
Abstract
A truncated gene (designated S1) encoding the receptor-binding domain (RBD) in the spike (S) protein of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) was amplified by PCR. The gene was cloned into prokaryotic expression vector pGEX-6P-1, resulting in a recombinant plasmid pGEX-SARS-S1. Subsequently, pGEX-SARS-S1 was transformed into host cells BL21(DE3)pLysS, and the expression of the S1 protein was induced by isopropyl β-D-thiogalactoside (IPTG). Polyclonal antibody against SARS-CoV S1 protein was generated in a rabbit immunized with the purified S1 protein. The reactivity of the antibody to the SARS-CoV S1 protein was confirmed by Western blot analysis. ELISA indicated that the antibody against SARS-CoV S1 protein had no cross reaction with S1 proteins of transmissible gastroenteritis virus, a porcine coronavirus, and infectious bronchitis virus, an avian coronavirus. The SARS-CoV S1 protein and its antibody are valuable reagents for related studies.
Introduction
The S protein of SARS-CoV is incorporated into the viral envelope and it is 1255 amino acids long, with low (20–27%) amino acid similarity among other coronaviruses. The low sequence similarity of the S protein implies that the S protein of SARS-CoV may have additional functions other than the usual functions of coronavirus spike proteins.(18) The S protein of SARS-CoV binds to angiotensin-converting enzyme 2 (ACE2), the function receptor, on the host cell.(10,19) The receptor-binding domain (RBD) is between residues 303 and 537 in the S protein.(20) The RBD controls what cells can be targeted for infection by SARS-CoV.(21)
Since the viral S protein is a main surface antigen and functional protein, the availability of SARS-CoV S protein benefits preparation of viral diagnostic reagents and development of vaccines. Nevertheless, isolation of S protein from the crude virus and artificial synthesis of the S protein may have some concerns on the bio-safety and production cost. In this study, we constructed a recombinant plasmid encoding the major receptor-binding domain in the S protein (designated S1) of SARS-CoV. The high level expression of the S1 protein was achieved in Escherichia coli system. Anti-S1 antibody was generated in a rabbit immunized with the purified S1 protein. The reactivity of the antibody to the SARS-CoV S1 protein was confirmed by Western blot analysis. ELISA indicated that the anti-SARS-CoV S1 antibody may be used as a diagnostic reagent in differentiating SARS-CoV S1 protein from S1 proteins of transmissible gastroenteritis virus (TGEV), a porcine coronavirus, and infectious bronchitis virus (IBV), an avian coronavirus. The current study offers necessary experimental materials for probing the molecular characteristics of SARS-CoV.
Materials and Methods
Construction of expression plasmid encoding the RBD of SARS-CoV S protein
A recombinant plasmid containing full-length S gene of SARS-CoV strain CUHK-W1 (GenBank accession no. AY278554) was used as PCR template.(12) Sense primer (CoVS1) 5′-GGGGggattcATGGGTTTTAACACTTTG and anti-sense primer (CoVS2) 5′-CCCCgaattcCTTGTTGAAATGGTTGAAA were designed to amplify a truncated S gene (nucleotides 664-1656) covering the RBD in the 5′ end part of SARS-CoV S protein. The lowercase letters included BamH I and EcoR I restriction enzyme sites, respectively. The PCR system consists of 1 μL template DNA (1 μg), 1 μL of each primer (50 pmol/each), 5 μL of dNTP mixture (TaKaRa, Dalian, China), 10X Ex-Taq Buffer (5 μL, TaKaRa), and 36.5 μL sterile H2O. The amplification condition includes 95°C for 10 min, 30 cycles of 94°C for 1 min, 56°C for 1 min, 72°C for 90 s, and a final extension of 72°C for 10 min. The resulting PCR product was purified with a DNA purification kit (KeyGen Biotech, Nanjing, China) and then inserted into Bam H I and EcoR I sites of a prokaryotic expression vector, pGEX-6P-1 (Amersham Biosciences, New York, NY). The recombinant plasmid designated as pGEX-SARS-S1 was identified by PCR and restriction enzyme digestion and visualized by ethidium bromide-containing agarose gel electrophoresis prior to DNA sequencing.
Expression and purification of SARS-CoV S1 protein
The recombinant plasmid pGEX-SARS-S1 was transformed into host cells E. coli BL21(DE3). Expression of the SARS-CoV S1 protein was induced using 1.0 or 0.5 mM isopropyl β-D-thiogalactoside (IPTG) at 37°C. The induced cells were pelleted at 10,000 rpm for 2 min at 4°C. The samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and the gels were stained with Coomassie Brilliant dye after the pellets were treated with 2 × SDS loading buffer. The purification of inclusion bodies and renaturation of fusion protein by dialysis were performed as previously described.(22) The protein of interest was designated as SARS-CoV S1 protein.
Preparation of specific polyclonal antibody
The preparation of specific antibody against SARS-CoV S1 protein was performed according to previous reports with modifications.(23,24) Briefly, a New Zealand rabbit was inoculated with 1 mL purified S1 protein (1 mg/mL) emulsified with equal amounts of Freund's complete adjuvant via subcutaneous injection. The immunization of the rabbit was boosted four times by inoculation of the same antigen (1 mL) mixed with equal volume of Freund's incomplete adjuvant at 1-week intervals. Antiserum was isolated from peripheral blood of the immunized rabbit.
Titration of the antibody using ELISA
The reactivity between the SARS-CoV S1 protein and its polyclonal antibody was analyzed using indirect ELISA according to previous reports with modifications.(22,23) Briefly, ELISA plates were coated with 100 μL renatured protein (0.5 μg) in carbonate-bicarbonate buffer (15 mM Na2CO3, 35 mM NaHCO3 [pH 9.6]) at 4°C overnight followed by blocking with 5% non-fat dry milk in PBS-0.05% Tween-20 (PBST) at 37°C for 2 h. The wells were incubated with the serially diluted polyclonal antiserum or control serum from a non-immunized rabbit at 37°C for 1 h after washing three times with PBST. Following three washings with PBST, the plates were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (1:2000 diluted in PBST; Boster, Wuhan, China) at 37°C for 1 h. The wells were incubated with OPD (o-phenylenediamine dihydrochloride) substrate (100 μL/well) for 15 min after complete washing with PBST. The optical density (OD) value was read at 490 nm using an ELISA reader, after stopping the reaction with 50 μL stop buffer (2 M H2SO4).
Western blot analysis
The bacteria bearing either pGEX-SARS-S1 or empty vector were induced using IPTG and the bacterial lysates were subjected to 12% SDS-PAGE and transferred to a nitrocellulose (NC) membrane. The NC membrane was blocked overnight at 4°C using 5% non-fat dry milk in PBST followed by incubation with the anti-SARS-CoV S1 antibody (1:3000 dilution in PBST) at 37°C for 1 h. After three washings with PBST, the membrane was incubated with HRP-conjugated goat anti-rabbit IgG (1:4000 dilution in PBST) at 37°C for 1 h. The protein bands were visualized using OPD substrate.
Specificity of the anti-SARS-CoV S1 antibody
The specificity of the anti-SARS-CoV S1 antibody was determined using ELISA according to a previous report with modification.(24) Briefly, ELISA plates were coated with purified SARS-CoV S1 protein; TGEV S1 protein (approximately half in the N terminal part of S protein) and IBV S1 protein (approximately half in the N terminal part of S protein) were used as coating antigens (0.5 μg/well), respectively. These antigens were diluted in carbonate-bicarbonate buffer (15 mM Na2CO3, 35 mM NaHCO3 [pH 9.6]) and the plates were kept at 4°C overnight. The next day, the wells were blocked with 5% non-fat dry milk in PBST at 37°C for 2 h. The wells were successively incubated with anti-SARS-CoV S1 antibody (1:100 dilution) and HRP-conjugated secondary antibody (1:2000 dilution) at 37°C for 1 h. OD490 value was read using an ELISA reader.
Results and Discussion
Construction of expression plasmid bearing the SARS-CoV S1 gene
Using conventional molecular techniques, a truncated gene encoding the RBD of SARS-CoV was cloned into a prokaryotic expression vector. The resulting recombinant plasmid pGEX-SARS-S1 was identified with BamH I, EcoR I, and PCR (Fig. 1). Subsequent sequencing result showed that the gene sequence is identical to that of the parental gene, indicating that there is no mutation in the process of gene recombination.
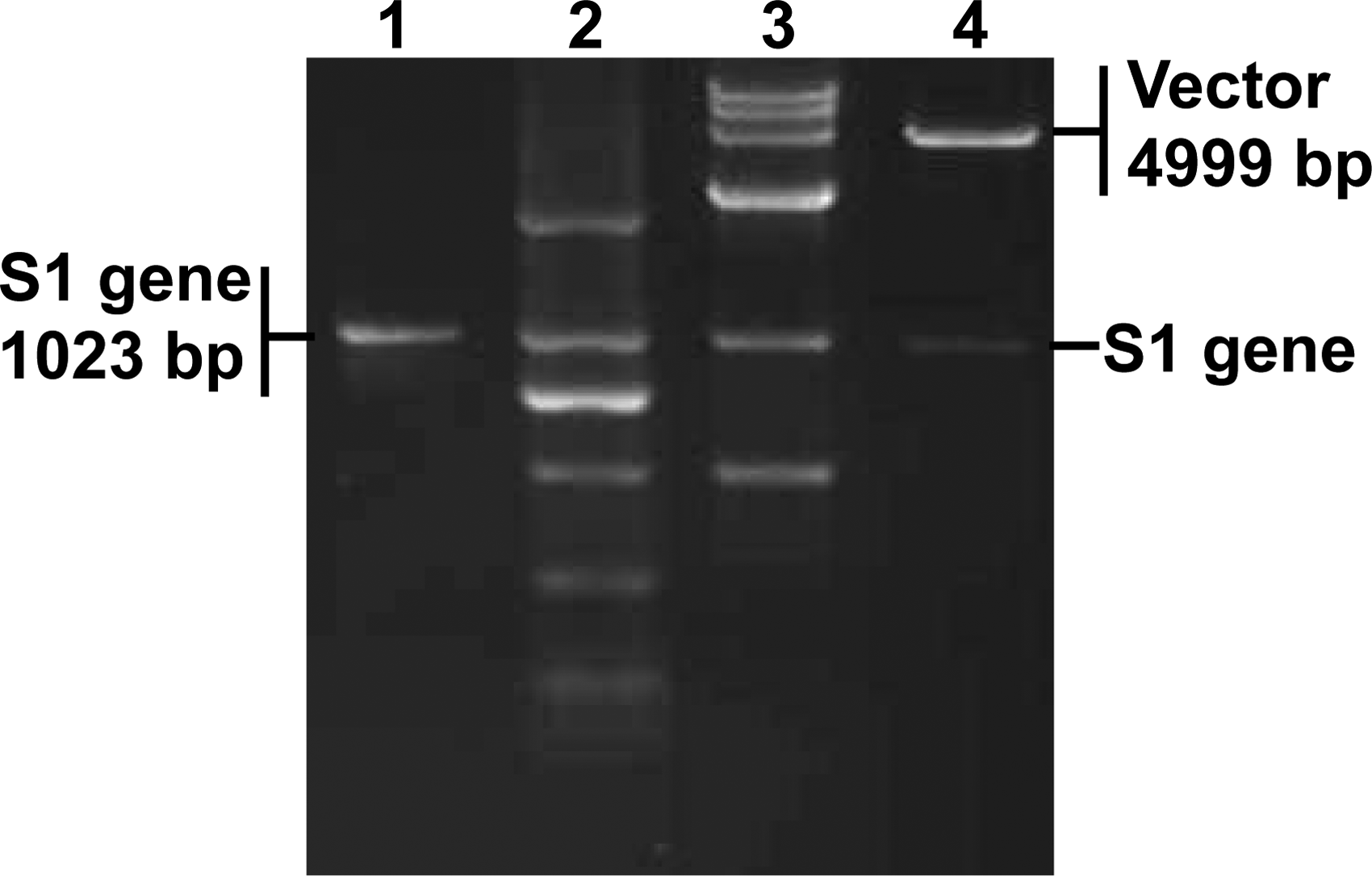
Identification of pGEX-SARS-S1. The recombinant plasmid pGEX-SARS-S1 was identified with PCR and restriction enzyme digestion. Lane 1, PCR amplification of the S1 gene; lanes 2 and 3, DL2000 and DL15,000 DNA marker (TaKaRa, Dalian, China); lane 4, digestion of pGEX-SARS-S1 with BamH I and EcoR I.
Expression and purification of recombinant SARS-CoV S1 protein
Following the construction of the recombinant pGEX-SARS-S1 gene, the protein expression was induced with IPTG at 37°C. To achieve a high level expression, the final concentrations of IPTG were adjusted to either 1 or 0.5 mM. Our result showed that the expression of the SARS-CoV S1 protein was significant in the presence of 0.5 mM IPTG (Fig. 2). Many successful examples have proven that the bacterial expression system is optimal for expressing heterologous proteins,(22–26) since the system has advantages in production cost, manipulation convenience, and expression efficacy.(27) The current study confirms that the system is suitable for producing SARS-CoV S glycoprotein. SDS-PAGE indicated that the S1 protein was expressed as early as 1 h post-induction of IPTG; however, there was no significant increase in terms of the protein production at a concentration of 1 mM IPTG. In contrast, the expression amount of the protein increased at 4 h post-induction of 0.5 mM IPTG. In the future, more experiments are needed to analyze the toxicity of IPTG and its impact on protein expression efficiency.
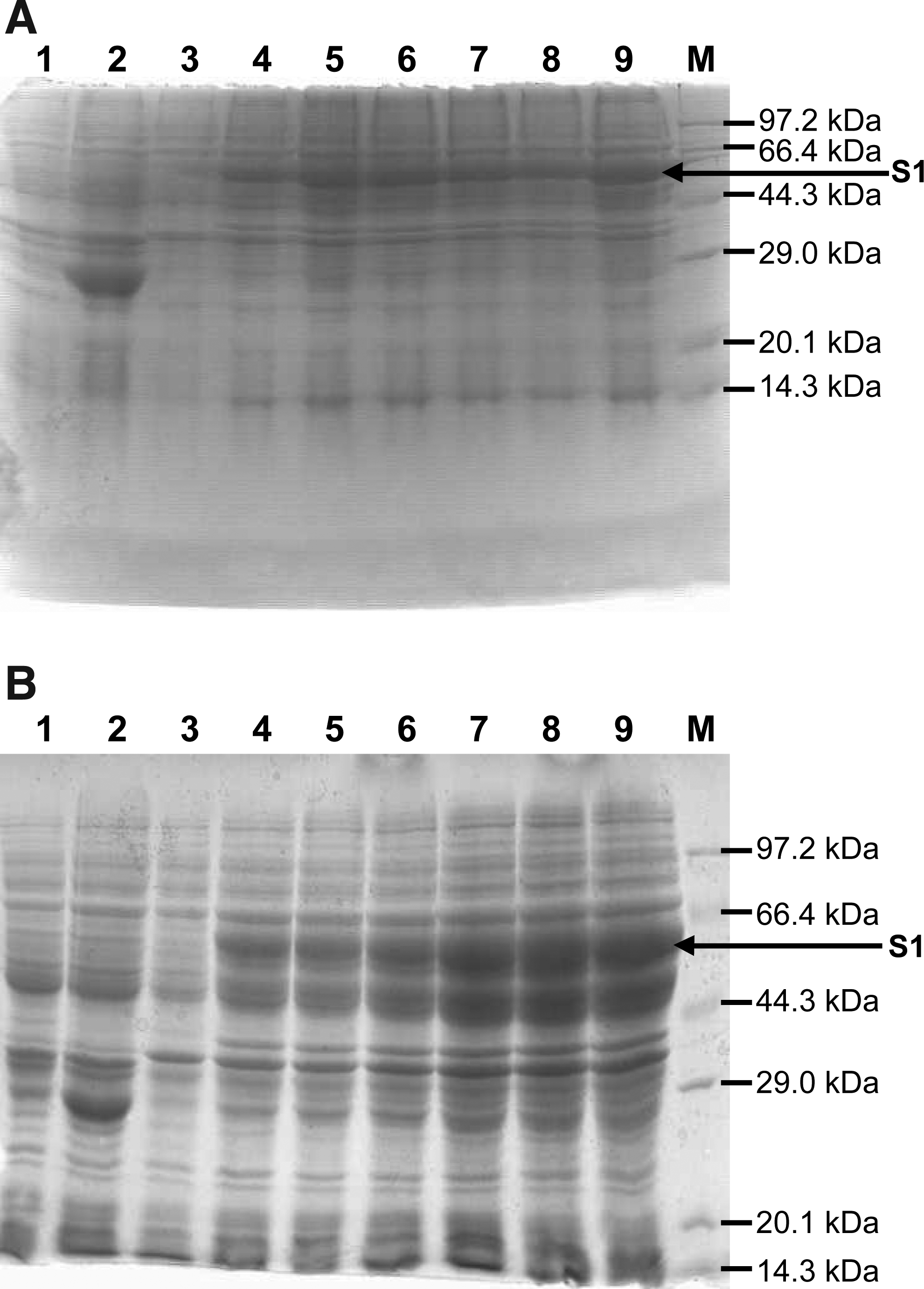
SDS-PAGE of bacterially expressed SARS-CoV S1 protein. The bacteria harboring the pGEX-SARS-S1 or empty vector were induced with IPTG, and the bacterial protein was analyzed by SDS-PAGE. Lane 1, uninduced empty vector transformed bacteria; lane 2, empty vector transformed bacteria at 4 h post-induction; lanes 3–9, pGEX-SARS-S1 transformed bacteria at 0–6 h post-induction, respectively; lane M, protein molecular weight marker. The expression of the S1 protein induced with 1 and 0.5 mM IPTG is shown in
Purification of the S1 protein and preparation of polyclonal antibody
In this study, the SARS-CoV S1 protein was purified by gel purification and the SDS-PAGE result is shown in Figure 3. The gel purification method is easy and inexpensive compared with the affinity column method. In particular, the former is more suitable for purification of proteins expressed in the form of inclusion body.(22,23) Subsequently, we used the purified SARS-CoV S1 protein to immunize a rabbit to generate a specific polyclonal antibody. ELISA indicated the titer of the antibody was 1:212, and the OD490 value of antiserum (P) was that of pre-immunized serum (N) ≥2 under such dilution (Fig. 4). Our previous and current results confirm that the bacterially expressed heterologous proteins are functional immunogens for generating both monoclonal and polyclonal antibodies.(22–25,28)
Purification of SARS-CoV S1 protein. The unpurified and gel-purified SARS-CoV S1 protein was analyzed by SDS-PAGE. Lane 1, purified S1 protein obtained at 5 h post-induction; lane 2, unpurified S1 protein obtained at 5 h post-induction; lane M, protein molecular weight marker.
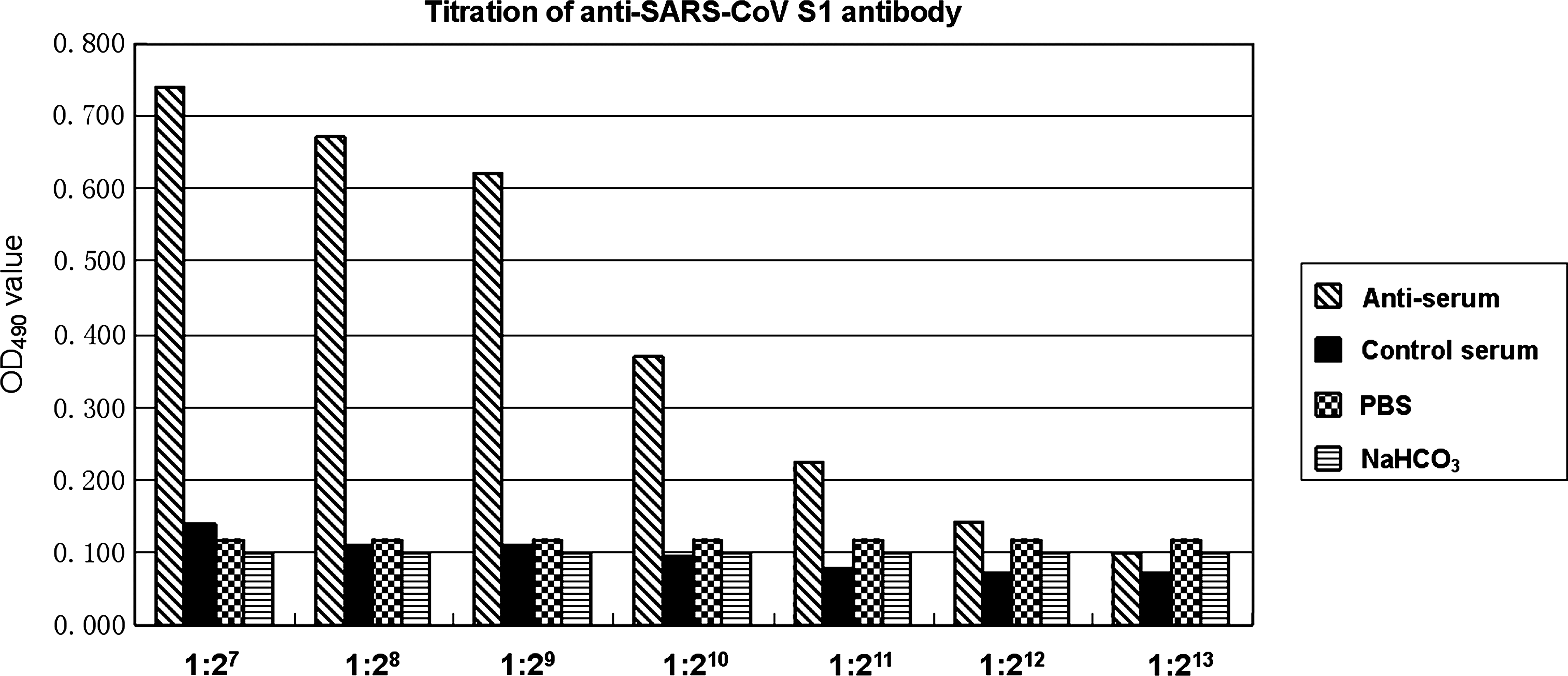
Titration of anti-SARS-CoV S1 antibody. The binding between the serially diluted antibody against SARS-CoV S1 protein and the S1 protein was analyzed by ELISA. The antibody dilution (x axis) and the OD490 value (y axis) are provided.
Biological activity of the SARS-CoV S1
To confirm the recognizing ability of the polyclonal antibody, the antibody was used as primary antibody to detect the SARS-CoV S1 protein. Western blot analysis showed that the polyclonal antibody reacted with the S1 protein (Fig. 5). However, an unspecific band was found in both the S1-expressing bacteria and the empty control bacteria. More experiments have to be done in the future to clarify the unspecific reaction between bacterial protein and the anti-S1 antibody. To further characterize the utility of the antibody, two S1 proteins of TGEV and IBV were used as control in ELISA. Our results showed that the anti-SARS-CoV S1 protein antibody reacted exclusively with the SARS-CoV S1 protein rather than with the other S1 proteins, indicating the specificity of the antibody (Fig. 6). The reactivity of the antibody to SARS-CoV is under investigation.

Western blot analysis of the polyclonal antibody. After the bacterial proteins of bacteria harboring either plasmid encoding pGEX-SARS-S1 or empty vector were transferred on a nitrocellulose membrane, a conventional immunoblotting was performed using the polyclonal antibody as primary antibody. Lane 1, empty vector control; lane 2, pGEX-SARS-S1-expressing bacteria. The molecular weight of the S1 protein of SARS-CoV is approximately 64 kDa.
Specificity of the anti-SARS-CoV S1 antibody. The anti-SARS-S1 antibody was used as primary antibody to detect the S1 proteins of SARS-CoV, TGEV and IBV in ELISA. PBS buffer was used as coating antigen control. The antibody was 1:212 diluted. The recognizing ability of the antibody to the proteins is indicated by comparison of OD490 values.
In conclusion, we have expressed the SARS-CoV S1 protein in a bacterial expression system and generated anti-SARS-CoV S1 antibody. The protein and the specific antibody may be used as diagnostic reagents for related research on SARS-CoV.
Footnotes
Acknowledgments
We acknowledge Prof. Georg Herrler (Institute of Virology, University of Veterinary Medicine, Hannover, Germany) for providing the SARS-CoV S gene. We also acknowledge funding support from the National Natural Science Foundations of China (nos. 30700590, 30972195); the Program for New Century Excellent Talents in Heilongjiang Provincial University (no. 1155-NCET-005) and the Program for Innovative Research Team of Northeast Agricultural University, China (no. CXZ008-1).
Author Disclosure Statement
The authors have no financial conflicts to disclose.