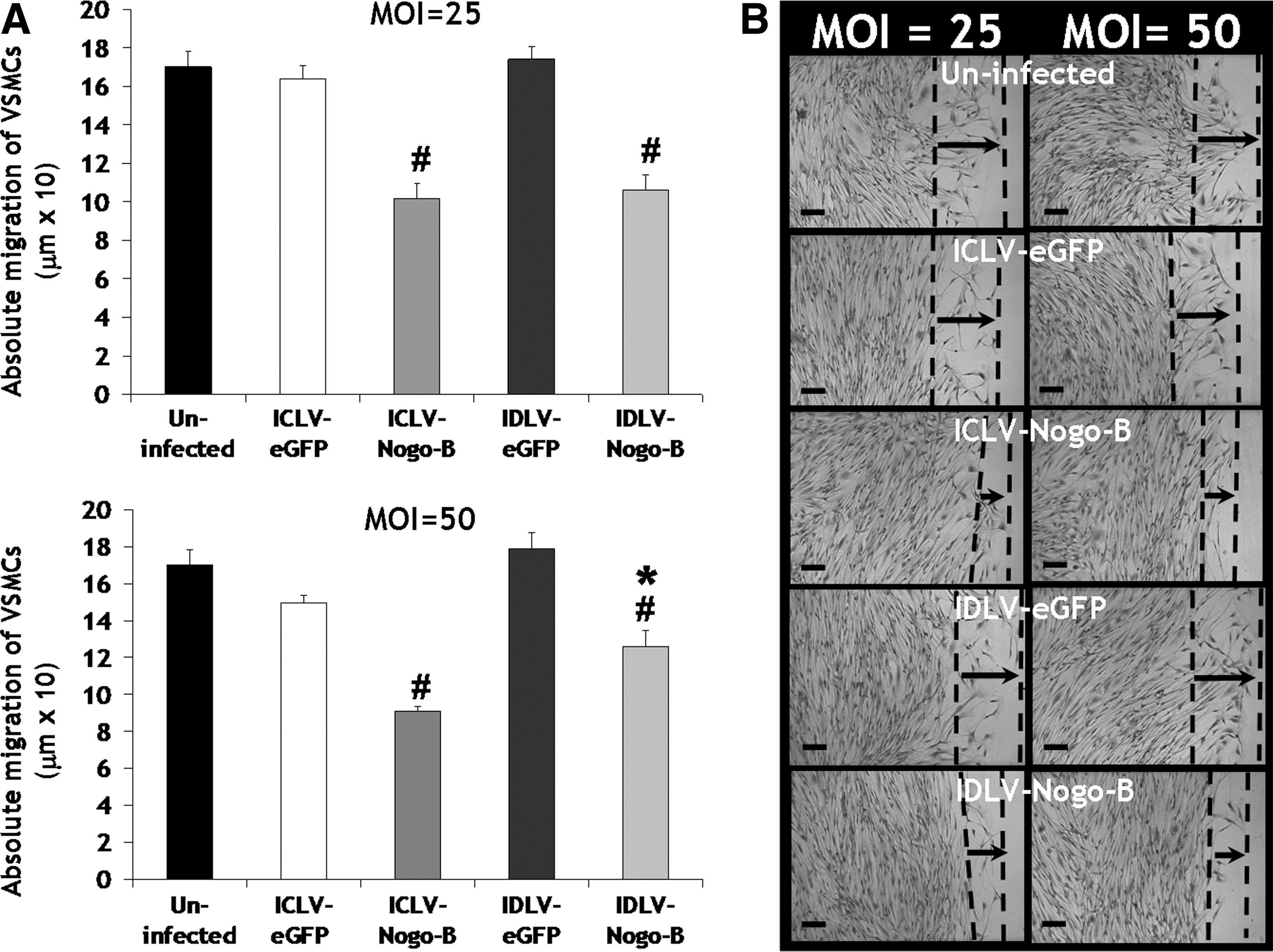
IDLV-Nogo-B reduced VSMC migration.
IDLV-Nogo-B-mediated human VSMC proliferation
Overexpression of Nogo-B negatively regulates VSMC proliferation in response to serum and PDGF-BB stimulation, as indicated by previous studies using adenovirus-based strategies (Kritz et al., 2008). We therefore investigated the effect of IDLV-Nogo-B on VSMC proliferation. IDLV-Nogo-B significantly reduced VSMC proliferation following 5 days post infection at MOIs of 25 and 50, compared to IDLV-eGFP control and uninfected VSMCs (MOI 25: 0.14±0.0084 optical density [OD] at 570 nm vs. 0.29±0.01 OD [52% significant reduction in VSMC proliferation] or 0.33±0.0038 OD [58%], p<0.001, respectively, and MOI 50: 0.074±0.0011 OD vs. 0.2±0.013 OD (63%) or 0.35±0.015 OD [79%], p<0.001, respectively) (Fig. 3A). Representative images are also shown at MOI of 50 (Fig. 3B). In addition, these data also demonstrated that IDLV-Nogo-B was as effective as ICLV-Nogo-B in reducing VSMC proliferation (Fig. 3A).
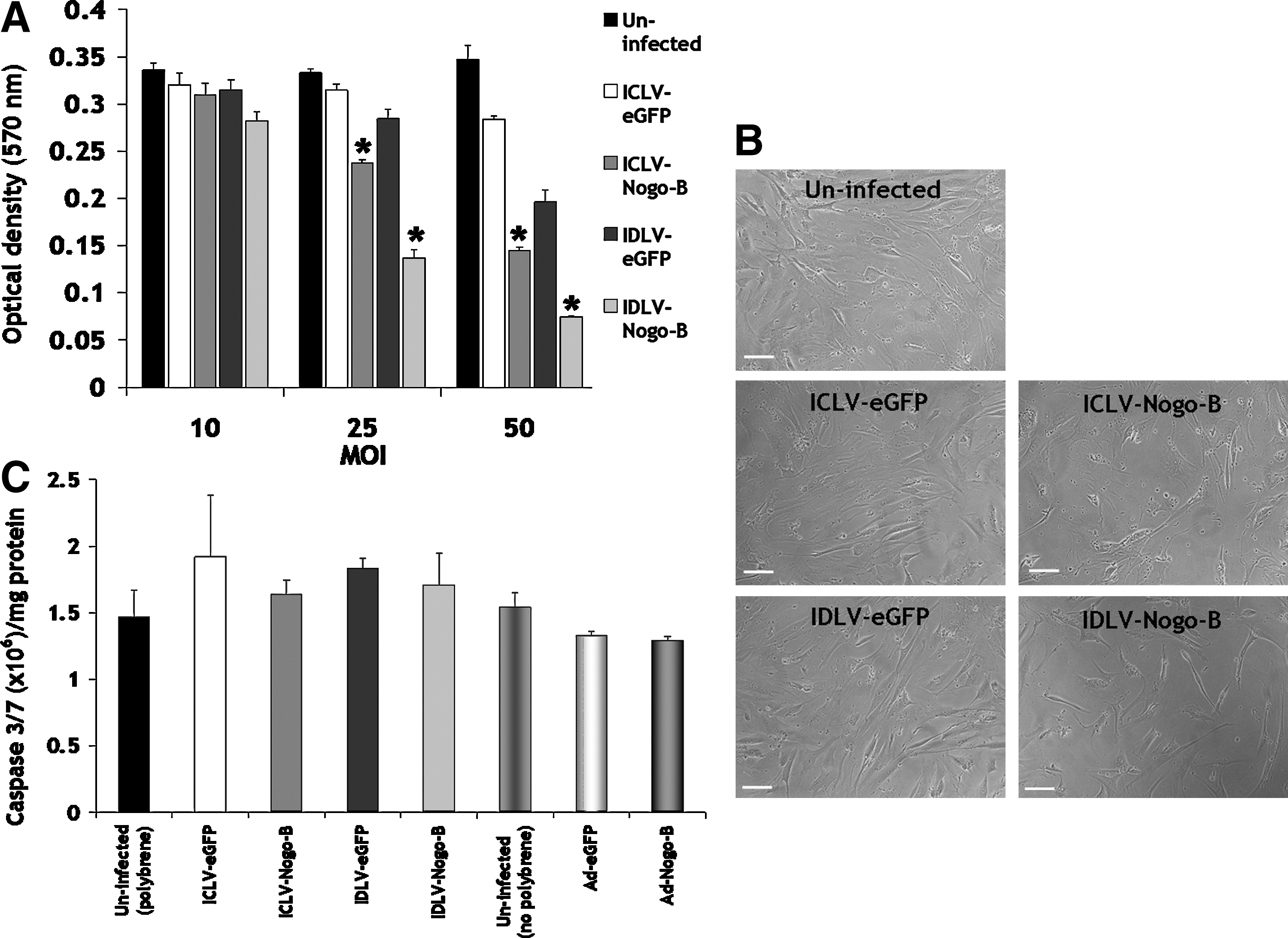
IDLV-Nogo-B reduced VSMC proliferation.
Previous reports have implicated that overexpression of Nogo-B can act as a pro-apoptotic protein in various cancer cells (Li et al., 2001; Tambe et al., 2004; Kuang et al., 2006). However, we have previously reported that the overexpression of Nogo-B facilitated by adenoviral vectors in the vasculature did not induce apoptosis (Kritz et al., 2008). We therefore assessed the induction of apoptosis mediated by LVs overexpressing Nogo-B in VSMCs. VSMC apoptosis was determined by a caspase-3/7 activity assay 5 days post-transduction. Results indicated that IDLV-Nogo-B and ICLV-Nogo-B at an MOI of 50 did not significantly induce casapse-3/7 activation (Fig. 3C). Therefore, IDLV-mediated elevation in Nogo-B blocks VSMC proliferation and migration in the absence of an effect on VSMC apoptosis.
Lentiviral integration site selection in human VSMCs
The above data suggests that IDLVs are effective in the context of VSMC gene transfer. Vector safety in the clinical setting is a major factor in the advancement of genetic therapies. To ascertain whether an improved safety profile of IDLV compared to ICLV could be observed in the context of vascular gene transfer, we next performed large-scale IS analyses of ICLV and IDLV vectors following infection of primary human VSMCs, and harvested DNA at 14 days post-transduction. We assessed integration by LAM-PCR and 454 pyrosequencing (Schmidt et al., 2007; Gabriel et al., 2009; Paruzynski et al., 2010).
ICLV and IDLV vectors were used to transduce three independent human VSMC batches isolated from three different individuals at MOIs of 10, 25, and 50. Subsequent sequencing of LAM-PCR amplicons resulted in the identification of 10,752 unique ICLV IS and 1,086 multiple mappable IS sequences, mainly due to location in repetitive elements of the human genome (Table 1). IS analyses on IDLV-transduced VSMCs identified 196 unique mappable and 118 multiple mappable IS sites, clearly verifying the reduced integration frequency of IDLV-based vectors (Table 1). Strengthening the observation that most integrated IDLV genomes result from a mechanism inconsistent with residual integrase activity (see Matrai et al., 2011), 28% of retrieved IDLV vector–cellular junctions contained deletions in the LTR, whereas from all ICLV insertion sites only 3% harbored deletions in the LTR. In addition, mapping of IDLV IS showed a close-to-random distribution in respect to integration within RefSeq genes (Fig. 4A). In contrast, ICLV IS showed the expected preference to integrate into RefSeq genes, independent of the different MOI used. Therefore, the sequences of the individual replicates were combined for further analyses (Fig. 4A and B).
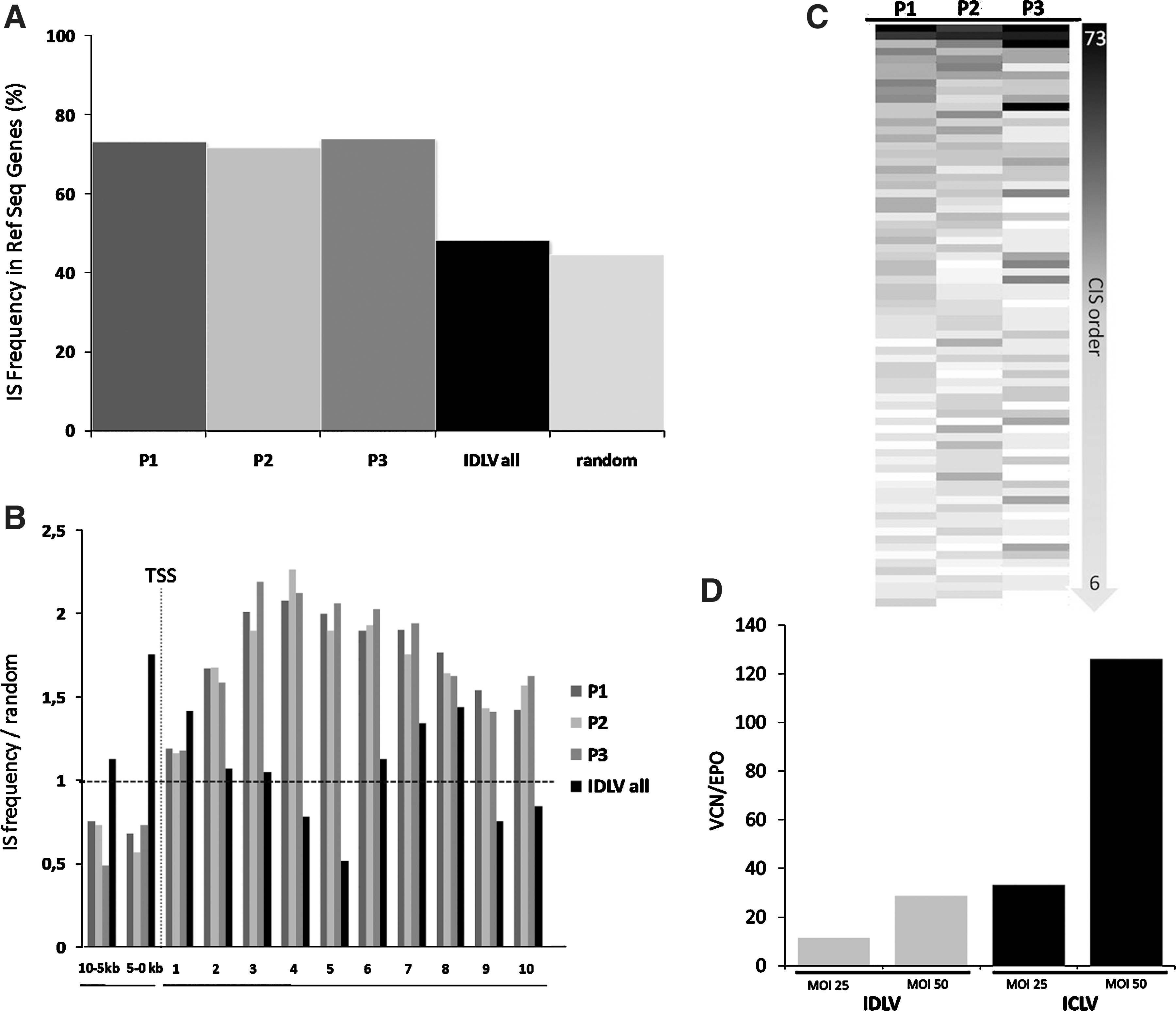
Distribution of ICLV and IDLV insertion sites in human vascular smooth muscle cells.
MOI, multiplicity of infection; IS, integration site; IDLV, integrase-deficient lentiviral vectors; ICLV, integrase-competent lentiviral vectors; n.d, no data.
We further analyzed the propensity of ICLV IS to be located in CIS. CIS are defined as chromosomal regions where multiple IS sites in the host genome are nonrandomly enriched. The order of CIS is defined by the number of individual IS located in a specific genomic window size (Abel et al., 2011). We did not detect any CIS while analyzing the IDLV IS; however, the analyses of the ICLV IS in patient (P)1–P3 revealed that 15 to 19% (p<0.0001) of total IS were located in CIS of higher orders (>6th order) (Fig. 4C). Strikingly, IS in CIS were present in all three samples analyzed to a similar intensity, indicating that an intrinsic IS bias may be responsible for this observed nonrandom distribution (Fig. 4C). A total of 708 RefSeq genes were associated with these CIS, of which 24% are also listed as cancer genes (
Discussion
The main aim of this study was to construct and assess a suitable candidate IDLV for potential application to vascular gene therapy, and to assess the safety implications in the context of integration events in primary human vascular cells. For the first time, this study reported the important feasibility of IDLVs for use in vascular cell gene transfer studies, which provided efficient transgene expression in VSMCs and led to effective phenotypic effects on VSMC migration and proliferation in vitro, concomitantly associated with very low integration frequencies. Our data supports the use of IDLV in general and the potential of Nogo-B as a candidate gene for therapeutic application to acute vascular injury using this system. It has already been reported that LVs have the potential to mediate gene transfer of transgenes into primary vascular cells within a short time exposure (minutes of exposure), which is required in this clinical setting (Dishart et al., 2003; Cefai et al., 2005). IDLVs would be highly advantageous in vascular gene therapy based on the improved safety profile. Notably, our results here indicated efficient transgene expression in VSMCs can be achieved by IDLVs. However, IDLVs did not provide enhanced efficient and long-term eGFP expression as well as their integrase-competent counterparts in VSMCs. IDLVs expressed lower transgene expression than their integrating counterparts, which is in correlation with earlier studies (Bayer et al., 2008; Matrai et al., 2011).
In addition, these findings corroborated with those of previous studies in dividing cells, which reported that IDLV-mediated transient transgene expression was due to episomes lacking replication signals and thus diluting as a consequence of cell division (Nightingale et al., 2006; Philippe et al., 2006; Apolonia et al., 2007; Matrai et al., 2011). In the vascular setting in vivo, this is worthy of further evaluation since blockade in vascular phenotypes in the short-term post-acute injury using adenoviruses have been shown to provide long-term protection (George et al., 2011). Clearly, strategies that slow or block VSMC proliferation in vivo would result in sustained transgene expression in the vessel wall. In addition, VSMCs have relatively low rates of proliferation in a disease state (Gordon et al., 1990), and during the acute vascular injury response ( Westerband et al., 1997; Hilker et al., 2002) with a proliferative index of approximately 1.34% (Westerband et al., 1997). Therefore, it is suggested that the IDLV-mediated episomal transgene would dilute slowly, but persist for the duration required (George et al., 2006; White et al., 2007). In addition, one can speculate that Nogo-B's effect on the inhibition of VSMC proliferation would also assist with the longevity of transgene mediated by IDLV-based episomal genomes.
Previous studies have shown IDLVs, especially the D64V mutants, to have important implications relevant for gene delivery into nondividing cells within different organs (eye, muscle, central nervous system, and liver of rodents), by retaining transduction efficiency of their wild-type integrase versions with minimal host genome integration in vitro and in vivo (Yanez-Munoz et al., 2006; Apolonia et al., 2007; Rahim et al., 2009; Matrai et al., 2011). In relation to these preclinical reports, results from this in vitro study implied efficient gene transfer and transgene expression mediated by IDLV-Nogo-B in human VSMCs, and as consequence was as efficient in modulating phenotypic effects of VSMCs. Therefore, the data reported herein denotes the use of IDLVs as an alternative and promising gene transfer vector system. However, the lack of ability to produce the high titres for LVs (Matrai et al., 2010) required for in vivo vascular gene transfer provides a substantial barrier and disadvantage for this vector system to be used in further vasculature studies in vivo at present.
We demonstrate that IS selection of ICLV in human VSMCs follows typical lentiviral target site patterns; however, regional hotspots resulting in higher order CIS have been detected. The propensity of ICLV insertions to be located in such CIS is significantly higher than expected by chance. Given that most genotoxic side effects in gene therapy trials using gammaretroviral vectors have been triggered by insertional activation of cellular genes located in close proximity to gammaretroviral CIS, it has recently been suggested that LV hotspots reflect a benign integration bias rather than oncogenic selection (Biffi et al., 2011). We further show that IDLV-based vectors integrate at very low frequency and randomly in human VSMCs, and therefore reduce the risk of insertional mutagenesis. This finding is consistent with a previous report (Matrai et al., 2011) and further strengthened by q-PCR, where we observed no major differences regarding VCN between IDLV (MOI of 50) and ICLV (MOI of 25). However, with LAM-PCR, we retrieved much higher integrated vector copies supporting the observed reduced integration frequency despite possible competition of episomal persisting IDLV (Fig. 4A, B, and D; Table 1).
We conclude that IDLV provide differences in VCN compared to ICLV but still attain similar phenotypic effects. In relation to these IS results, active transcriptional units of nondividing cells possess fewer LV-mediated integration frequencies, with a similar association to a random distribution of integration, unlike that of rapidly dividing cells. Moreover, LV-mediated integration sites were most commonly found in nonexpressed genes of postmitotic cells (Bartholomae et al., 2011). This could have possible implications in LV-transduced VSMCs (Gordon et al., 1990; Westerband et al., 1997; Hilker et al., 2002).
We note that IS patterns obtained from dividing VSMCs in vitro may not reflect the pattern of integration mediated by ICLV in VSMCs in vivo as described in other tissues previously (Bartholomae et al., 2011). In a recent study, IDLV-mediated gene delivery in hepatocytes gave rise to random integrations without obvious skewing toward gene-coding regions (Matrai et al., 2011). Therefore, it is possible that ICLVs and most importantly IDLVs could give rise to a lesser likelihood of genotoxicity in VSMCs (Bartholomae et al., 2011; Matrai et al., 2011).
Taken together, this study addresses the potential of IDLVs for future use in vascular cell gene delivery studies with associated low-integration frequencies, with justification to take forward this new candidate IDLV-Nogo-B into in vivo models of acute vascular injury.
Footnotes
Acknowledgments
We thank C. Weber for technical assistance. This work was supported by the British Heart Foundation (BHF) studentship grant FS/07/022/22946.
Author Disclosure Statement
Authors declare no conflict of interest.