
Abstract
Abstract
Background:
Predicting local lung tissue pharmacodynamic (PD) responses of inhaled drugs is a longstanding challenge related to the lack of experimental techniques to determine local free drug concentrations. This has prompted the use of physiologically based pharmacokinetic (PBPK) modeling to potentially predict local concentration and response. A unique opportunity for PBPK model evaluation is provided by the clinical PD data for salbutamol, which in its inhaled dosage form (400 μg), produces a higher bronchodilatory effect than in its oral dosage form (2 mg) despite lower drug concentrations in blood. The present study aimed at evaluating whether inhalation PBPK model predictions of free drug in tissue would be predictive of these observations.
Methods:
A PBPK model, including 24 airway generations, was parameterized to describe lung, plasma, and epithelial lining fluid concentrations of salbutamol administered intratracheally and intravenously to rats (100 nmol/kg). Plasma and lung tissue concentrations of unbound (R)-salbutamol, the active enantiomer, were predicted with a humanized version of the model and related to effect in terms of forced expiratory volume in 1 second (FEV1).
Results:
In contrast to oral dosing, the model predicted inhalation to result in spatial heterogeneity in the target site concentrations (subepithelium) with higher free drug concentrations in the lung as compared with the plasma. FEV1 of inhaled salbutamol was accurately predicted from the PK/PD relationship derived from oral salbutamol and PBPK predictions of free concentration in airway tissue of high resistance (e.g., 6th generation).
Conclusion:
An inhalation PBPK-PD model was developed and shown predictive of local pharmacology of inhaled salbutamol, thus conceptually demonstrating the validity of PBPK model predictions of free drug concentrations in lung tissue. This achievement unlocks the power of inhalation PBPK modeling to interrogate local pharmacology and guide optimization and development of inhaled drugs and their formulations.
Introduction
I
Retention of total drug in the lung, however, as determined from preclinical in vivo experiments or clinical pharmacokinetic (PK) studies, does not provide any information regarding the actual levels of free and pharmacologically active drug in the lung tissue.(4) Neither does it reveal how the exposure varies between the different lung regions and, most importantly, how this compares with the free levels in blood, that is, regional targeting. In fact, there are no experimental methodologies available to do this, although indirect approaches such as target occupancy have been employed.(3,5)
The inability to define the target site concentration(s) of inhaled drug even in preclinical species clearly poses a huge challenge for developing inhaled drug treatments with the objective of optimizing drug potency and targeting of relevant lung structures. Currently used methodologies to evaluate inhaled drug pharmacology and predicting therapeutic dosing in man seem to still largely depend on dose–response studies in preclinical species with functional readouts at the level of the lung as a whole.(6,7) Although the utility for corticosteroids and bronchodilators is retrospectively demonstrated, this empirical approach provides little detail as to which drug or formulation properties need optimization and why.
Moreover, there are significant translational gaps to be considered: How well does inhalation PK in an animal model represent that of the human patient? With changes occurring in drug material or formulation, what is the anticipated impact on local pharmacology? How well does the animal model reflect human disease in terms of which lung structures that are required to treat? Clearly, addressing these questions as such require a more mechanistic approach to study and optimize inhaled drugs, including quantitative mathematical frameworks such as that provided by PK and PK/pharmacodynamic (PD) modeling.
As reviewed by Borghardt et al.,(8) a number of inhalation whole-body physiologically based pharmacokinetic (PBPK) models have been described in recent years,(7,9–13) including the commercially available platform GastroPlus.(14) These models all seek to integrate mechanistic descriptions of drug disposition in the lung with the systemic PK, although with different regional descriptions of the lung and emphasis on different aspects of lung biopharmaceutics and PK. The GastroPlus model has 3 regions (tracheobronchial, bronchiolar, and alveolar/interstitial tissue), the model by Martin and Finlay(13) has 14 tracheobronchial regions and a single alveolar region, whereas the Boger et al.(9) and Caniga et al.(7) models use only 2 regions (tracheobronchial and alveolar).
The inhalation PBPK models also differ with respect to the type of data used for their evaluation. The typical evaluation of predictivity is plasma PK following inhalation as done for budesonide with the GastroPlus model.(11) Furthermore, changes in plasma PK by different formulations/devices have been accurately predicted for AZD5423(12) and ciprofloxacin.(13) The rat version of the Boger model was additionally validated for dry powder inhalation of fluticasone propionate with in vivo measured glucocorticoid receptor occupancy as a means of evaluating the pharmacologically active free concentration in tissue.(9)
Despite the abovementioned, none of the PBPK models to date has been evaluated with clinical data of free tissue exposure, obviously because of the difficulty in generating such data. An evaluation of that kind would provide the required confidence in taking the next steps toward extending inhalation PBPK modeling to predict also local PD responses through integrated inhalation PBPK/PD modeling.
Interestingly, studies of inhaled(15) versus oral(16) dosage forms of salbutamol have shown that 400 μg inhaled salbutamol produces a more pronounced effect on forced expiratory volume in 1 second (FEV1) than does a 2-mg oral dose, despite plasma concentrations of the two enantiomers being an order of magnitude lower (Fig. 4a). These data represent the best, if not the only, dataset to evaluate inhalation PBPK modeling with respect to predictions of free tissue concentrations. This is possible because after oral dosing the free drug concentration at the level of the subepithelial smooth muscle can be assumed, based on the free drug hypothesis, to be the same as that measured in blood plasma.
The objective of the present study was to assess, as a concept test, how well a PBPK/PD approach could predict the time-course of FEV1 change induced by inhaled salbutamol. A development of the Boger model was made to accommodate 24 airway generations each consisting of compartments representing epithelial lining fluid (ELF), epithelium, and subepithelial tissue. The model was first parameterized using preclinical in vivo PK data and then translated to human with incorporation of known human PK parameters. Finally, it is shown how the FEV1 PK/PD relationship of (R)-salbutamol (the active enantiomer) derived from orally dosed salbutamol is used together with inhalation PBPK/PD modeling to accurately predict FEV1 following inhalation.
Materials and Methods
Chemicals
Salbutamol-D3 was purchased from CDN Isotopes, Inc. (Quebec, Canada) and Dulbecco's Phosphate-Buffered Saline (DPBS) (CaCl2 and MgCl2) from Life Technologies (Carlsbad, CA). Salbutamol was sourced from compound management at AstraZeneca R&D Gothenburg. Chemicals were of analytical grade and all solvents of HPLC grade.
Animals
Male Wistar Han rats (Harlan, Horst, The Netherlands) weighing 320–385 g were used for the in vivo study. The animals were group-housed at 18°C–22°C under a 12-hours light/12-hours dark cycle with free access to food and water for at least 5 days before the experiment. The study was approved by the Animal Ethics Committee of Gothenburg (135-2014) and was carried out at AstraZeneca R&D Gothenburg, whose facilities are accredited by the American Association for the Accreditation of Laboratory Animal Care.
Pharmacokinetic study
As liquid chromatography–tandem mass spectrometry can separate salbutamol-D3 from salbutamol, the PK for two different administration routes could be investigated in the same animal. First, salbutamol-D3 was given intravenously to the animals (n = 21) through the tail vein (100 nmol/kg, 1 mL/kg). The animals were thereafter lightly sedated by inhalation of 5% isoflurane supplemented with air and placed slanting in a supine position on a tilted board to enable administration of salbutamol through intratracheal (IT) instillation (100 nmol/kg, 0.5 mL/kg). All animals returned to their cages after IT dosing. The vehicle used for both salbutamol-D3 and salbutamol was saline.
Blood samples (100 μL/sample) were repeatedly collected from vena jugularis in catheterized animals (n = 3) at the following time points: 0.033, 0.083, 0.167, 0.25, 0.5, 1, 2, 4, 6, 7, and 24 hours. At the last time point, the lungs were lavaged three times with a solution (4 mL, DPBS) to collect bronchoalveolar lavage (BAL) fluid and the lungs were subsequently dissected. An additional group of animals (n = 18) were dedicated to terminal sampling of blood, BAL fluid, and lungs. These animals were sacrificed at the following time points: 0.05, 0.5, 1, 2, 4, and 7 hours (n = 3/time point). The terminal blood samples (5 mL) were collected from the aorta.
Analytical procedures
Samples of plasma, lung and BAL fluid were treated and analyzed for concentrations of urea, salbutamol, and salbutamol-D3 as detailed in the Supplementary Data (Supplementary Data are available online at www.liebertpub.com/jamp).
By assuming that the concentration of urea is the same in the plasma and ELF,(17) the ELF concentration of salbutamol and salbutamol-D3 (CELF) can be calculated as:
where CBAL is the drug concentration in the BAL fluid, Curea,pl is the urea concentration in plasma, and Curea,BAL is the urea concentration in the BAL fluid.
Inhalation PBPK model setup
The general workflow of the present study is outlined in Figure 1. Technical details are described in the sections hereunder.

Study flow chart for PBPK/PD modeling. The PBPK model is first set up for the rat and fit to data with estimation of certain lung-related drug parameters. The model is then translated to man to enable prediction of FEV1 following inhalation from an established oral PK/PD relationship. Prediction accuracy is seen as an indication of validity for PBPK predictions of free tissue concentration following inhalation. FEV1, forced expiratory volume in 1 second; IT, intratracheal; PBPK/PD, physiologically based pharmacokinetic/pharmacodynamic.
Model structure
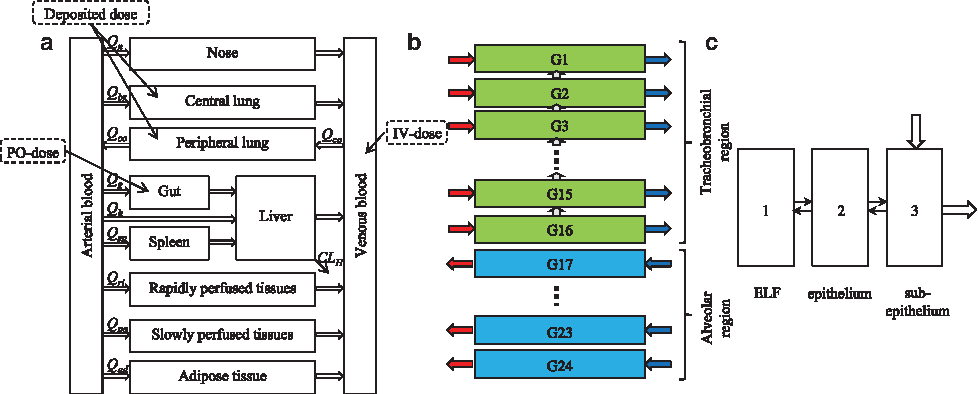
A whole-body PBPK model, which places emphasis on pulmonary drug disposition was implemented in MATLAB R2013a (Mathworks, Inc., Natick, MA). For the rat, the lung was divided into 24 airway generations and one extrathoracic region according to the morphometry presented by Lee and Wexler.(18) For humans, the same structural representation was used and the airway morphometry was described according to Weibel's model.(19)
In this article, airway generation 1 refers to the trachea. The structural model is illustrated in Figure 2. Blood flows and tissue volumes of the included organs are presented in Tables 1 and 2 for rat and man, respectively. Details regarding calculations of the regional surface areas as well as volumes of the ELF (VELF), the epithelium (Vep), and the subepithelium (Vsub) are provided in the Supplementary Data.
Poorly perfused = 1–other tissues; **Richly perfused = richly perfused + brain + kidney froma; QCO = cardiac output, 20.77 L/(h·kg).a
References: aGearhart et al.(41); bArundel(42); cBrown et al.(43); dInternal AstraZeneca data, han Wistar (n = 100); eBoger et al.(9); fCampbell et al.(44); and gChen and Kaufman.(45)
BW, body weight; NA, not applicable.
Poorly perfused = 1–other tissues; **Richly perfused = heart + brain + kidney froma; ***The nose was excluded from the human structural model since nasal drug deposition does not take place after oral inhalation; QCO = cardiac output, 5.2 L/min.a
Each airway generation is divided into three compartments: (1) the ELF, (2) the epithelium, and (3) the subepithelium (Fig. 2c). The airway generations either belong to the tracheobronchial (generations 1–16) or the alveolar region (generations 17–24). The tracheobronchial region is perfused by the bronchial blood flow (Qbr) and the alveolar region by the entire cardiac output (QCO). The local bronchial blood flow to a tracheobronchial generation i (Qbr,i) can be calculated using the following relationship:
where Vsub,i is the subepithelial volume of generation i,
and
where Di is the airway diameter (mm). To the best of our knowledge, no such relationship has been established for the alveolar region. The local blood flow to an alveolar region i (QCO,i) was therefore assumed to be constant in terms of flow per tissue volume, that is,
Perfusion rate-limited distribution was assumed to apply for all tissues in the PBPK model. For compartment i, the rate of change of quantity was described as(21):
where Vi is the tissue volume, Ci is the drug tissue concentration, Qi is the blood flow to the tissue, CA is the arterial drug concentration, R is the blood/plasma ratio, and Kp,i is the tissue–plasma partition coefficient.
As salbutamol is a rapidly dissolving compound, adding a dissolution-step did not alter the output (simulations not shown). Therefore, in the interest of computational speed, it was judged redundant to account for drug dissolution and mucociliary clearance. All ordinary differential equations (ODEs) used in the model are provided in the Supplementary Data.
Regional scaling of the permeability
As salbutamol is held to be mainly transported through the paracellular route, regional scaling of the permeability should primarily consider this mechanism. The airway epithelium is said to be leakier than the alveoli(22,23) and the permeability of paracellular markers, such as mannitol, have been estimated to be >30 times lower in the alveolar region.(24) Clearly, this number is associated with an uncertainty. Nevertheless, in the absence of more information, the alveolar permeability of salbutamol is said to be 30-fold lower than the permeability in the tracheobronchial region.
ODEs describing the permeability
As the transport rate of salbutamol across a cell layer has demonstrated a time dependence in vitro,(25) where the maximum transport rate was achieved firstly after 25 minutes, the model structure needs to account for this time dependency. To describe the time delay, the epithelium was included as a state, where its primary function is to act as a transit compartment. The flux from the ELF to the transit compartment (epithelium) in region j was described as:
where Vfluid,j is the fluid volume in region j, CELF,j is the drug concentration in the ELF in region j, P is the permeability, αj is the scaling of the permeability (α = 1 for j = 1, …, 16, and α = 1/30 for j = 17, …, 24), Aj is the surface area in region j, fu,fluid,j is the unbound fraction in the ELF in region j, Cep,j is the drug concentration in the transit compartment (epithelium) in region j, Vu,lung is the unbound lung volume of distribution, dfj is the deposition fraction in region j, and DIT is IT dose.
In the rat, the dose volume (Vdose) cannot be said to be negligible as compared with the calculated VELF (Vdose = 500 μL/kg and VELF≈180 μL/kg). Neglecting Vdose would thus significantly overestimate the initial CELF. Nevertheless, the lung can effectively clear fluid by several mechanisms.(26) To more accurately mimic the experimental situation after IT instillation, Vfluid,j was therefore set to have a time dependence:
Vdose was assumed to decline linearly such that all dose fluid had vanished 10 minutes after dosing. During this time interval, Equation (7) was modified to ensure that mass balance was maintained.
PBPK model parameterization
Systemic PK parameters
Modeling of the plasma PK after intravenous (IV) administration of salbutamol to rats provided estimates of the plasma clearance (CLp) and the steady-state volume of distribution (Vss). This is described in detail in the Supplementary Data (Supplementary Table 1 and Supplementary Fig. 1).
Tissue-to-plasma partition coefficients
The Kp for the lung (Kp,lung) was calculated from the plasma and lung concentration profiles after IV administration according to:
where the two integrals were obtained from an NCA conducted in Phoenix™ WinNonlin® 6.3.0 (Pharsight, Sunnyvale, CA). The unbound lung volume of distribution (Vu,lung) was subsequently calculated as:
where fu,p is the unbound fraction in plasma.
All other Kp-values were predicted in SimCYP Simulator v. 15 by the Rodgers methods,(27) which uses the following input: pKa, logP, fu,p, and molecular weight. All predicted Kp-values were subsequently corrected by a Kp-factor to add up to the observed Vss as described previously by Boger et al.(9) The final Kp-values are presented in Table 3 and all other drug-specific parameters in Table 4.
Kp,richly is the mean value of the predicted Kp-values of the heart, kidney, and brain; **Kp,poorly is the mean value of the predicted Kp-values of bone and muscle.
CL for (R)/(S)-salbutamol after intravenous dosing. Salbutamol has enantioselective CL in humans and thus varies with administration route, which is described in detail in the Supplementary Data.
fu,fluid currently cannot be measured. Since salbutamol has a high fu,p, fu,fluid was assumed to be 1.
References: aInternal AZ-data; bPK-study; cEquation (10); dEstimated parameter; eMorgan et al.(47); fRacemic F from Ward et al.(29); and gEstimated based on data in Morgan et al.(47)
β, estimated parameter for the pulmonary permeability; CLB, blood clearance; CLP, plasma clearance; F, oral bioavailability; fu,p, fraction unbound in plasma; fu,fluid, fraction unbound in epithelial or nasal lining fluid; ka, oral absorption rate constant; Papp, apparent permeability; Vss, volume of distribution at steady state; Vu,lung, unbound lung volume of distribution.
Parameter estimation of β
The effective permeability (Peff) was calculated based on an empirical relationship derived by Sjögren et al.,(28) where the apparent permeability (Papp) is used as input. However, this relationship has been derived for orally administered drugs and Peff for the epithelium in the gastrointestinal tract is not necessarily translatable to the epithelium in the lung. The permeability (P) in the lung was therefore set to:
where β was estimated by minimizing the weighted sum of squares based on predicted and observed values of Cp and Clung after IT instillation. More details on the parameter estimation are provided in the Supplementary Data.
Systemic PK parameters in man
Salbutamol has been demonstrated to exhibit enantioselective clearance (CL) in humans (46.77 and 14.70 L/h for (R)- and (S)-salbutamol, respectively) and thus also different oral bioavailabilities (F, 0.094 and 0.687 for (R)- and (S)-salbutamol, respectively).(29) As the current study considers the PK of (R)/(S)-salbutamol, both for the in vivo studies and the in silico modeling, the CL should reflect a weighted average of the two enantiomers. Since the rat PK parameters were estimated based on (R)/(S)-salbutamol levels and there is no evidence of enantioselective lung metabolism of salbutamol,(29) the CL estimated in this study is used for simulating both IV and IT delivery in rats.
In contrast, a weighted average of CL for oral and inhaled drug delivery needs to be calculated for humans as drug absorption will take place from the gut. Details thereof are provided in the Supplementary Data, which also contains detailed descriptions of the estimations of Vss and the oral absorption rate constant (ka) from clinical data.
PBPK modeling and simulations
Simulations in rats
Administration through the IV and IT routes (100 nmol/kg) were simulated using the drug-specific parameters for salbutamol (Tables 3 and 4). The deposition pattern after IT dosing is described in detail in the Supplementary Data (Supplementary Fig. 2). The simulations were subsequently compared with experimental data on Clung, CELF, and Cp generated in this study. All Cp measurements made at t = 24 hours were below the lower limit of quantification. CBAL measurements were unrealistically high at t = 24 hours (10–60 times higher than the corresponding observations at t = 7 hours), suggesting an experimental artefact. Clung and CELF from this time point were therefore excluded.
Human predictions
The PK was translated from animal to man by: (1) switching to human physiological parameters (Table 2), (2) using a human deposition pattern and human PK parameters (details provided in the Supplementary Data: Supplementary Table 2), and (3) assuming that the permeability was conserved across species.
Inhaled (400 μg) and oral administration (2 mg) were simulated using drug-specific parameters for salbutamol (Tables 3 and 4). The model predictions were compared with clinical data on Cp and FEV1 from Refs.(15,16,29) As the Cp measurements in Ward et al.(29) had been made after an inhaled dose of 1200 μg, these observations were divided by three to reflect an inhaled dose of 400 μg.
To relate the free drug concentration to the pharmacological readout (FEV1), only the active enantiomer (R)-salbutamol should be considered. As the salbutamol doses consider a racemic mixture, the PK of (R)-salbutamol could be simulated by giving 50% of the dose (200 μg and 1 mg after inhaled and oral delivery, respectively) and using the enantioselective CL and F. The relevant target site was identified in two steps. First, as β-receptor agonists act on smooth muscle, the subepithelial layer was identified as the relevant compartment. Second, a relevant airway generation (6th generation) was selected by considering the contribution of different airway generations to the airway resistance (Supplementary Fig. S4 in Supplementary Data).
A time-independent sigmoid maximal response (Emax) PK/PD model was used to relate the oral salbutamol FEV1 data to the time-matched PBPK model predictions of free (R)-salbutamol in plasma. (For oral dosing free (R)-salbutamol in plasma is predicted almost identical to free (R)-salbutamol in subepithelial tissue of e.g., the 6th airway generation). Further details on PK/PD parameter estimation are provided in the Supplementary Data (Supplementary Table 3). Subsequently, the equivalent PBPK model predictions for inhaled salbutamol were used in the PK/PD model to predict the time course of FEV1.
Sensitivity analysis
A sensitivity analysis was performed to evaluate how the system responds to changes in: (1) the estimated parameter β, and (2) the regional deposition pattern. This was done by simulating inhalation of (R)/(S)-salbutamol in man (400 μg) using different sets of β and deposition patterns. Two outputs judged as important for the conclusion of this study were considered: PK/PD predictions of FEV1 based on the free target site exposure of (R)-salbutamol and Cp of (R)/(S)-salbutamol. A detailed description can be found in the Supplementary Data.
Results
Simulations in rats
Following estimation of β (4.25 ± 0.41), the model predictions of Clung and Cp were consistent with experimental data after IT instillation capturing a slow drug absorption to plasma and a prolonged lung retention as compared with IV dosing (Fig. 3a, b). Furthermore, the model well described the corresponding concentrations after IV administration, which were characterized by a high peak concentration and a rapid initial decline in both Clung and Cp (Fig. 3b).
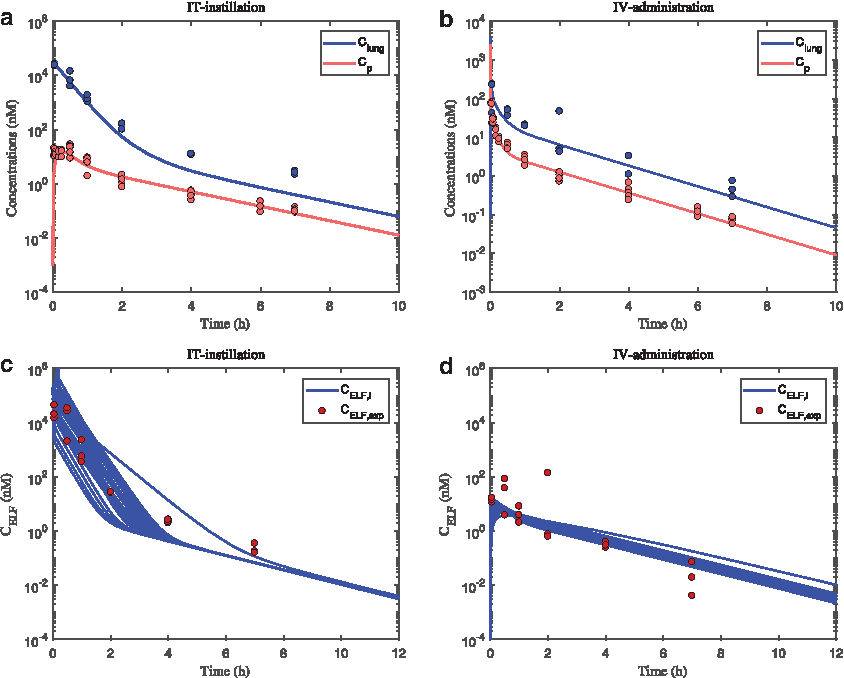
Predicted and observed plasma (Cp, red) and lung concentrations (Clung, blue) after:
The measurements of CELF reflect an average concentration as the BAL procedure will retrieve drug from all airway generations and thus not reflect regional concentrations. As shown in Figure 3c, d, distinctly higher CELF was observed initially after IT as compared with IV dosing. This behavior was captured by the model predictions. Furthermore, the observed steeper decline in CELF than Clung was captured by the model predictions, highlighting that flux of drug within the lung is well described.
Human predictions
By using β and Vu,lung estimated in the rat and reported human-specific systemic PK parameters, it was possible to make predictions of plasma concentrations of (R)/(S)-salbutamol. To accurately describe the totality of the inhaled oral and inhalation data it was required to account for the higher clearance and hence hepatic first pass metabolism of (R)-salbutamol, which renders the proportions of (R)- and (S)-salbutamol to be different following inhalation and oral administration. When accounted for, the model well described the observed difference in plasma exposure of (R)/(S)-salbutamol after oral and inhaled dosing in man (Fig. 4a).
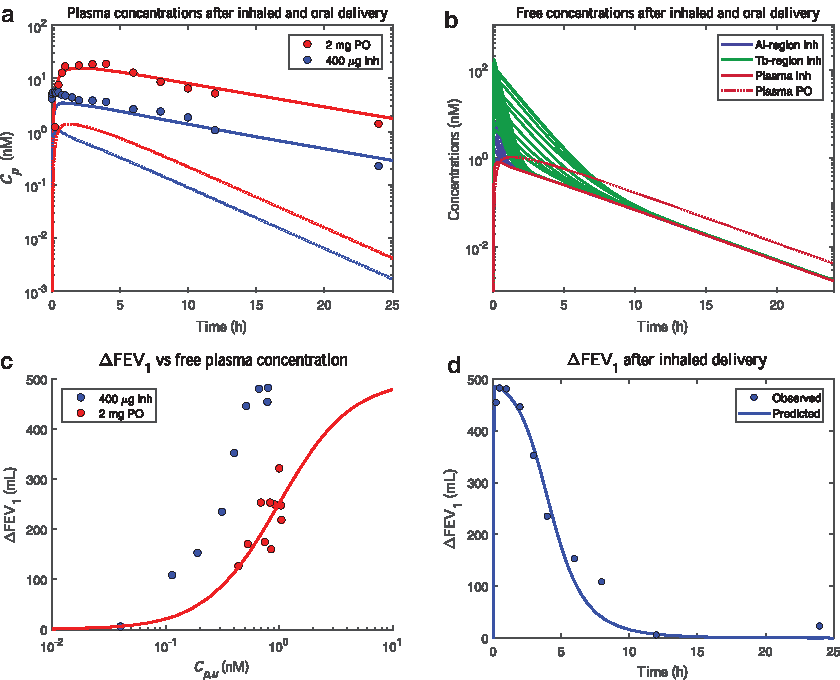
Model predictions (lines) and observations (circles) after inhaled (400 μg, blue) and oral delivery (2 mg, red) of (R)/(S)-salbutamol in man. The subfigures show:
A sensitivity analysis showed that the model predictions were consistent with the plasma observations after increasing/decreasing β by 20% as well as after modifying the deposition pattern by increasing/decreasing the alveolar deposition by twofold (Supplementary Fig. S5 in Supplementary Data). It was also noted that the difference in systemic exposure to (R)-salbutamol (inhaled vs. oral) was smaller than for (R)/(S)-salbutamol (Fig. 4a). This can be explained by the lower oral bioavailability of (R)-salbutamol (0.094 and 0.687 for (R)- and (S)-salbutamol, respectively(29)), thus leading to a lower exposure of this enantiomer after oral delivery. This phenomenon is not as pronounced after inhalation since no enantioselective lung metabolism has been observed,(29) that is, the pulmonary bioavailability is expected to be the same for both enantiomers.
With respect to the subepithelial lung tissue, the model interestingly predicted a spectrum of free concentrations of (R)-salbutamol after inhalation, which initially exceeded the corresponding free concentrations after oral delivery (Fig. 4b). In contrast, the predictions for oral salbutamol showed the same free concentration of (R)-salbutamol in all tissue compartments.
Finally, to evaluate the utility of the PBPK model predictions, the relationships between FEV1 and different measures of exposure were investigated. Clearly, changes in FEV1 are associated with higher free plasma concentrations of (R)-salbutamol after oral dosing than after inhalation, suggesting better effect for less drug in plasma for inhalation (Fig. 4c). A PK/PD model was used to describe the oral data with parameter estimates for EC50, γ, and Emax being 0.99 ± 0.09 nM, 1.36 ± 0.43, and 500 mL (fixed), respectively (Fig. 4c and Supplementary Fig. 3).
For inhaled salbutamol, substitution of the free plasma concentration with PBPK predictions of a relevant target site concentration (subepithelial layer in the 6th airway generation), resulted in predictions of FEV1 that were closely in line with observed data (Fig. 4d). A sensitivity analysis showed that this relationship remained when β and the deposition pattern were modified (Supplementary Fig. S5 in Supplementary Data).
Discussion
Being able to assess the free drug exposure at relevant pharmacological target sites in the lung is crucially important throughout inhaled drug discovery and development. Not only does the local and systemic PK need optimization to achieve targeting of the right lung structures, but also the dose required to elicit this localized response needs to be sufficiently low to effectively avoid undue developability problems. Therefore, it is pivotal in a clinical investment decision to have confidence in adequate drug exposure in the target tissue.(30,31) In addition to functional readouts in preclinical studies that suggest that a localized exposure has been achieved, PBPK modeling could provide further clarity and underpinning of the prediction of therapeutic dosing in humans.
Equivalently, in the early stages of clinical development, demonstration of target engagement as a proof of mechanism is typically done to support progression into larger and more costly clinical trials that aim to demonstrate drug efficacy.(32) However, a target site in the lung may be inaccessible for sampling and in such instances a PBPK model prediction informed by the observable pharmacokinetic data would provide additional confidence to support the progression of a drug project.
Although recent years have seen an accelerated development of inhalation PBPK models, a seemingly unresolvable issue remains—validation with clinical data on free drug concentrations in the tissue. Because these data cannot currently be obtained experimentally, we have instead conducted an indirect evaluation of PBPK predictions of free drug concentrations by first characterizing the FEV1 PK/PD relationship for oral salbutamol (Fig. 4c) and then using this to make a PBPK/PD model prediction for inhaled salbutamol to compare with clinical data (Fig. 4d).
In the process of setting up the inhalation PBPK model, we took a similar approach as would be applied in a drug discovery or development setting. This is typically to generate the required drug-specific preclinical data (in vitro and in vivo) as well as to incorporate any relevant available literature and clinical data (Fig. 1). The model structure (Fig. 2) was derived from morphometry and the histological arrangements of pulmonary epithelium and parameterized with available data on regional blood flow and tissue dimensions.
The parameterization was first done in the preclinical species (the rat), which allows more detailed evaluation of important lung pharmacokinetic processes. In this study, we estimated experimentally Kp,lung and the permeability factor β by using an IT/IV dual delivery setup with salbutamol (inhaled) and stable-labeled salbutamol-D3 (IV) to define lung tissue, BAL, and plasma PK for both administration routes. Next, we ascertained that the PBPK model structure and estimated parameters allowed a sufficient description of the preclinical data to support making the translation to humans. For instance it was crucially important to use the particular structure for epithelium (depicted in Fig. 2) to describe the temporal aspects of salbutamol transport between ELF and lung tissue of this study as well as the delayed onset of salbutamol transport observed in epithelial cell cultures.(25)
On the other hand, a dissolution submodel as used previously for fluticasone propionate(9) was not required for the model fit of the highly soluble salbutamol. When it happens in systems modeling (such as PBPK modeling) that a provided model structure is incapable of describing key features of a dataset at hand, it signals that some process is not properly accounted for and that predictions from that model should be treated with less confidence. If, on the other hand, a mechanistically justified model modification can be made to resolve the issue, there is not only better certainty in the predictions, but also new mechanistic understanding has been obtained.
Accurate quantification of regional deposition was initially regarded a major challenge for predicting regional salbutamol exposure in the lung, especially as regional deposition fractions do not have absolute experimental estimates.(4) Because deposition models require the availability of pharmaceutical specifications, such as droplet/particle size distribution, etc. and the inhalation maneuver, we instead parsimoniously used the deposition pattern previously described for pMDI administration of salbutamol.(33) The distribution of drug deposition fractions across the 24 generations was approximated from the reported proximal and distal lung deposition fractions. Sensitivity analysis (Supplementary Fig. S5 in Supplementary Data), however, showed minimal impact of sizeable (twofold) changes in proximal/distal deposition on predictions of drug concentrations in plasma and in fact also on the predicted FEV1.
Thanks to the long clinical use of salbutamol, the systemic PK, including oral absorption, was well described and became very useful. In fact, some studies had even used enantioselective quantification of (R)- and (S)-salbutamol.(29) Simultaneous PBPK model description of systemic PK for both oral and inhaled salbutamol was possible only when taking into account the higher hepatic clearance and first pass of (R)-salbutamol relative to the (S)-enantiomer. For the rat studies, it was not required to apply enantioselective quantification since it was assumed, based on reports of lung PK studies in the horse that the lung distribution PK is the same for both enantiomers.(34)
Only once the systemic PK was adequately described was it considered relevant to evaluate the predictions of local concentrations in the lung. The most stunning aspect of these predictions is perhaps the heterogeneity in exposure across the airway generations. There is a spectrum of concentrations from very high in the trachea to close to free plasma levels in the alveoli (Fig. 4b). This regional difference should not be unexpected, as it results from the simultaneous effect of how the drug deposits, permeates the epithelium, and is being cleared by the regionally varying blood flow.
Having accurately described the observed (R)/(S)-salbutamol plasma concentrations following inhaled and oral dosing with the associated predictions of the active (R)-enantiomer concentrations (Fig. 4a), we went on to investigate the relationships with the FEV1 response. The separated concentration–response relationships for inhaled and oral salbutamol (Fig. 4c) reveal a distinct, but <10-fold, advantage of inhalation in terms of avoiding systemic exposure for the same effect level. While this advantage is evidently sufficient for patients and physicians to favor the inhaled dosage form, it also points to a finite gain in safety margin.
For orally dosed salbutamol, the concentration of free (R)-salbutamol is practically identical in plasma and tissue of all the lung regions. Hence, plasma could be used to derive the PK/PD relationship for oral salbutamol. The determined human in vivo potency of (R)-salbutamol (1 nM) was markedly lower than reported EC50 derived from in vitro tissue preparations, which are typically in the range of 100 nM or above.(35,36) It is noted, however, that the basal contraction in these experimental setups may be exaggerating the muscle tone of the asthmatic smooth muscle, shifting the determined EC50 values toward the higher range. Interestingly, in a study with horse muscle strips, where a relatively small level of contraction was applied, the EC50 was as low as 5 nM for the racemic salbutamol,(37) which resonates better with the 1 nM EC50 for the active enantiomer as estimated in the present study.
Ahead of exploring inhaled salbutamol PK/PD, we considered how to define the airway generation or range of airway generations in which β-agonist bronchodilator drugs elicit the effects that become manifest in the FEV1 measurement. To the best of our knowledge, such definition is not described in literature. We therefore considered the estimated contribution to airway resistance from each of the airway generations by referring to the pioneering work of Green.(38) He had derived from the physics of laminar airflow and Weibel's morphometric model of the lung(39) that the contribution to airway resistance is at its maximum in the 6th generation and with minimal contribution from the 11th generation and downward (Supplementary Fig. S4 in Supplementary Data). This prediction was later experimentally confirmed by Macklem and Mead.(40)
On this basis, while realizing that a single airway generation is not solely responsible for the FEV1 effect, we used the 6th generation for PBPK/PD predictions of inhaled salbutamol FEV1. As these predictions were very close to the observed data (Fig. 4d), we had met the objective of the study and made it plausible that the PBPK predictions of free drug concentrations in subepithelial tissue of the larger airways are accurate, if not also in other regions.
With the successful completion of this evaluation, one can now with better confidence begin applying inhalation PBPK to make model estimations of free drug exposure in the lung and link these to observed pharmacological responses or the lack thereof. We anticipate that inhalation PBPK/PD modeling is becoming an integral part of drug development as a research tool to reason around and predict regional lung targeting as well as therapeutic dosing. It will facilitate optimization of inhaled drug compounds and formulations and ultimately inform decisions to progress the right drug candidates into clinical development.
With that said, further development and validation work of PBPK/PD models needs to take place to establish the applicability beyond the β-adrenergic bronchodilator target site. Other target sites of importance for respiratory disease include the pulmonary epithelium, small airway tissue, and immune cells resident at the luminal surface of the epithelium.
Hence, as alluded to above, a plethora of inhalation PBPK models is anticipated to result from attempts to handle various drug properties and biological scenarios. Pivotal in expanding the general applicability of inhalation PBPK/PD modeling will be to experimentally determine, with microscopic resolution, the spatial distribution of inhaled drug and the spatial aspects of its functional response. Moreover, advancing the mathematics in the models may open new avenues to explore in greater detail and realism the various concentration gradients in the lung and multilevel heterogeneity of drug exposure and response.
In conclusion, we have completed the first evaluation of an inhalation PBPK model with respect to estimating free drug concentrations in tissue. This demonstrates the concept and utility of using PBPK/PD modeling to optimize inhaled drug treatments of respiratory disease by design and development of inhaled drugs targeting defined structures of the lung.
Footnotes
Acknowledgments
Britt-Marie Fihn and Astrid Collin are acknowledged for their skillful in vivo work, as are Marie Brännström and Anna Carlsson for bioanalytical work. This study was funded by AstraZeneca R&D.
Author Disclosure Statement
E.B. and M.F. are full-time employees at AstraZeneca R&D. M.F. is affiliated with Uppsala University.
Reviewed by:
Per Backman
Benjamin Weber
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.