
Abstract
Spinal muscular atrophy (SMA) is a kind of neuromuscular disease characterized by progressive motor neuron loss in the spinal cord. It is caused by mutations in the survival motor neuron 1 (SMN1) gene. SMN1 has a paralogous gene, survival motor neuron 2 (SMN2), in humans that is present in almost all SMA patients. The generation and genetic correction of SMA patient-specific induced pluripotent stem cells (iPSCs) is a viable, autologous therapeutic strategy for the disease. Here, c-Myc-free and non-integrating iPSCs were generated from the urine cells of an SMA patient using an episomal iPSC reprogramming vector, and a unique crRNA was designed that does not have similar sequences (≤3 mismatches) anywhere in the human reference genome. In situ gene conversion of the SMN2 gene to an SMN1-like gene in SMA-iPSCs was achieved using CRISPR/Cpf1 and single-stranded oligodeoxynucleotide with a high efficiency of 4/36. Seamlessly gene-converted iPSC lines contained no exogenous sequences and retained a normal karyotype. Significantly, the SMN expression and gems localization were rescued in the gene-converted iPSCs and their derived motor neurons. This is the first report of an efficient gene conversion mediated by Cpf1 homology-directed repair in human cells and may provide a universal gene therapeutic approach for most SMA patients.
Spinal muscular atrophy (SMA) is a kind of neuromuscular disease characterized by progressive motor neuron loss in the spinal cord. It is caused by mutations in the survival motor neuron 1 (SMN1) gene. SMN1 has a paralogous gene, survival motor neuron 2 (SMN2), in humans that is present in almost all SMA patients. The generation and genetic correction of SMA patient-specific induced pluripotent stem cells (iPSCs) is a viable, autologous therapeutic strategy for the disease. Here, c-Myc-free and non-integrating iPSCs were generated from the urine cells of an SMA patient using an episomal iPSC reprogramming vector, and a unique crRNA was designed that does not have similar sequences (≤3 mismatches) anywhere in the human reference genome. In situ gene conversion of the SMN2 gene to an SMN1-like gene in SMA-iPSCs was achieved using CRISPR/Cpf1 and single-stranded oligodeoxynucleotide with a high efficiency of 4/36. Seamlessly gene-converted iPSC lines contained no exogenous sequences and retained a normal karyotype. Significantly, the SMN expression and gems localization were rescued in the gene-converted iPSCs and their derived motor neurons. This is the first report of an efficient gene conversion mediated by Cpf1 homology-directed repair in human cells and may provide a universal gene therapeutic approach for most SMA patients.
Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disease due to the loss of α-motor neurons in the spinal cord, with an occurrence of 1/6,000–1/10,000 live births and a carrier frequency of 1/40–1/60. 1,2 Mutations in the survival motor neuron 1 (SMN1) gene are responsible for SMA. 3 SMN2, a gene paralogous to SMN1, has high sequence homology to SMN1. A silent mutation in exon 7 (840 C>T) of SMN2 causes a splicing defect that leads to the omission of exon 7 in most of the mRNA transcripts, and 90% of the yield is an unstable, truncated protein (SMNΔ7). 4 The SMN2 copy number inversely correlates with the disease severity. 5 More than two partially functional copies of the SMN2 gene are present in most SMA patients. Therefore, SMN2 has long been identified as an ideal target for SMA therapy.
Several therapeutic studies have been done to improve SMN protein levels via increasing exon 7 inclusions in SMN2 transcripts. Researchers have employed different methods such as compounds, 6 bifunctional oligonucleotides, 7 or anti-sense oligonucleotide (ASO), 8 –10 and the SPINRAZA, 11 an ASO drug, has been approved by the U.S. Food and Drug Administration (FDA). However, these approaches cannot correct SMN1 mutations or permanently restore SMN expression. Gene conversion of SMN2 to SMN1 in SMA skin fibroblast 12 and SMA patient-specific induced pluripotent stem cells (iPSCs) using single-stranded oligodeoxynucleotide (ssODN) has been demonstrated, in which the genetic modification turned out to be permanent and heritable. 13 However, the low efficiency and tumorigenic risk of c-Myc-iPSCs limits their application.
The clustered regularly interspaced short palindromic repeats (CRISPR) system has emerged as a powerful tool for efficient genome editing in cells and organisms. 14 –16 The CRISPR-associated 9 (Cas9) nuclease can be targeted to specific genome sites with a specificity 20-nt targeting sequence single-guide RNA (sgRNA) to generate a blunt end. 14 Previous studies have demonstrated that CRISPR/Cas9 can efficiently correct mutations in human cells. 17 –19 Recently, a new CRISPR from Prevotella and Francisella 1 (Cpf1), which recognizes a thymidine-rich protospacer-adjacent motif (PAM) and generates a sticky end distal to the PAM site guided by a single CRISPR RNA (crRNA), 20 was shown to be effective in mammalian genome editing. 20 –23 Although Duchenne muscular dystrophy mutation correction via non-homologous end-joining (NHEJ) using Cpf1 in iPSCs has been successful, 24 its potential usefulness in the perfect correction of genetic mutations in mammalian cells of disease has yet to be demonstrated.
Here, c-Myc-free and non-integrating iPSCs were generated from urine cells of an SMA patient using an episomal iPSC reprogramming vector. It was demonstrated that Cpf1 provides a robust and efficient RNA-guided genome editing system for seamless genetic conversion of SMN2 to an SMN1-like gene, thereby restoring SMN expression in the gene-converted iPSCs and their derived motor neurons (iMNs).
Materials and Methods
Reprogramming of human urine cells
Sterile urine was collected from a 22-year-old male diagnosed with homozygous deletion of exon 7 and exon 8 of the SMN1 gene following informed consent. The outgrowth renal tubular epithelial cells (urine cells) were collected for iPSC generation between passages 1 and 3. Cells (∼4–6 × 105) were nucleofected using an Amaxa™ Basic Nucleofector™ Kit for primary mammalian epithelial cells (Lonza) and a Nucleofector II (Lonza) running program T020. For each experiment, 3.5 μg of pEP4E02SET2K and 2 μg of the pCEP4-hsa-miR302-367 were used. The transfected cells were plated on a Matrigel™ (BD Biosciences) coated six-well plate in medium from a REGM Bullet Kit. After 8–12 h, the medium was replaced by mTeSR1 (Stemcell Technologies). Twenty days after transfection, embryonic stem cell (ESC)-like clones were picked up and expanded in mTeSR1 medium for further characterization. This study was approved by the Ethics Committee of Central South University, and all participants provided written informed consent.
CRISPR design, assembly, and efficiency evaluation
CRISPR RGEN Tools (
The pCAG-EGxxFP (plasmid #50716) reporter was obtained from Addgene. The short polymerase chain reaction (PCR) product flanking the crRNA target site was cloned into the multiple cloning site of pCAG-EGxxFP by BamHI and EcoRI, yielding pCAG-EGxxFP-SMN2T/C. For the gene disruption assay, HEK293T cells were transfected with 500 ng of CRISPR/AsCpf1, 250 ng of SMN2 gene-specific crRNA, and 500 ng of pCAG-EGxxFP-SMN2T/C using jet-PRIMER (Poly Plus). The expression of restored enhanced green fluorescent protein (EGFP) was measured by fluorescence microscopy and flow cytometry 72 h after transfection.
Nucleofection and drug selection
Conversion of T to C in exon 7 of the SMN2 gene with ssODN was performed in accordance with the following procedure. SMA-iPSCs on a Matrigel-coated 60 mm dish were used for transfection. Before transfection, cells were incubated with 10 μM of ROCK Inhibitor (Stemcell Technologies) for 2 h. Then the cells were disassociated into single cells by trypLE™ Select (Life Technologies) and counted. Subsequently, 6 μg of AsCpf1 plasmid, 3 μg of crRNA1 vector, and 1 μL of 40 μM ssODN were mixed with 1.6 × 106 SMA-iPSCs using 100 μL of Human Stem Cell Nucleofector Kit 2 (Lonza) and nucleofected with Nucleofector II under program B016. Cells were plated on a Matrigel-coated six-well plate in mTeSR1 containing 10 μM of Y27632 for 24 h. Then, 50 μg/mL of G418 was used to select cells for 3 days. Another 3 days later, resistant cells were detached with trypLE™ Select and counted. One thousand cells were re-plated on mouse embryonic fibroblasts feeder cells in ES medium supplemented with 10 μM of Y27632 for 24 h and changed to ES medium without Y27632 every day until clones picking.
PCR detection of gene-converted clones
Genomic DNA was isolated using phenol/chloroform extraction. PCR was performed using LA Taq DNA polymerase (TaKaRa) according to the manufacturer's instructions. For each PCR, 200 ng of genomic DNA was used with Hpy188I-F and Hpy188I-R to amplify a 190 bp product of the SMN2 gene. Then, the PCR products were digested by Hpy188I (New England Biolabs) and analyzed by polyacrylamide gel electrophoresis (PAGE). The positive PCR products were cloned and sequenced by Sanger sequencing.
iPSC differentiation into motor neurons
Differentiation of MNs from iPSCs was performed, as previously described. 25 Briefly, iPSCs were dissociated with dispase (1 mg/mL) and cultured in MN-induction medium, including Dulbecco's modified Eagle's medium (DMEM)/F12, Neurobasal medium at 1:1, 0.5 × N2, 0.5 × B27, 0.1 mM of ascorbic acid (Sigma–Aldrich), and 1 × Glutamax (all others from Life Technologies). Different combinations of CHIR99021, DMH1, SB431542, retinoic acid (RA) (Sigma–Aldrich), purmorphamine (Pur), valproic acid (Sigma–Aldrich), and DAPT (all others from Selleck) were added to the medium at different stages (Fig. 4A).
For neuromuscular junction assessment, C2C12 cells were cultured in DMEM (1 g/L glucose) with 10% fetal bovine serum. For differentiation, cells were seeded in plates coated with Matrigel and maintained in DMEM containing 2% horse serum for 5 days. 26 MNX1+ iMNs were plated onto myotubes for 10 days, and neuromuscular synapses were labeled by choline acetyltransferase (ChAT) and α-bungarotoxin (BTX; Thermo Fisher Scientific).
Quantitative reverse transcription PCR analysis
Total RNA was isolated with Trizol reagent (Sigma–Aldrich). RNA was treated with DNase (Thermo Fisher Scientific) to eliminate any residual DNA. Quantitative reverse transcription PCR (qRT-PCR) was performed using the primers, as previously reported,
27
with a HiScript II One Step qRT-PCR SYBR Green Kit (Vazyme) according to the manufacturer's instructions in a Bio-Rad CFX96 Touch q-PCR system. The hypoxanthine phosphoribosyl transferase (HPRT) gene was amplified as an endogenous control. One microliter of each PCR product was analyzed by PAGE. All the primers are shown in Supplementary Table S1 (Supplementary Data are available online at
Immunocytochemistry
Cells were plated in Matrigel-coated 24-well chamber slides. At 48–72 h post plating, the cells were washed with Dulbecco's phosphate-buffered saline (DPBS) and fixed for 20 min with 4% paraformaldehyde. After washing, the cells were permeabilized with DPBS-0.1% Triton X-100 for 15 min and blocked for 30 min in 5% bovine serum albumin (BSA). Cells were then incubated with first antibodies in 5% BSA at 4°C overnight. Cells were washed for the appropriate times with DPBS-0.1% Triton X-100. After incubating with secondary antibodies for 1 h, the samples were coverslipped with 4′,6′-diamidino-2-phenylindole (DAPI). Slides were examined, and images were captured under the microscope. To analyze the number of gems analysis, a previously described protocol was used. 28 At least 100 nuclei per sliced were surveyed via randomly selected field from each sliced by two independent researchers who were blinded to the status (untreated or treated). All antibodies are listed in Supplementary Table S2.
Western blot
Cells were lysed with RIPA supplemented with 1 × phenylmethanesulfonyl fluoride (1 mM) and proteinase inhibitor for 5 min on ice. The lysate was disrupted by ultrasound and heated to 90°C for 10 min. The samples were measured by the BCA assay using BSA standards (Thermo Fisher Scientific), and 15 μg of protein per group was resolved by 10% sodium dodecyl sulfate (SDS-PAGE) and transferred to a polyvinylidene fluoride membrane. After blocking with 5% nonfat milk in 0.1% TBST (TBS containing 0.1% Tween-20), the membrane was incubated overnight with mouse anti-human SMN and anti β-actin at 4°C. The membrane was then incubated with goat anti-mouse IgG horseradish peroxidase-linked secondary antibody (1:10,000; Sigma–Aldrich) for 1 h at room temperature and visualized using an ECL detection Kit (Thermo Fisher Scientific). Antibodies are listed in Supplementary Table S2.
Statistical analysis
Data were analyzed with GraphPad Prism v5.01. Student's t-test was used to compare data between two groups. One-way analysis of variance (ANOVA) was performed to compare data among three or more groups, followed by Tukey's multiple comparison test to compare between two groups. p-Values of <0.05 were considered statistically significant.
Results
Generation and characterization of c-Myc-free and non-integrating patient-specific iPSCs
Genomic DNA of a 22-year-old male patient with SMA type III was collected and used as template for multiplex ligation-dependent probe amplification (MLPA). Non-signal of the SMN1 exon 7 and exon 8 fragment was detected in the SMA diagnostic test, while three signals of SMN2 and a normal karyotype were detected (Fig. 1A), indicating the deletion of exon 7 and exon 8 of SMN1, along with three copy numbers of SMN2 in the patient.

Genotyping and c-Myc-free and non-integrating induced pluripotent stem cell (iPSC) generation.
Urine cells were collected from the patient, cultured, and nucleofected (Fig. 1B) with plasmids encoding OCT4, SOX2, SV40 large Tantigen, KLF4, and hsa-miR302-367cluster to replace c-Myc (Supplementary Fig. S1 and Supplementary Table S1). 29 These plasmids were progressively lost from cells, resulting in the c-Myc-free iPSCs without the integration of vectors and exogenous sequences. As a normal control, human iPSCs (hiPSCs) were also generated from the urine cells of a 26-year-old healthy male donor using the same method, which contained two copies of SMN1 and two copies of SMN2 (Supplementary Fig. S2A and B). After contiguous induction, human ESC-like clones were isolated for further characterization (Fig. 1C).
All the iPSC clones expressed pluripotent markers Oct3/4, Nanog, tumor-related antigen (TRA)-1-60, TRA-1-81, and surface antigens stage-specific embryonic antigen (SSEA)-4, while SSEA-1 could not be detected (Fig. 1D and Supplementary Fig. S2D). All iPSC lines maintained a normal karyotype (Fig. 1E and Supplementary Fig. S2C). All iPSC lines were demonstrated to form teratomas in vivo. After staining with hematoxylin and eosin, primitive gut (endoderm), cartilage (mesoderm), and neural tissue (ectoderm) were observed (Fig. 1F and Supplementary Fig. S2E) and free of reprogramming vectors (Fig. 1G).
Construction of CRISPR/AsCpf1 and activity assay
To convert the SMN2 gene 840 T to C in situ, two crRNAs and one sgRNA were designed that recognized the SMN2 exon 7 locus (Fig. 2A and Supplementary Table S1). The study used the pCAG-EGxxFP, which contained partially duplicated EGFP coding sequences and the crRNAs and sgRNA target sites, to validate the specific cleavage activities of the crRNAs and sgRNA. After cleavage by Cpf1 or Cas9 and the duplicated sequence-mediated repair, the EGFP expression was restored (Fig. 2B). Therefore, the AsCpf1-crRNA (or Cas9-sgRNA) and pCAG-EGxxFP-SMN2T were transfected into HEK293T cells. To confirm whether the RNA-guided endonucleases continuously recut the edited site, the AsCpf1-crRNA (or Cas9-sgRNA) was also co-transfected with pCAG-EGxxFP-SMN2C. The reconstituted EGFP fluorescence was detected by flow cytometry (Fig. 2C and Supplementary Fig. S3), showing that crRNA1 had high efficiency and specificity.
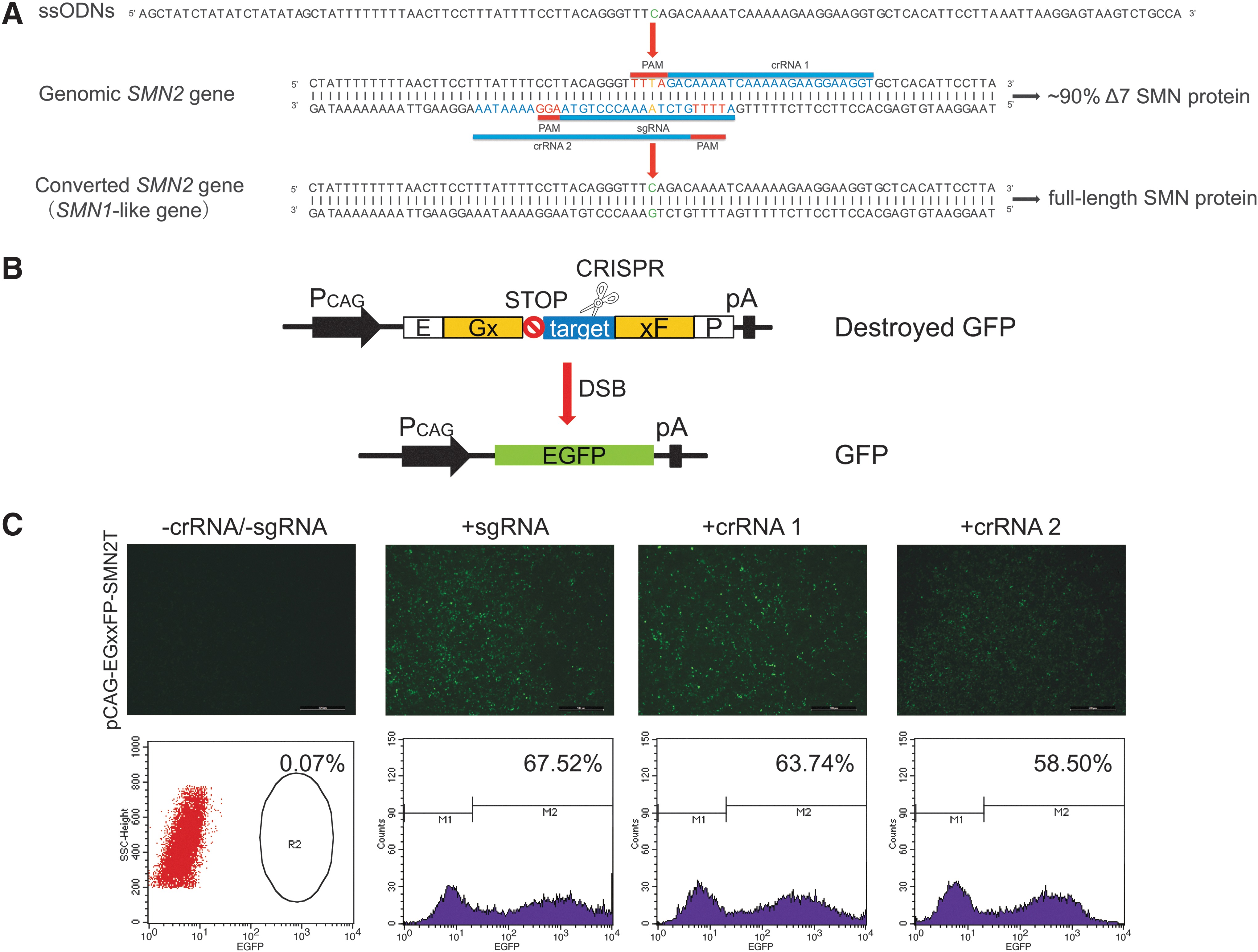
Site-specific CRISPR/Cpf1-crRNA or CRISPR/Cas9-sgRNA design and activity assay.
Gene conversion of SMN2 to SMN1-like gene using CRISPR/Cpf1 and ssODN
The direct exchange of T to C at position +6 of exon 7 promotes the inclusion of exon 7 in SMN2. 12,13 To convert the T to C of the SMN2 gene in SMA-iPSCs (Fig. 2A), a 120 nt ssODN was synthesized mapping to the specific SMN1 sequence. The SMA-iPSCs were transfected with SMN2-specific crRNA1, AsCpf1, and ssODN. Then, 36 mono-clones were picked up, and the gene-converted clones were identified and detected using PCR amplification with the primers Hpy188I-F/R and digestion with the endonuclease Hpy188I digesting. Of the 36 tested clones, the Hpy188I digest demonstrated four gene-converted clones (i.e., clones 4, 9, 24, and 36; Fig. 3A).

Gene conversion of SMN2 and SMN expression in iPSCs.
To detect whether these mono-clones had indels in the converted copies of SMN2, the encompassing region was PCR amplified, cloned, and sequenced, and more than nine sequences were analyzed. All the converted copies seemed to be exactly the same as SMN1, though clone 9 had two converted copies with another mutant, and clone 4 also had one converted copy and one mutant (Supplementary Fig. S4A). In addition, to detect whether the exogenous sequence residual was retained in the converted cells, all converted SMA-iPSCs (cSMA-iPSCs) clones were screened via highly sensitive primers, which could detect as little as 10 fg of CRISPR/Cpf1 plasmid DNA, and there were no detectable bands in these cSMA-iPSCs clones (Supplementary Fig. S4B). These results demonstrated that the conversion of SMN2 840 T to C is efficiently and seamlessly achieved by the combination of CRISPR/Cpf1 with ssODN.
SMN expression in converted iPSC clones
Next, the SMN expression was evaluated in converted iPSCs. The transcripts of full-length SMN (SMN-FL) and truncated SMN (Δ7 SMN) were detected by qRT-PCR, with the HPRT housekeeping gene used as a control. Each sample was run in triplicate. The SMN-FL transcription levels in converted iPSCs were higher than those in SMA-iPSCs (Supplementary Fig. S5A–C).
SMA patient cells are usually deficient in SMN sub-nuclear bodies or gems. To test whether the increased level of SMN after gene conversion could induce nuclear accumulation of gems, immunofluorescence analysis was performed of the converted iPSCs using untreated SMA-iPSC as a negative control and hiPSCs as a positive control. The presence of SMN protein in the nucleus and cytoplasm in converted clone 36 was accompanied by a profound increase in the number of gem structures, while few SMN gems were observed in untreated SMA-iPSCs (Fig. 3B and C), demonstrating that conversion of SMN2 to SMN1-like gene increases SMN localization to gems.
To validate the SMN protein level further, the cell lysates were collected, and the SMN protein was detected via Western blot. The SMN protein level in converted clone 36 was similar to that in hiPSCs and much higher than that in SMA-iPSCs (Fig. 3D). Together, these results indicate that the conversion of SMN2 840 T to C induced stable genetic correction in SMA-iPSCs.
To test the specificity of crRNA1 used in the experiment, the entire human reference genome was surveyed via the Cas-OFFinder (
Restoration of SMN expression in the converted iPSC-derived MNs
Next, the differentiation of c-Myc-free and non-integrating iPSCs into MNs was validated before and after the gene conversion, and the restoration of SMN expression was evaluated in the converted iMNs. iMNs from SMA-iPSCs, converted iPSCs (cSMA-iMNs), and normal control hiPSC (hiMNs) were differentiated by using a combination of small molecules that regulate multiple signaling pathways, as described by Zhong-Wei et al. 25 (Fig. 4A). After 12 days under differentiation conditions, a robust population of OLIG2+ motor neuron progenitors (MNPs) had multiplied (Fig. 4B), and these derived MNPs could expand more than eight passages (Supplementary Fig. S8A). Subsequently, the WNT agonist and dual SMAD inhibitor were withdrawn, and a high concentration of RA with 0.1 μM of Pur, an SHH signaling agonist, were added into the culture system. After 6 days, nearly pure ISLET1-positive iMNs were obtained at day 18 (Fig. 4B and Supplementary Fig. S8B). Then, DAPT, a NOTCH signaling inhibitor, was applied to enhance the MN maturation. After a further culture for 1 week, most MNs expressed ChAT and were positive for SMI32 (Fig. 4B). During MN maturation, it was found that if plated at low density, the SMA-iMNs could hardly survive. qRT-PCR revealed that ChAT (Fig. 4C) and vesicular acetylcholine transporter (VAChT; Fig. 4D) expression were markedly reduced in SMA-iMNs, which is consistent with previous reports, 31 but they were restored in cSMA-iMNs, suggesting that transmitter synthesis and release may be rescued in cSMA-iMNs.

Differentiation of iPSCs into motor neuron.
To analyze the ability of iMNs to form neuromuscular junctions, the iMNs were co-cultured with C2C12-derived myotubes for 10 days. More aggregated BTX-positive acetylcholine receptors were observed on myotubes overlapping with ChAT+ iMNs in cSMA-iMNs or hiMNs compared with SMA-iMNs (Fig. 5A) and the detectable gems significantly increased in the nuclei of the cSMA-iMNs (Fig. 5B). In addition, the cell lysates of ChAT+ iMNs were collected to estimate SMN expression. SMN expression in cSMA-iMNs was higher than that in SMA-iMNs and similar to that in hiMNs (Fig. 5C). These results demonstrated that SMN expression was restored in the converted iPSC-derived MNs.

SMN expression in motor neuron.
Discussion
SMA is a devastating neuromuscular disorder. The carrier risks for the parents is as high as 1/43 in China. 32 Stem cells, especially iPSCs, have proven to be a useful in vitro disease model and potential therapeutic cell source. To facilitate iPSCs to be more suitable for clinical application, some methods for generating patient-derived iPSCs that are free of exogenous residuals and/or tumorigenic risk factors should be explored. Corti et al. previously described the generation of iPSCs with no exogenous gene residues using the OriP/Epstein–Barr nuclear antigen-1-based vector containing c-Myc, OCT4, Klf4, and Sox2. 13 However, c-Myc increases tumor formation in iPSC-derived chimeric mice, as described by Yamanaka et al. 33 Thus, the use of c-Myc has greatly hindered the clinical application. Here, c-Myc-free and non-integrating iPSCs were generated from an SMA patient via an episomal iPSC reprogramming vector, replacing c-Myc with hsa-miR302-367cluster. Obviously, being free of the tumorigenic risk factor c-Myc makes iPSCs more suitable for basic research and future clinical applications.
Homologous recombination-mediated gene targeting has been particularly difficult in human pluripotent stem cells. The engineered nucleases ZFN, TALEN, and CRISPR provide opportunities for accurate and efficient genome editing in stem cells. Several publications have described combining engineered nucleases with a donor vector for gene modification in hemophilia A 34,35 and sickle cell disease. 36 After selection, the selection marker remains 34 or is removed from the genome using a recombinase. 35 Either way, some residual sequences may increase the risk of genome instability in the corrected iPSCs and interfere with the expression of the corrected gene. 36 In the present study, this issue was addressed while avoiding overly time-consuming and laborious methods by using ssODN as the donor template. Furthermore, the strategy of combining CRISPR/Cpf1 with ssODN could provide a robust platform for gene correction in vitro and in vivo.
With its T-rich PAM sequence, Cpf1 further expands the genome editing range of RNA-guided genome editing nucleases, which allows for applications in the correction of other disease-related mutations that do not contain G-rich PAM sequences for Cas9. Moreover, Cpf1 is more specific than Cas9. 21 In the present study, introducing a mutation simultaneously in the PAM (5′-TTTN-3′ to 5′-TTCN-3′) sufficiently prevented CRISPR/AsCpf1 from re-editing (Supplementary Fig. S3) once the SMN2 840 T was converted to C, 20,21 addressing the risk of further targeting and re-cutting by CRISPR/Cas9, even after the desired edit had been introduced in a previous study (data not shown). It is therefore suggested that “blocking mutations” should be simultaneously introduced in the 5′-NGG-3′ PAM or guide RNA binding sequence to prevent re-editing by CRISPR/Cas9. 37
Two categories of methods are used in SMA therapy. The first category includes small-molecule compounds to stimulate the basal level of SMN2 transcription, 38 small interfering RNAs to correct SMN2 splicing, 9,39 or approaches to improve SMN protein stabilization. 40 However, these strategies require repeated treatment. The second category comprises gene therapy, usually relying on viral vectors such as scAAV9s and lentiviruses to insert SMN cDNA 41 –43 or replace exon 1 of the SMN1/2 genes with functional SMN cDNA in the host genome. 44 Unfortunately, these viral approaches still have limitations in that an unwanted insertional mutation may be induced or that side effects may result from immune responses. It is well known that every SMA patient has at least one copy of the SMN2 gene, and the copy number of SMN2 is negatively correlated with SMA disease severity. 45 Although SMN1 and SMN2 encode identical SMN proteins, a single nucleotide, SMN2 840 T, leads to the modification of a splicing modulator and the omission of exon 7 in 90% of SMN2 mRNA transcripts, yielding a defective, exon 7-skipped protein isoform. 4 Therefore, the conversion the SMN2 gene to an SMN1-like gene by means of a single nucleotide mutation of T to C in exon 7 of SMN2 is an ideal gene therapy approach for SMA, though previous studies have shown that conversion efficiencies were very low (∼4%). 13 This study confirmed that a robust and efficient conversion of SMN2 to an SMN1-like gene can be achieved via HDR mediated by the newly discovered CRISPR/Cpf1 nucleases in patient-derived iPSCs. It was found that the conversion of SMN2 840 T to C corrects the mRNA splicing to include exon 7 and leads to higher levels of full-length SMN mRNA (Supplementary Fig. S5A), restoring SMN expression (Fig. 3D). The results show the success of this proof-of-concept experiment. This strategy is specific, efficient, and applicable for other human genetic diseases.
It is noted that through the multistage differentiation, the gene-converted iPSCs efficiently differentiated into MNPs and further into MNs. In this way, under the same genetic background, the multistage comparison of MN before and after gene conversion is more conducive to research on the pathogenesis of SMA. Furthermore, the differentiated MNPs expansion can be sustained for multiple passages, providing the possibility for in vivo transplantation and in vitro drug screening.
In summary, an efficient and seamless gene conversion of SMN2 to SMN1 in SMA-iPSCs was achieved using CRISPR/Cpf1 and ssODN. The SMN expression and gems localization were significantly rescued in the gene-converted iPSCs and their derived motor neurons. This is the first report of an efficient gene conversion mediated by Cpf1 HDR in human cells and may provide a universal gene therapeutic approach for almost all SMA patients.
Footnotes
Acknowledgments
We thank the patients or health control for their participation in this study, and thank Weijuan Wu and Huimin Zhu for genotyping by MLPA. This work was supported by the National Natural Science Foundation of China (81400928), the National Key R&D Program of China (2016YFC0905102) and the Fundamental Research Funds for the Central Universities of Central South University (2016zzts166 and 2016zzts168).
Author Disclosure
We declare that there are no conflicts of interest or financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.