
Abstract
Duchenne muscular dystrophy (DMD) is a severe inherited, muscle-wasting disorder caused by mutations in the DMD gene. Gene therapy development for DMD has concentrated on vector-based DMD minigene transfer, cell-based gene therapy using genetically modified adult muscle stem cells or healthy wild-type donor cells, and antisense oligonucleotide-induced exon-skipping therapy to restore the reading frame of the mutated DMD gene. This study is an investigation into DMD gene targeting-mediated correction of deletions in human patient myoblasts using a target-specific meganuclease (MN) and a homologous recombination repair matrix. The MN was designed to cleave within DMD intron 44, upstream of a deletion hotspot, and integration-competent lentiviral vectors expressing the nuclease (LVcMN) were generated. MN western blotting and deep gene sequencing for LVcMN-induced non-homologous end-joining InDels (microdeletions or microinsertions) confirmed efficient MN expression and activity in transduced DMD myoblasts. A homologous repair matrix carrying exons 45–52 (RM45–52) was designed and packaged into integration-deficient lentiviral vectors (IDLVs; LVdRM45–52). After cotransduction of DMD myoblasts harboring a deletion of exons 45 to 52 with LVcMN and LVdRM45–52 vectors, targeted knock-in of the RM45–52 region in the correct location in DMD intron 44, and expression of full-length, correctly spliced wild-type dystrophin mRNA containing exons 45–52 were observed. This work demonstrates that genome surgery on human DMD gene mutations can be achieved by MN-induced locus-specific genome cleavage and homologous recombination knock-in of deleted exons. The feasibility of human DMD gene repair in patient myoblasts has exciting therapeutic potential.
Introduction
Gene targeting is a powerful tool for creating genetic modifications as a gene repair strategy for a variety of genetic diseases (Urnov et al., 2005; Arnoud et al., 2007; Lombardo et al., 2007; Grizot et al., 2009; Martin et al., 2010; Carroll, 2011; Jensen et al., 2011; Li et al., 2011). Gene targeting and homologous recombination (HR) efficiency can be greatly enhanced by using endonucleases (meganucleases [MNs], zinc finger nucleases [ZFNs], transcription activator-like effector nucleases [TALENs], and clustered regularly interspaced palindromic repeats/CRISPR-associated [CRISPR/Cas] nucleases) to cleave at specific chromosomal loci where HR is to take place. The resulting double-strand break (DSB) can be repaired by two main mechanisms: HR or nonhomologous end-joining (NHEJ) (Longhese et al., 2010). HR requires the presence of an identical or nearly identical sequence to be used as a template for repair of the DSB (Jensen et al., 2011). MNs can be used to stimulate HR up to 10,000-fold in cultured cells in comparison with HR at a noncleaved site (Rouet et al., 1994; Choulika et al., 1995; Paques and Duchateau, 2007). As a genome surgery repair strategy, NHEJ, by the nature of its DSB repair mechanism, is limited to correction of the disrupted reading frame of mutated genes by introducing InDels (microdeletions or microinsertions), and unlike HR, cannot correct deletions by cDNA knock-in or other targeting strategies. In the context of DMD, gene-targeting studies have been limited to NHEJ repair of the DMD reading frame. In an artificial model system, an engineered MN restored the normal reading frame through NHEJ repair of dog microdystrophin sequences carrying a frameshift mutation (Chapdelaine et al., 2010). The MN induced InDels in the microdystrophin so that truncated dystrophin was expressed, both in myoblasts in vitro and in muscle fibers in vivo (Chapdelaine et al., 2010).
The aim of the study described here was to establish whether gene-targeted correction of the mutated DMD gene via HR is possible using a target-specific MN together with a specific repair matrix. We present here a novel system able to repair human DMD gene mutations on the X chromosome by MN-induced locus-specific genome cleavage, and HR to knock-in deleted exons in human DMD patient cells. We demonstrate for the first time targeted cDNA knock-in and human DMD gene repair through HR repair of an MN-induced DSB by a targeting repair matrix. This suggests exciting therapeutic potential for the development of permanent gene correction therapy in DMD patient cells and tissues.
Materials and Methods
Cells
Immortalized human DMD myoblasts, carrying a deletion of exons 45–52, and a second human DMD cell line carrying a deletion of exons 48–50 (Chaouch et al., 2009), together with primary human skeletal muscle cells (hSkMCs) (TCS Cellworks, Buckingham, UK) and 293T cells, were cultured according to published protocols (Popplewell et al., 2010, 2011) and used in experiments as indicated.
Construction of lentivirus-based MN and targeting matrix plasmids
A meganuclease (MN-DMD31), derived from I-CreI and engineered as described previously (Smith et al., 2006; Grizot et al., 2011; Daboussi et al., 2012), has been designed and developed to target intron 44 within the DMD gene, upstream of a mutation hotspot. Single-chain MN constructs of the two variants of DMD31 (3631 and 3633) were subcloned into the MluI/SwaI-digested lentiviral (LV) transfer plasmid (pRRLscSegfpCncs1w), with enhanced green fluorescent protein gene (eGFP) coexpression. The resulting plasmids, pLV-MN3631-eGFP and pLV-MN3633-eGFP, contain two expression cassettes; one encodes eGFP under the control of the spleen focus-forming virus (SFFV) promoter, and the other contains a cytomegalovirus (CMV) promoter and MN-DMD31. Both plasmids contain the woodchuck hepatitis virus (WHV) posttranscriptional regulatory element (WPRE) sequence downstream of the MN gene.
The targeting matrix was designed to contain arms of homology to intron 44 flanking a cDNA block encoding exons 45–52; a synthetic splicing donor and acceptor derived from human immunoglobulin and human β-globulin gene, respectively; and an intronic splicing enhancer sequence from the rat Fgfr2 gene (DISE [downstream intronic splicing enhancer] element) (Seth et al., 2008). The synthetic donor and acceptor sequences used were designed through in silico analysis to optimize splice signal strength (Shapiro and Senapathy, 1987), and are available on request. The various elements within the targeting matrix were designed to be flanked by unique restriction enzyme sites to allow the modification of the matrix as required. The flanking arms needed to be completely homologous to intron 44 on either side of the DSB produced by MN-DMD31 cleavage. The degree of polymorphism in the ∼3-kb sequence on either side of the DSB was elucidated by PCR amplification of harvested genomic DNA from the three cell types (DMD del45–52, DMD del48–50, 293T) with overlapping primers. Genomic DNA was purified with a DNeasy blood and tissue kit (Qiagen, Crawley, UK), and PCR was performed with 2×PCR master mix (GeneSys, Camberley, UK). The PCR products were sequenced (GATC Biotech, Konstanz, Switzerland), and the sequences were aligned to show the position of polymorphic residues, using Vector NTI software (Invitrogen, Paisley, UK). Two targeting matrices were synthesized (by GeneART, Regensburg, Germany) with arms homologous to DMD del45–52: one targeting matrix (S1) having 1-kb arms of homology, and the other (S2) had 1.5-kb arms of homology. The synthesized targeting matrices were then inserted invertedly into the AgeI/ApaI-digested lentivector backbone (pRRLsinPPT-ISce-IT), generating pLV-RM45–52 S1 and pLV-RM45–52 S2. Correct synthesis and subcloning were confirmed by restriction enzyme digestions according to the manufacturer's instructions (New England BioLabs, Hitchin, UK). The sequence of targeting matrix constructs and primer sequences used for studying polymorphism within intron 44 are available on request.
LV vector production and titration
For the delivery of targeting matrices, integration-deficient lentiviral vectors, pseudotyped with the vesicular stomatitis virus envelope glycoprotein (VSV-G), were used (referred to as LVdRM45–52 S1 and LVdRM45–52 S2), whereas integration-competent lentiviral vectors were used for the expression of the meganuclease (referred to as LVcMN3631 and LVcMN3633); these were produced as previously described (Yáñez-Muñoz et al., 2006; Jacome et al., 2009; Pichavant and Tremblay, 2012). Briefly, lentiviral vectors were generated by transient transfection of HEK293T cells with pMD2.VSV-G, pRSV.REV and pMDLg/pRREintD64V (for integration-deficient), or pMDLgpRRE (for integration-competent) packaging plasmid, and the relevant transfer plasmid. At set time points after transfection, the harvested HEK293T cell medium was centrifuged at 690×g for 10 min at room temperature and then filtered through a 0.22-μm filter (Nalgene, Rochester, NY) to remove cell debris. The filtered medium was then harvested and transferred to high-speed polyallomer centrifuge tubes (Beckman Coulter, Brea, CA) and centrifuged at 50,000×g in an SW32Ti rotor (Beckman Coulter) for 2 hr at 4°C. The vector was then resuspended in Dulbecco's modified Eagle's medium (Invitrogen), centrifuged at 1400×g for 10 min, and incubated with DNase I (5 U ml−1; Promega, Madison, WI) and 10 m
For viral titration, HeLa cells were transduced with serial dilutions of vector stock in the presence of Polybrene (8 μg/ml). eGFP expression controlled by the SFFV promoter in LV-MN was evaluated by flow cytometric analysis. eGFP+ cells were scored by flow cytometric analysis 72 hr after transduction. Real-time PCR titration of total vector DNA by late reverse transcript amplicon quantification was also performed on HeLa cell samples harvested 24 hr after transduction. Briefly, AL buffer (Qiagen) was added to cells and incubated for 10 min at 56°C. The number of LV reverse-transcribed copies was determined by quantitative-PCR (qPCR), using TaqMan detection of PCR products in real time with the MyiQ single-color detection system (Bio-Rad, Hercules, CA). qPCR values were normalized using β-actin gene quantification in the DNA extracts with a SYBR green detection system. qPCR titers of 6–9×108 were routinely achieved. It should be noted that as a general rule, eGFP titers were 100-fold lower than those obtained by qPCR.
Western blot analysis of meganuclease expression
Total protein extraction from del45–52 human DMD myoblasts 2 days and 5 days after transduction with LVcMN3631 and LVcMN3633 was performed in radioimmunoprecipitation assay (RIPA) buffer (150 mM sodium chloride, 1% Nonidet P-40 [NP-40], 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 50 mM Tris, pH 8.0). Ten micrograms of total protein extract was electrophoretically separated on 3–8% Tris–acetate gels (Invitrogen) and then transferred onto nitrocellulose (Invitrogen). MN expression was revealed with a specific I-CreI mouse monoclonal antibody. α-Tubulin antibody (ab4074) (Abcam, Cambridge, UK) was used as a loading control.
Assessment of MN activity by deep gene sequencing
To evaluate the efficiency of MN-DMD31 variants to cut DNA at their target site, del45–52 human DMD myoblasts were transduced with LVcMN3631 and LVcMN3633 at eGFP multiplicities of infection (MOIs) of 1 and 0.5. Five days after transduction, genomic DNA was purified with a DNeasy blood and tissue kit (Qiagen), and a ∼400-bp product was amplified around the MN target site by PCR, using 2× PCR master mix (GeneSys). The purified products were deep sequenced (by GATC Biotech) to assess the mutation rate as a result of NHEJ repair of the DSB induced by MN-DMD31 at its target site in intron 44 of the DMD gene, compared with nontreated controls.
Detection of corrected dystrophin genomic DNA in del45–52 human DMD myoblasts
Del45–52 human DMD myoblasts were transduced in 24-well plates with LVcMN3631 (qPCR MOI, 1000) together with either LVdRM45–52 S1 or LVdRM45–52 S2 (qPCR MOI, 1000). After 72 hr, genomic DNA was harvested with a DNeasy blood and tissue kit (Qiagen). Seminested PCR using LongAmp Taq polymerase (New England BioLabs) was performed to examine HR between intron 44 sequences in the targeting matrix and endogenous intron 44. The products were separated on 1% agarose gels in Tris–borate/EDTA buffer, and HyperLadder I (Bioline, London, UK) was used as marker.
Detection of corrected dystrophin mRNA in del45–52 human DMD myoblasts
Transduction of del45–52 human DMD myoblasts with LVcMN3631 (qPCR MOI, 1000) together with either LVdRM45–52 S1 or LVdRM45–52 S2 (qPCR MOI, 1000) was performed in 24-well plates. After 72 hr, RNA was harvested with a QIAshredder and an RNeasy extraction kit (Qiagen), and nested RT-PCR was performed to examine corrected mRNA expression, using a GeneScript RT-PCR system kit (GeneSys) for the first round and 2× PCR master mix (GeneSys) for the second round. For analysis of the left side of knock-in mRNA, first-round primers from exons 43 (endogenous) and 48 (matrix) were used, and from exons 44 (endogenous) and 46 (matrix) in the second round. For the right-hand side of knock-in, primers from exons 50 (matrix) and 55 (endogenous) were used in the first round, and from exons 51 (matrix) and 54 (endogenous) in the second round. The products were separated on a 1.2% agarose gel in Tris–borate/EDTA buffer and HyperLadder IV (Bioline) was used as marker. Details of primers and RT-PCR, qPCR, and PCR protocols used in this study are available on request.
Results
Generation of DMD-specific meganucleases
A number of I-CreI-derived MNs have been designed and developed to target various introns within the DMD gene, upstream of mutation hotspots. The MNs developed and details of their target sites are listed in Table 1. The work described in this paper concentrates on the development of MN-DMD31, the MN that targets intron 44 of the DMD gene. More than 25% of the mutations that cause DMD arise from this intron, and therefore this MN would have the potential to treat the highest proportion of patients with DMD. In-depth BLAST analysis was performed across the human genome to reveal that the target site of MN-DMD31 maps to position >chromosome:GRCh37:X:32364354:32364377, 2.233 kb downstream from exon 44. No other sequences within the human genome showed complete identity with the MN-DMD31 target site, implying that the nuclease will specifically target this locus. Importantly, most potential off-target sites are outside of known genes, further reducing the chances of insertional mutagenesis (summarized in Table 2).
The intron targeted by each MN is listed together with the percentage of DMD mutations arising from the intron targeted.
Building of lentiviral vectors expressing MN
To enable genome correction ex vivo, contemporaneous and transient expression of relevant MNs is required along with delivery of targeting matrices. Single-chain MN constructs of the two variant isoschizomers of MN-DMD31 (3631 and 3633) have been subcloned into an LV transfer plasmid with eGFP coexpression to monitor gene expression. The eGFP gene was controlled by the internal SFFV promoter, and MN-DMD31 by the immediate-early CMV promoter. The plasmid also contains the WHV WPRE sequence to enhance translation. Successful subcloning was confirmed by restriction enzyme and sequence analysis (data not shown). Integration-competent lentiviral vectors (ICLVs) expressing either of the two variants of MN-DMD31 (referred to throughout as LVcMN3631 and LVcMN3633) were generated and infectious viral particle titers were measured by qPCR and flow cytometric analysis based on eGFP expression. HeLa cells transduced with LVcMN3631 or LVcMN3633 at eGFP MOIs of 1 and 2 led to eGFP expression in 95–98.2% of cells (Fig. 1a and b), suggesting high transduction efficiencies of the vectors at the MOIs used. No assessment of cellular toxicity was made because none was visibly evident.
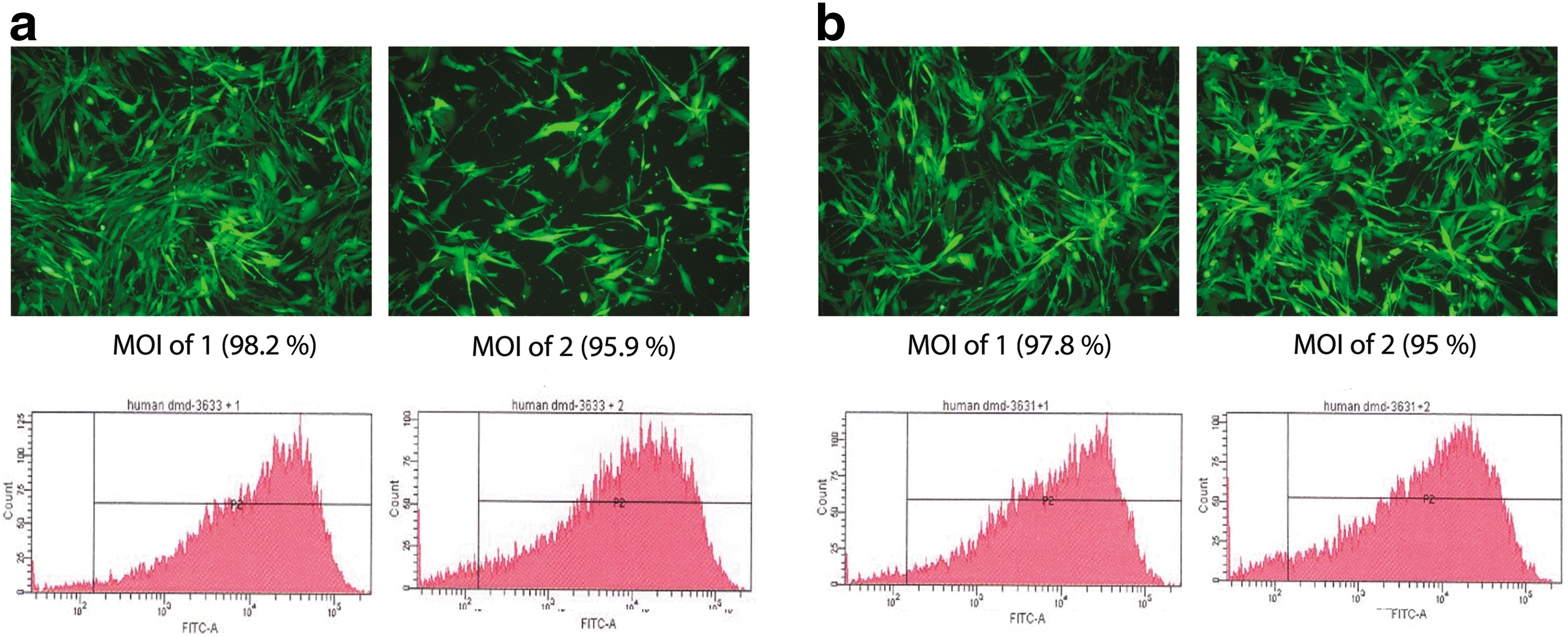
eGFP expression in HeLa cells infected with bicistronic LV expressing eGFP and MN. HeLa cells were transduced with LVcMN3631 or LVcMN3633. After 72 hr, eGFP expression controlled by the SFFV promoter in LVcMN3631 and LVcMN3633 was evaluated by fluorescence microscopy and flow cytometric analysis. Results for LVcMN3631 are shown in
Assessment of lentivirus-based MN expression
To examine the expression of MN protein in human DMD patient myoblast cells harboring exon 45–52 deletion (del45–52 human DMD cells), western blot analysis was performed with a specific I-CreI antibody at 2 and 5 days after transduction with LVcMN3631 and LVcMN3633. α-Tubulin was used as an internal control, and results are shown in Fig. 2a. The highest normalized MN expression is seen with LVcMN3633 at an eGFP MOI of 1. Normalized MN expression for the different variants, different MOIs, and different time points is therefore expressed as a percentage of that seen with LVcMN3633 at an MOI of 1 (Fig. 2a). Strong MN protein expression is equally evident for both variants, with a clear correlation between MOI used and level of MN protein expression for both variants.
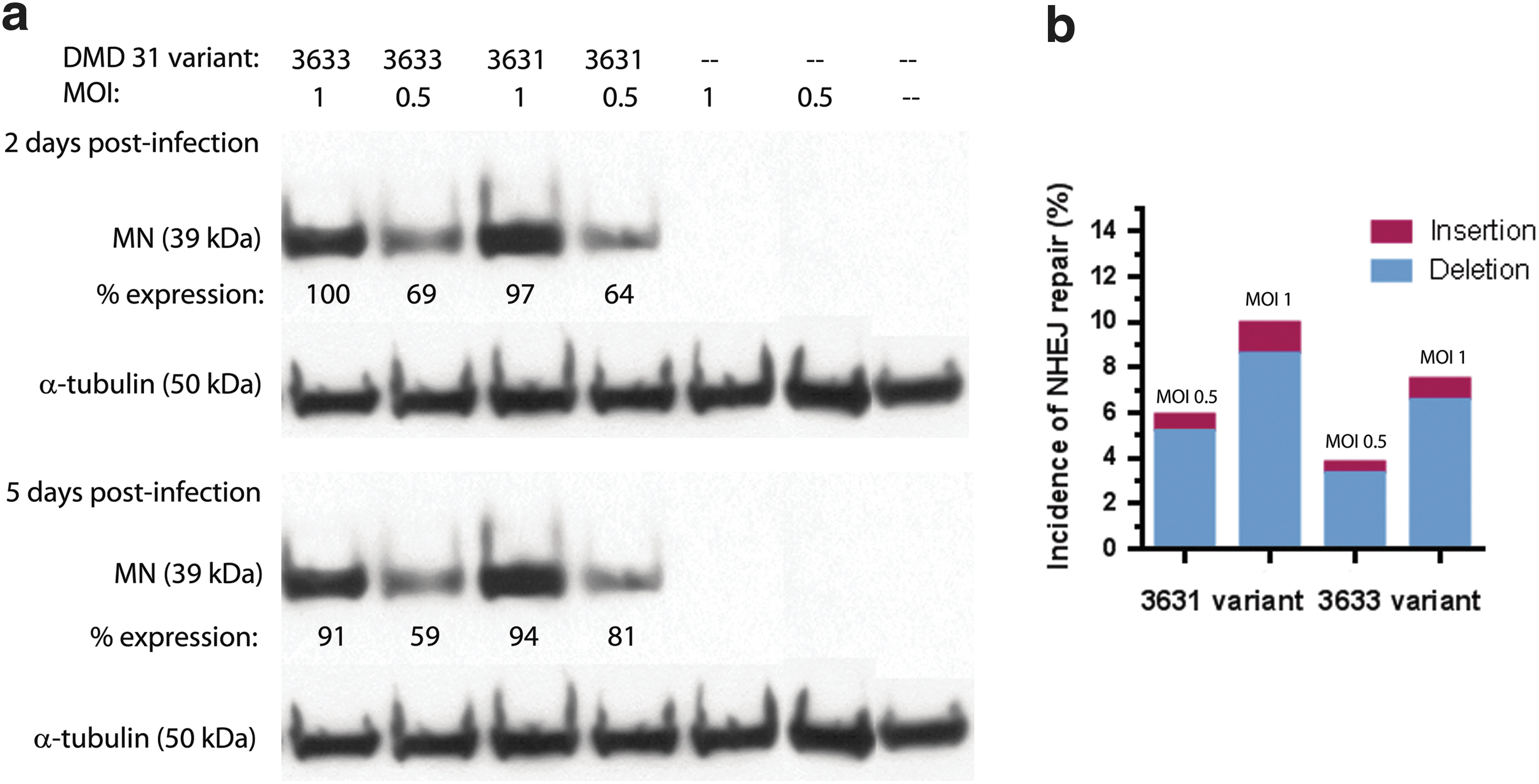
Analysis of MN expression in human del45–52 DMD cells transduced with LVcMN.
To evaluate the efficiency of MN-DMD31 (two variants) to induce a DSB at its target site on the chromosomal locus in intron 44 of the DMD gene, del45–52 human DMD cells were transduced with LVcMN3631 and LVcMN3633 at eGFP MOIs of 1 and 0.5. Five days after transduction, genomic DNA was purified and a ∼400-bp product was amplified around the MN target site by PCR. The purified PCR product population was then subjected to deep gene sequencing to assess the frequency of InDels resulting from NHEJ repair of DSBs induced by LVcMN3631 and LVcMN3633 at the MN target site. The results are presented in Fig. 2b and show that in the del45–52 DMD cells transduced by LVcMN3631 and LVcMN3633 up to 10% of X chromosomes exhibit detectable InDels specifically at the target site. This was shown to be specific to the MN-treated cells, because InDel frequency was negligible in nontreated controls (data not shown). There is a correlation between the level of InDels arising from NHEJ repair and MOI of LV used: an MOI of 0.5 produced 5.25 and 3.52% NHEJ repair for MN3631 and MN3633, respectively, whereas an MOI of 1 produced 9.12 and 6.79% NHEJ repair for MN3631 and MN3633, respectively. These results suggest that variant MN3631 is more active than MN3633 despite similar levels of expression between MN-3631 and MN-3633 (Fig. 2b). This variant (MN3631) of MN-DMD31 was therefore used in the subsequent gene correction experimentation.
Building of an homologous repair matrix (RM45–52) targeting the MN cleavage site
The aim of MN-mediated gene correction of the DMD gene is the insertion of a cDNA block of deleted exons into the intron harboring the deletion junction. To drive effective splicing of the inserted cDNA with the endogenous mRNA, flanking splice sites and other regulatory sequence elements are required around the cDNA block. Arms of homology are required to drive HR between genomic DNA and the RM45–52 homologous repair matrix at the MN-induced DSB site, and subsequent cDNA knock-in. To establish complete homology and to elucidate positions of any polymorphic residues, sequencing of ∼3 kb upstream and downstream of the MN target site within intron 44 was performed on PCR amplicons produced using overlapping primers on genomic DNA from human embryonic kidney 293T cells, del45–52 DMD cells, and del48–50 DMD cells. Alignment of portions of sequence data showing polymorphisms in the three different genomic DNAs is shown in Fig. 3, where polymorphic residues are shaded and the MN-DMD31 target site is boxed (solid line). It should be noted that although a number of extra primers were specifically designed and used for sequencing, the number of residues in the poly(T) sequence (boxed with dotted line) just downstream from the MN target site could not be resolved. The right-hand arm of homology was therefore designed to be downstream of this poly(T) sequence within intron 44. The left-hand arm of homology was designed to be upstream of the MN target site. At polymorphic positions, the arms of homology within the repair matrix were synthesized to be homologous to the sequence present in genomic DNA of del45–52 human DMD cells.

Designing the flanking arms of homology for the exon 45–52 repair matrices. To establish complete homology, sequencing of ∼3 kb of intron 44 upstream and downstream of the meganuclease target site was performed on PCR amplicons produced by PCR with overlapping primers on genomic DNA from 293T cells (sequence A), del45–52 DMD cells (sequence B), and del48–50 DMD cells (sequence C). Alignment of those portions of sequence data showing polymorphisms in the three different genomic DNAs is shown. The residues showing polymorphism are shaded, and the DMD31 target site is boxed (solid line). It should be noted that although a number of extra primers were specifically designed and used for sequencing, the number of residues in the poly(T) sequence (boxed with dotted line) just downstream from the target site could not be resolved. The arm of homology was therefore designed to be 3′ of this poly(T) sequence.
The cDNA block, flanking splice sites, and arms of homology have been incorporated into the RM45–52 targeted repair matrices (Fig. 4). Two basic RM45–52 repair matrix designs were constructed, one with 1-kb arms of homology (RM45–52 S1) and the other with 1.5-kb arms of homology (RM45–52 S2). In both cases the arms of homology flank a cDNA encoding the block of exons 45–52, which are deleted in del45–52 human DMD cells. This cDNA block is flanked on either side by synthetic splice donor and acceptor sites derived from the human immunoglobulin and human β-globulin gene, respectively, and other regulatory sequences to enhance splicing events (i.e. synthetic branch point, polypyrimidine tract, and intronic splicing enhancer sequence from rat Fgfr2 gene [DISE element]) (Seth et al., 2008). The synthesized targeting matrices were then inserted in the reverse orientation into an IDLV backbone (pRRLsinPPT-ISce-IT) to avoid aberrant and unexpected splicing of the mRNA viral genome. Correct synthesis and subcloning were confirmed by restriction enzyme digestion (data not shown). For the two targeting matrices, IDLV vectors were generated by standard transient transfection technology in 293T cells (LVdRM45–52 S1 and LVdRM45–52 S2). Real-time PCR was used to accurately quantify viral genomes (data not shown). Titers of 5×108/ml were typically achieved.
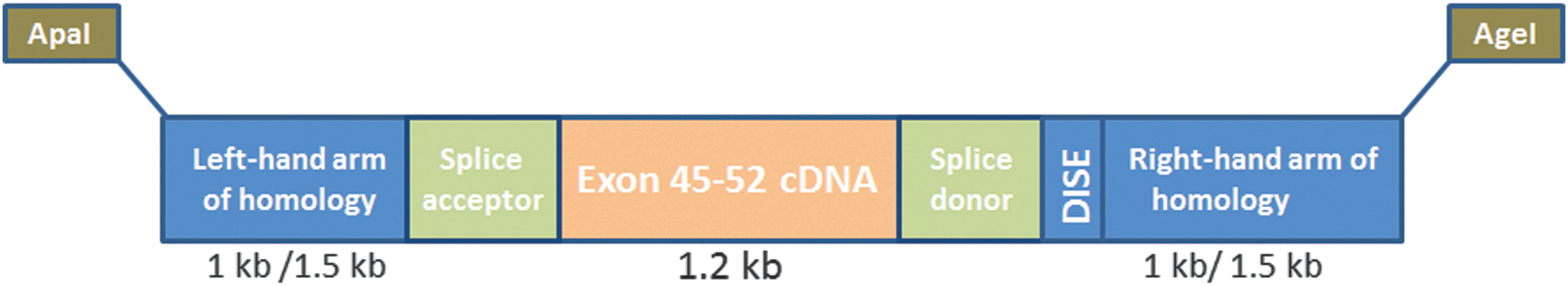
Schematic showing design of targeting matrix for exon 45–52 cDNA knock-in. The exon 45–52 cDNA block with flanking artificial splice sites (synthetic branch point, polypyrimidine tract and splice acceptor site, synthetic splice donor, and intronic splicing enhancer sequence from rat Fgfr2 gene [DISE element]) and arms homologous to sequences neighboring the MN target site within intron 44 have been incorporated into a repair matrix.
Targeted exon knock-in and gene correction in DMD myoblasts by cotransduction with MN and repair matrix vectors
To evaluate the potential of combined MN-induced DSBs and the presence of targeting matrices to induce gene repair by HR, del45–52 human DMD cells were cotransduced with the MN vector (LVcMN3631) and one or the other of the two RM vectors (LVdRM45–52 S1 or LVdRM45–52 S2). After 72 hr, genomic DNA and total RNA were harvested for analysis. In the case of genomic DNA, semi-nested PCR using LongAmp Taq polymerase was performed to assess the incidence of HR repair on both sides of the DSB induced by MN-3631. For the left-hand side of the DSB, this was examined with nested forward primers to amplify from endogenous intron 44 of the DMD gene upstream of the 1.5-kb left arm of homology to exon 45 within the targeting matrices (same reverse primer in both rounds) (Fig. 5a). An amplicon would be detectable only where HR has occurred because exon 45 is not present in nontreated genomic DNA of del45–52 human DMD cells. In addition, no amplicon would be produced from the repair matrix itself because the forward primers used are upstream of the 1.5-kb left arm of homology within the targeting matrix. Similarly, for the right-hand side of the DSB, HR occurrence was examined by amplification of a product from exon 51 (same forward primer in both rounds) to endogenous intron 44 of DMD gene downstream of the 1.5-kb right arm of homology (nested reverse primers) (Fig. 5b). An amplicon would be detectable only where HR has occurred because exon 51 is not present in nontreated genomic DNA of del45–52 human DMD cells. In addition, no amplicon would be produced from the repair matrix itself because the reverse primers used are downstream of the 1.5-kb right arm of homology within the targeting matrix. Highlighted products in Fig. 5 show that HR has occurred between endogenous intron 44 and the right and left arms of homology within the targeting matrix (RM45–52 S2), as a specific consequence of MN3631 cleavage and DSB production. It should be noted that longer arms of homology (1.5 kb) within the targeting matrix appear to drive more efficient homologous recombination, because the targeting matrix with shorter (1-kb) arms of homology did not produce any detectable evidence of HR (data not shown). This suggests that the length of homology within the targeting matrix influences the efficiency of the HR event between endogenous genome and targeting matrix. Although the primers used here would not detect off-target insertion events, the likelihood of the cDNA block being inserted into a nonspecific site is small. BLAST analysis of the whole human genome shows that intron 44 of the DMD gene is the only sequence that should be targeted by MN3631 (see Table 2). In addition, the cDNA block is bounded by two 1.5-kb arms that are homologous to the DMD intron 44 sequence around the MN target site, which ensures that the cDNA block will drive HR and that this will occur only within intron 44 of the DMD gene.
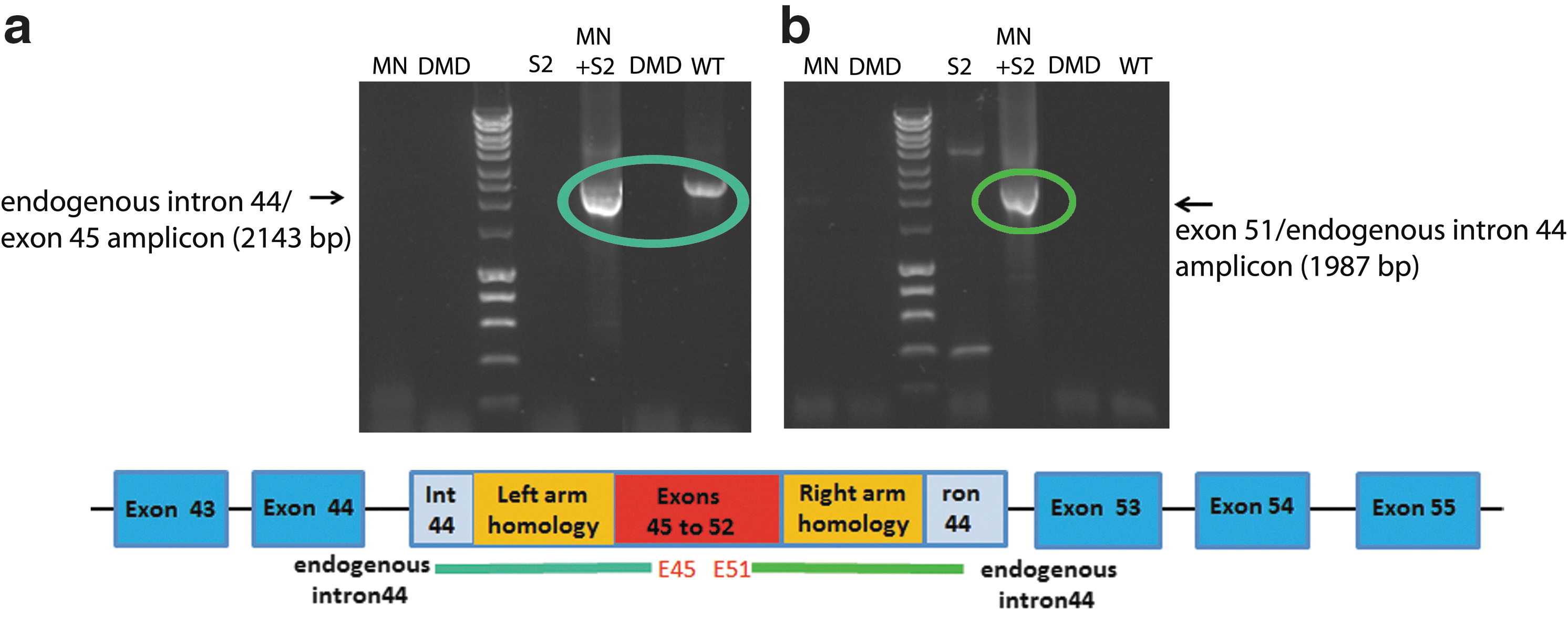
Homologous recombination of the targeting matrix and endogenous intron around the MN cleavage site. Del45–52 human DMD myoblasts were cotransduced in 24-well plates with LVcMN3631 (qPCR MOI, 1000) and LV-RM45–52 S2 (qPCR MOI, 1000). After 72 hr, genomic DNA was harvested and seminested PCR using LongAmp Taq polymerase was performed to examine homologous recombination between intron 44 in the targeting matrix and endogenous intron 44 in del45–52 human DMD myoblasts. PCR was performed with primers to amplify from endogenous intron 44 upstream of the 1.5-kb left arm of homology to exon 45
To evaluate the potential of the cDNA knock-in into DMD patient genomic DNA to be transcribed into full-length dystrophin mRNA, total RNA was harvested from the cotransduced DMD cells and nested RT-PCR was performed. Second-round primers were used to amplify from exon 44 (endogenous) to exon 46 (matrix) (Fig. 6a), and to amplify from exon 51 (matrix) to exon 54 (endogenous) (Fig. 6b). The highlighted products in Fig. 6 show that exon 45–52 cDNA from the targeting matrix has been successfully knocked-in between endogenous exons 44 and 53 and that this corrected genomic DNA was successfully transcribed into mRNA, demonstrating that the corrected DMD gene is functional. However, it has not yet been possible to demonstrate that the corrected mRNA leads to dystrophin protein expression. Without any form of enrichment of corrected cells by selection or clonal dilution, the levels of dystrophin protein expressed are likely to be too low to be detectable by western blot or by immunocytochemical staining for dystrophin protein in cultured muscle cells.
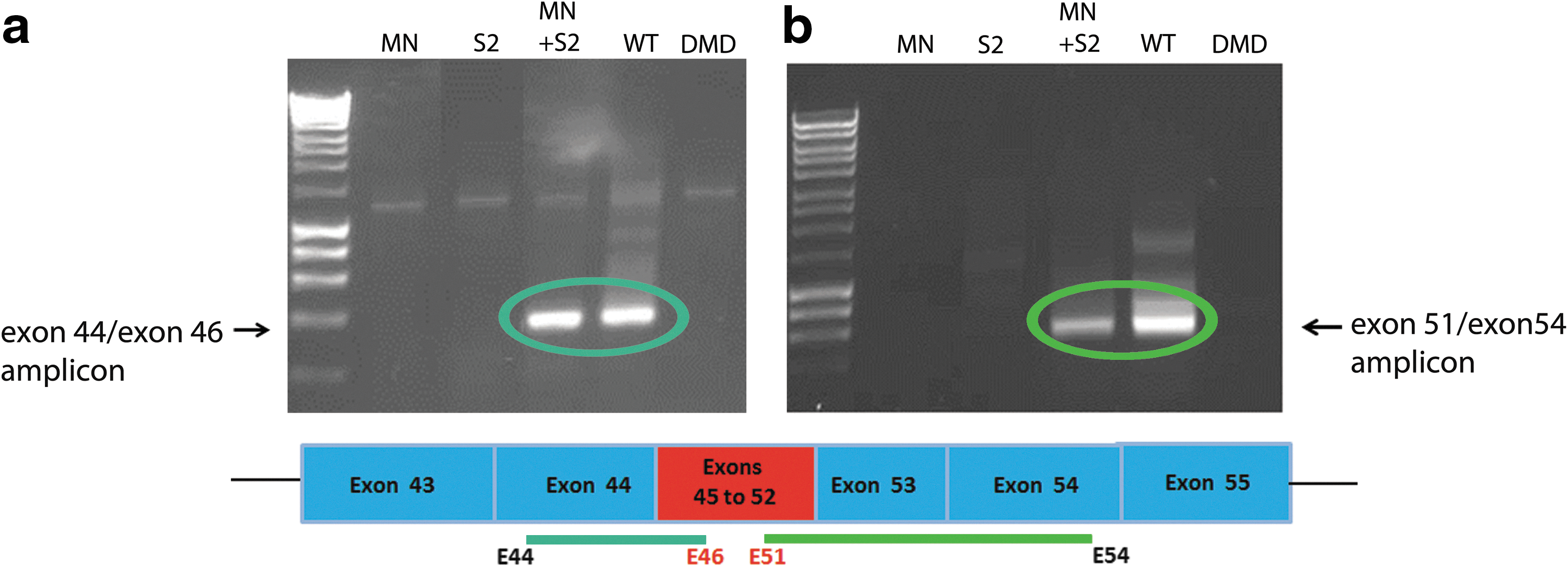
Exon 45–52 restoration in dystrophin mRNA by homologous recombination of the targeting matrix on MN cutting. LVcMN3631 (qPCR MOI, 1000) and LVdRM45–52 S2 (qPCR MOI, 1000) were used to cotransduce del45–52 human DMD myoblasts in 24-well plates. After 72 hr, RNA was harvested and nested RT-PCR was performed to examine corrected mRNA expression. In
Discussion
The work reported here is an investigation into DMD gene correction of deletions in human patient myoblasts, using a meganuclease (MN), a synthetic target-specific DNA endonuclease, together with a specific homologous recombination repair matrix. We clearly demonstrate that for the first time, in situ repair of human DMD gene deletions on the X chromosome can be achieved by MN-induced locus-specific genome cleavage, and homologous recombination to knock-in deleted exons by gene-targeting strategies, suggesting exciting therapeutic potential for developing permanent gene correction therapy in DMD patient cells and tissues. At present, transplantation of zinc finger nuclease (ZFN)-mediated genetically modified T cells into recipients in a phase 1 clinical trial has been completed with successful results in patients with HIV infection (clinicaltrial.gov; identifier NCT00842634), suggesting the great potential of gene-targeting strategies in the clinic.
As a gene therapy, genome correction using MNs and targeting repair matrices would induce permanent changes in the DMD gene, removing the need for repeated long-term administration of a drug or a vector. The benefit of this approach as a clinical therapy in terms of reducing the cost and immunological and toxicological risk is obvious. Current preclinical and clinical studies for DMD have largely concentrated on the use of AAV vector delivery of microdystrophin (Schnepp et al., 2003; Liu et al., 2005; Wang et al., 2007a,b), exon skipping induced by antisense oligonucleotides (Jearawiriyapaisarn et al., 2008; Cirak et al., 2011; Goemans et al., 2011; Goyenvalle et al., 2012), and use of ataluren, a novel small molecular agent designed to induce translational read-through of premature termination codons (PTCs) (Welch et al., 2007; Wilton, 2007). These strategies face a number of potential limitations that must be overcome before they can be considered a general treatment for DMD. These hurdles include potential immunological (Herzog, 2007; Wang et al., 2007a,b) and toxicological risks, the need for repeated administration, high dosing costs, and clinical applicability being restricted to certain subsets of patients with DMD. Genome correction has the major advantage that with the right sequence-specific MN and targeting repair matrix, any DMD mutation would be treatable, unlike with ataluren, which is applicable only in patients with PTCs resulting from nonsense mutations, and exon skipping, which is applicable only in patients with frame-shifting deletions. In addition, genome correction therapy for DMD would produce full-length dystrophin protein, whereas only truncated protein expression is possible with exon-skipping and AAV vector-based strategies, leading to the milder Becker muscular dystrophy phenotype.
We have been involved in the development of a battery of MNs targeting various deletion hotpots in the DMD gene. Here we report work using two nuclease variants targeting intron 44. We show that lentivectors carrying MNs are expressed efficiently in human DMD patient cells. MN expression leads to significant cutting of the target locus in these cells, with the frequency of InDels (diagnostic of cutting at the target site) reaching 10%. When used in combination with a repair matrix designed to mediate knock-in of exon 45–52 cDNA, the expected targeted modification was detected at the genomic DNA level in human DMD patient cells. This correlated with expression of dystrophin mRNA including the knocked-in exons, demonstrating that genome-corrected DNA is functionally transcribed.
We have clearly demonstrated the proof of principle that a genome correction for DMD via HR, using this strategy, is possible; the potential to correct permanently all DMD deletions could be possible in future. Although the MN used in this study had a cutting efficiency of 10%, the overall frequency of correction seems too low to allow the detection of dystrophin protein. Work will now concentrate on steps to improve the efficiency of HR repair by using strategies such as pharmacological inhibition of DNA-PK, a DNA repair enzyme that stimulates NHEJ and blocks HR (Peng and Lin, 2011; Li et al., 2012). We are also currently developing a linear amplification-mediated PCR (LAM-PCR) reaction to allow quantification of repaired genomic DNA. In line with the work of Lombardo and colleagues (2007) and Jacome and colleagues (2009), future studies will concentrate on the development of nonintegrating MN-expressing vectors to make the gene correction strategy clinically applicable, and on the isolation of corrected human DMD myogenic cells ex vivo. Steps are currently being taken to isolate corrected cells by dilution cloning, and to introduce a marker into the repair matrix that will allow selection of corrected cells. These corrected cell clones will be used to demonstrate dystrophin protein production in culture, for transplantation into immunodeficient dystrophic mice, and for development of appropriate in vivo studies to establish further the therapeutic potential of genome correction as a treatment for DMD.
Footnotes
Acknowledgments
The authors thank the Association Français contre les Myopathies for funding this work and the platform for human cell immortalization of the Myology Institute in Paris for DMD patient myoblast cell lines. Authors at RHUL (L.P., G.D., T.K., O.I., and R.J.Y.M.) designed and conducted the experimental work, and contributed to the choice of genomic target sequences in the human DMD gene, analyses of data and writing the manuscript. Authors at AIM (X.L., K.M., T.V., and V.M.) contributed to work involving immortalized DMD cells, and analyses of data. Authors at Cellectis (A.D., A.G., F.P., and F.C.) contributed to the choice of genomic target sequences and design of engineered meganucleases.
Author Disclosure Statement
Authors A.D., A.G., F.P., and F.C. are employees of Cellectis. Authors L.P., G.D., and R.J.Y.M. are registered as co-inventors on a patent application for protection of the repair matrix design.