
Abstract
Abstract
Background:
Fluticasone propionate (Fp) is an inhaled corticosteroid with well-established safety and efficacy profiles. This study evaluated the systemic pharmacokinetics of Fp inhaled from a novel, inhalation-driven multidose dry powder inhaler (MDPI) that does not require coordination of actuation with inhalation.
Methods:
This was a single-center, open-label, randomized, 3-period crossover, single-dose study in healthy Japanese and Caucasian subjects aged 20–45 years, inclusive. Subjects were randomized to one of six treatment sequences including combinations of four inhalations of Fp MDPI 100 μg (400 μg total dose), Fp MDPI 200 μg (800 μg total dose), and Fp Diskus® 100 μg (400 μg total dose). The primary objective was to assess pharmacokinetics (maximum plasma concentration [Cmax] and area under concentration-vs.-time curve [AUC]) for each treatment. Safety and tolerability were also assessed.
Results:
Thirty subjects (15 Caucasian, 15 Japanese) met entry criteria and were randomized; all 30 subjects completed the study. At the inhaled Fp total doses evaluated (400 and 800 μg), the shapes of plasma concentration-vs.-time curves and systemic exposure (AUC0-t and Cmax) were similar in Japanese and Caucasian subjects. Geometric mean ratios (Japanese/Caucasian) for AUC0-t ranged from 1.11 to 1.15, and for Cmax ranged from 0.90 to 1.05, with no substantial differences between ethnic groups. In both ethnic groups, and in the combined population, systemic exposure (AUC0-t and Cmax) was highest for Fp MDPI 800 μg, followed by Fp MDPI 400 μg, and last by Fp Diskus 400 μg. No clinical laboratory, vital signs, or physical examination findings were considered clinically significant.
Conclusions:
Systemic exposure following inhaled single doses of Fp was comparable in healthy adult Japanese and Caucasian subjects for each total dose and inhaler. The new MDPI provided more efficient drug delivery than Diskus, suggesting that Fp MDPI may provide similar clinical efficacy at a lower inhaled dose compared with Diskus. Single-dose inhaled Fp (400–800 μg) was generally well tolerated in healthy adults.
Introduction
D
A novel, inhalation-driven, multidose dry powder inhaler (MDPI; Teva Branded Pharmaceutical Products R&D, Inc., Frazer, PA) for the delivery of inhaled asthma medications has been developed that does not require patients to coordinate actuation with inhalation. As this device has been developed for the delivery of both albuterol and Fp, the goal is to provide patients with a simplified asthma management approach that utilizes the same delivery device for both rescue and controller medication. Inhaled albuterol delivered from this MDPI has been shown to be effective and well tolerated in patients with persistent asthma(6,7) and to provide protection from exercise-induced bronchoconstriction.(8)
This study characterized plasma pharmacokinetic profiles after inhalation of Fp from the MDPI device compared with the current product Fp Diskus® (Flutide® Diskus®, GlaxoSmithKline K. K., Japan) marketed in Japan. The primary objective was to assess the pharmacokinetic profiles of single doses of Fp administered from Fp MDPI (400 μg and 800 μg total doses) and Fp Diskus (400 μg total dose) in both Japanese and matched Caucasian healthy adult subjects. Safety and tolerability of Fp administered via the two different inhaler devices were assessed as secondary objectives.
Methods
This was a single-center, open-label, randomized, three-period crossover, single-dose pilot study in healthy Japanese and matched Caucasian subjects. The study was conducted in 2012 at one study site (WCCT Global, LLC, Cypress, CA). All study procedures were conducted in full accordance with the International Conference on Harmonisation Good Clinical Practice Consolidated Guideline (E6) and any applicable national and local laws and regulations. All protocols were approved by the appropriate institutional review board, and written informed consent was obtained from each subject before starting any study procedures or assessments.
Subjects
The study enrolled healthy male and female subjects aged 20 to 45 years, inclusive. Japanese subjects must have been born in Japan, had both parents and four grandparents of Japanese descent, possessed a Japanese passport, resided outside of Japan for no more than 5 years, and had no significant changes with regard to diet since leaving Japan. Non-Japanese subjects were required to have parents and grandparents of Caucasian descent and were matched individually (on a 1:1 basis) with respect to gender, age (within 5 years), and height (0.05 m) to a Japanese subject.
Additional inclusion criteria for all subjects included: body mass index (BMI) of 19 to 30 kg/m2, body weight ≥50 kg, resting sitting heart rate of >45 to <90 beats per minute, systolic and diastolic blood pressure of <140/90 mmHg, no smoking for ≥6 months before screening and a maximum smoking history of 5 pack-years, and good general health based on medical history, physical examination, 12-lead electrocardiogram (ECG), vital signs, and clinical laboratory results.
Subjects were excluded if they had a history of clinically significant cardiovascular, hepatic, renal, hematologic, neuropsychologic, endocrine, gastrointestinal, or pulmonary disease; glaucoma, cataracts, ocular herpes simplex, or malignancy other than basal cell carcinoma; respiratory disease (e.g., intermittent or persistent asthma, emphysema, chronic bronchitis); any disease/condition known to interfere with absorption, distribution, metabolism, or excretion of drugs; respiratory infection (including common cold and flu), sinusitis, or ear infection within 14 days of random selection; known or suspected hypersensitivity to any steroid, including Fp; use of an intranasal corticosteroid within 7 days, ICS within 30 days, systemic corticosteroid within 60 days, or any investigational drug within 30 days; or use of cytochrome P450 isoenzyme 3A4 (CYP3A4) inhibitors (eg, ritonavir, ketoconazole, itraconazole) within 4 weeks.
Eligible female subjects were not pregnant, breast-feeding, or attempting to become pregnant; had a negative serum pregnancy test result; and were either of non-childbearing potential or willing to commit to using a consistent and acceptable method of birth control for the duration of the study. Only subjects who demonstrated both an inspiratory flow rate of at least 60 L/min and proper use of the inhaler devices were randomly assigned to treatment.
Study design
A total of 30 eligible subjects (15 Japanese and 15 Caucasian) were randomly assigned to one of six treatment sequences (ABC, ACB, BAC, BCA, CAB, or CBA) in accordance with a randomization schedule generated using SAS® software 9.2 (SAS Institute, Cary, NC). Each treatment sequence contained three treatments: Treatment A: Fp MDPI 100 μg x four inhalations (400 μg total dose); Treatment B: Fp MDPI 200 μg x four inhalations (800 μg total dose); Treatment C: Fp Diskus 100 μg x four inhalations (400 μg total dose). Study design is depicted in Figure 1.
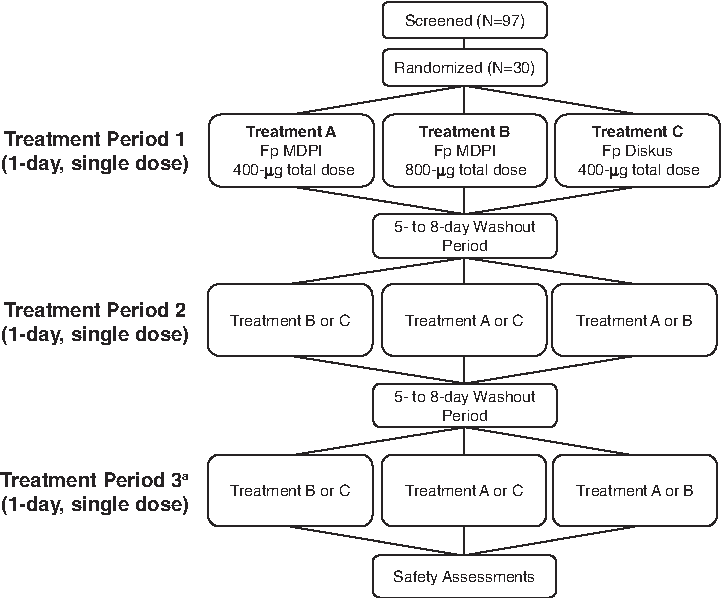
Study schema for this single-center, open-label, randomized, 3-period crossover, single-dose pilot study. Fp, fluticasone propionate; MDPI, multidose dry powder inhaler. aPatients were given a different treatment from period 2.
The study consisted of a screening visit (up to 28 days before treatment) and three treatment visits. On the day before each treatment, subjects were trained using the In-Check DIAL training device (Clement Clarke International, Harlow, Essex, UK) and were expected to demonstrate an inspiratory flow rate of ≥60 L/minute. Site personnel reviewed directions for use of each inhaler device with the subjects, who were required to demonstrate proper use of empty MDPI and Diskus devices at each training visit. The following day (treatment visit), subjects inhaled four times from the device to which they were randomized for each treatment period. Pharmacokinetic assessments of plasma Fp levels were performed before and after dosing on the day of each treatment. Treatment periods were separated by a 5- to 8-day washout period.
Pharmacokinetics
Blood samples (6 mL) for determining Fp plasma concentrations were obtained predose (within 10 min before inhalation); 2, 5, 10, 20, 30, and 45 min; and 1, 1.25, 1.5, 2, 3, 4, 6, 8, 12, 18, and 24 hours after dose administration during each of the three treatment periods. Plasma concentrations of Fp were analyzed using a validated high-performance liquid chromatography–tandem mass spectrometric method with a lower limit of quantitation of 1.0 pg/mL. Pharmacokinetic parameters for Fp were determined by noncompartmental methods(9) using a validated Pharsight Knowledgebase Server (PKS, version 3.1) and WinNonlin® software (Enterprise Version 5.1.1, 2006, Pharsight Corporation, Mountain View, CA).
The following pharmacokinetic parameters, when possible, were determined for each subject: area under the plasma concentration-vs-time curve from time zero to the time of the last quantifiable concentration (AUC0-t) by the linear trapezoidal rule; maximum plasma concentration (Cmax); time to Cmax (tmax); AUC from time zero extrapolated to infinity (AUC0-∞) as the sum of AUC0-t and the area extrapolated from the last measurable plasma concentration to infinity (Clast/λz); elimination rate constant (λz) estimated by linear regression of the terminal portion of the semi-logarithmic plasma concentration-vs-time curve; apparent terminal elimination half-life (t½) as ln(2) divided by λz; and % extrapolation of
Safety assessments
Safety was assessed by evaluating reported adverse events, hematology laboratory test results, vital signs measurements, concomitant use of medication, and physical examination findings. All adverse events were coded according to version 15.0 of the Medical Dictionary for Regulatory Activities.
Statistics
The pharmacokinetic analysis dataset included all randomly assigned subjects who received at least one dose of study drug, had at least one of the evaluable pharmacokinetic parameters, and had sufficient data to calculate AUC0-t and Cmax from any treatment period prior to experiencing a major protocol violation. The primary pharmacokinetic parameters were AUC0-t and Cmax for plasma Fp. The AUC0-t and Cmax data were natural log-transformed before statistical analysis. Comparisons of AUC0-t and Cmax between treatments and/or between Japanese and Caucasian subjects were carried out using a parametric analysis of variance (ANOVA) model with terms for sequence, period, treatment, ethnicity (Japanese or Caucasian), and treatment-by-ethnicity interaction and a random effect of subject within sequence. The ANOVA model was used to determine the difference between Japanese and Caucasian subjects overall and by treatment, and the associated p values and 95% confidence intervals on the log scale.
Differences between Japanese and Caucasian subjects and associated 95% confidence intervals estimated from the ANOVA model on the log scale were back-transformed to obtain the estimated ratio of geometric means and the 95% confidence intervals for this ratio. The same ANOVA model was used for comparison of AUC0-t and Cmax between pairwise treatments. Treatment differences and associated 90% confidence intervals were back-transformed to obtain the estimated ratio of geometric means and the 90% confidence interval for this ratio.
The safety analysis dataset included all randomized subjects who received at least one dose of randomized study drug.
Results
Study population
A total of 97 healthy subjects were screened, and 30 subjects (15 Caucasian and 15 Japanese) met entry criteria and were randomly assigned. Of the 67 subjects who were not enrolled, 45 were excluded based on the inclusion/exclusion criteria, 19 were excluded based on the decision of their physician, 2 withdrew consent, and 1 was not enrolled due to pregnancy. All 30 subjects completed the study and all were included in the safety analysis set and the pharmacokinetic analysis set.
Demographic characteristics of randomly assigned subjects are summarized by ethnic group in Table 1. Caucasian and Japanese ethnic groups were well matched with respect to mean age (31.3 years in the two ethnic groups) and gender (67% male in both ethnic groups). As expected, mean body weight and BMI were higher in Caucasian compared with Japanese subjects.
BMI, body mass index; SD, standard deviation.
Pharmacokinetics
At the inhaled Fp total doses evaluated (400 μg and 800 μg), the shapes of the mean plasma concentration-vs-time curves were similar in Japanese and Caucasian subjects (Fig. 2). Pharmacokinetic parameters for each ethnic group are shown by treatment in Table 2. Geometric mean ratios (Japanese/Caucasian) for AUC0-t ranged from 1.11 to 1.15, and for Cmax ranged from 0.90 to 1.05, with no substantial differences between Japanese and Caucasian subjects with any of the treatments.
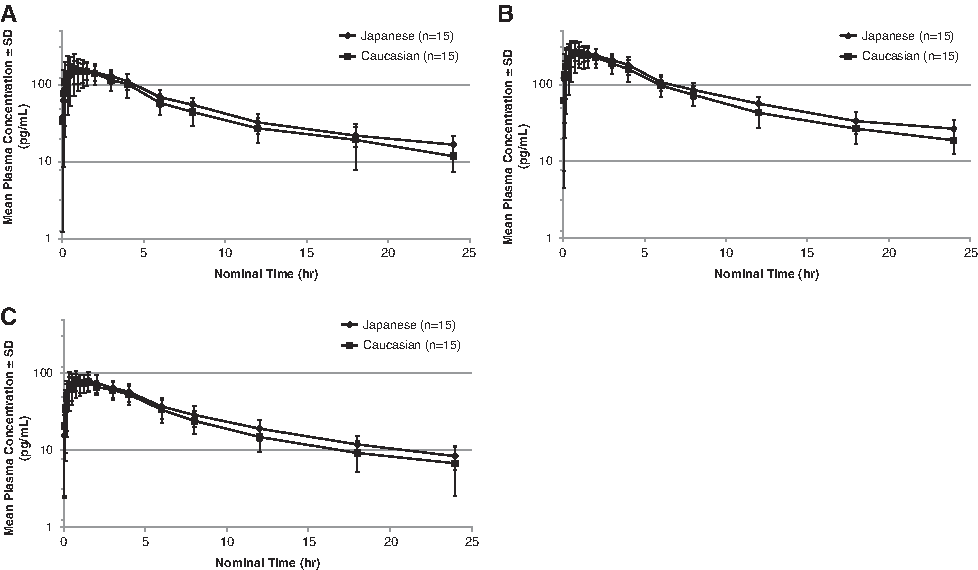
Mean ± standard deviation (SD) plasma concentrations of fluticasone propionate (Fp) over 24 hours after single-dose treatment with
Geometric mean ratio = geometric mean of Japanese/geometric mean of Caucasian.
P value for comparison between Japanese and Caucasian populations based on the ratio of the geometric means.
Patients were only included in this analysis if their AUC0-t was at least 80% of the
n = 10; en = 13; fn = 12; gn = 14.
λz, elimination rate constant;
In the combined population (Japanese and Caucasian), differences were observed between the three treatments, with plasma concentration-vs-time profiles being highest for the Fp MDPI 800 μg total dose and lowest for the Fp Diskus 400 μg total dose (Fig. 3). Pharmacokinetic parameters for the combined population are summarized in Table 3. Systemic exposure to Fp, as measured by AUC0-t and Cmax, was highest for the Fp MDPI 800 μg group, followed by Fp MDPI 400 μg group, and lastly by the Fp Diskus 400 μg group. Fp was rapidly absorbed following oral inhalation; median tmax ranged from 52.5 to 60.0 minu. Mean t½ ranged from 9.7 to 11.3 hours for all three treatments.
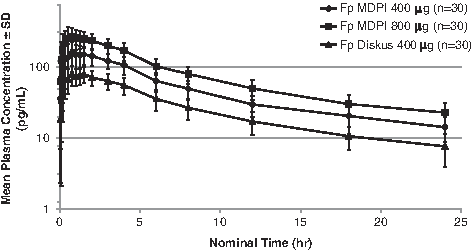
Mean ± standard deviation (SD) plasma concentrations of fluticasone propionate (Fp) over 24 hours by treatment for all subjects. MDPI, multidose dry powder inhaler.
Patients were only included in this analysis if their AUC0-t was at least 80% of the
n = 23; cn = 26; dn = 29.
λz, elimination rate constant;
Safety
There were no deaths, withdrawals due to adverse events, or serious adverse events during the study. All adverse events are summarized in Table 4. No clinical laboratory, vital signs, or physical examination findings were considered clinically significant.
Fp, fluticasone propionate; MDPI, multidose dry powder inhaler.
Discussion
For the inhaled Fp total doses evaluated (Fp MDPI 400 μg, Fp MDPI 800 μg, and Fp Diskus 400 μg total doses), the pharmacokinetic profiles of plasma Fp were similar in healthy Japanese and Caucasian subjects. In the present study, AUC0-t values for Fp 400 μg (MDPI and Diskus) and Fp 800 μg (MDPI only) were slightly (<15%) higher in Japanese subjects compared with Caucasian subjects. This finding is consistent with results from a recent study of single-dose and repeat inhalation of fluticasone furoate 800 μg from a dry powder inhaler that reported somewhat (<30%) higher plasma drug exposure (Cmax and AUC) in Japanese subjects compared with Caucasian subjects.(10) Systemic exposures between ethnic groups in the current study were similar, suggesting that clinical dose adjustments are not required in patients of Japanese descent.
In the individual ethnic groups, as well as when both ethnic populations were combined, there were clear differences between systemic exposures produced by each of the three treatments in this study, with AUC0-t and Cmax being highest for the Fp MDPI 800 μg total dose and lowest for the Fp Diskus 400 μg total dose. Systemic exposure to Fp was approximately two-fold higher for the Fp MDPI 400 μg total dose than for the Fp Diskus 400 μg total dose, suggesting more efficient delivery of Fp via the MDPI compared with Diskus. Some studies have observed lower Fp systemic exposure with Diskus compared with matched doses from other inhalers, including a pressurized MDI,(11) a reservoir powder inhalation device,(12) a unit-dose capsule-based dry powder inhaler (Rotacaps®/Rotahaler®),(13,14) and a Conix™ dry powder inhaler.(15)
Fluticasone plasma profiles represent systemic absorption of Fp deposited in the lungs because oral absorption of fluticasone is minimal (<1%).(16) Therefore, the systemic activity is predominantly dependent on the amount absorbed from the lung, which is the ultimate goal of an ICS. Factors that may contribute to pharmacokinetic variations between different inhalers include disparities in particle size,(15,17,18) airflow resistance,(13,14) and the patient/device interface (e.g., orientation of the mouthpiece, leading to differences in throat vs lung deposition).(12)
In addition, the bioavailability of Fp has been shown to be higher in healthy subjects compared with patients with asthma.(19) However, since the present study was conducted to detect differences between delivery of the same ICS using two different dry powder inhalers with healthy adults serving as their own controls, it is anticipated that the differences observed in healthy subjects would also be present in patients with asthma. Although this study was not designed to determine the reasons for the observed differences between the MDPI and Diskus, the data suggest that the increased efficiency of dose delivery from the MDPI may provide similar clinical efficacy with a lower inhaled dose compared with Diskus.
There were no new safety issues in this study. Inhaled single doses of Fp (400–800 μg total dose) were generally well tolerated when administered to healthy subjects aged 20 to 45 years of either ethnic group.
Conclusions
The pharmacokinetic results of this study demonstrated that systemic exposure to inhaled single doses of Fp was comparable between healthy adult Japanese and Caucasian subjects for each dose and inhaler evaluated. The new MDPI provided greater drug delivery based on systemic exposure compared with the Diskus inhaler, suggesting that the Fp MDPI may provide similar clinical efficacy at a lower inhaled dose compared with Diskus. Inhaled Fp total doses of 400 and 800 μg were generally well tolerated when administered to healthy adults of either ethnic group in this study.
Footnotes
Acknowledgments
This study was sponsored by Teva Branded Pharmaceutical Products R&D, Inc.
Medical writing assistance was provided by Lisa Feder, PhD, and Lela Creutz, PhD, of Peloton Advantage and was funded by Teva Branded Pharmaceutical Products R&D, Inc. Teva provided a full review of the article.
Author Disclosure Statement
Apinya Bee Vutikullird has no financial conflicts to disclose. Michael Gillespie and Sharon Song are employees of Teva Pharmaceuticals, Frazer, PA. At the time of manuscript preparation, Jonathan Steinfeld was an employee of Teva Pharmaceuticals, Frazer, PA.