
Abstract
The effects of interferons (IFNs) are mediated through the induction of around 2,000 IFN-stimulated gene (ISG) products. However, the majority of these ISGs do not directly instigate IFN-mediated states, such as the defining resistance to viral infection. Rather, most ISGs encode cell signaling molecules that enhance the responsiveness to pathogens, and systemically disseminate signals from localized sites of infection. Relatively few IFN effector proteins have been well characterized. The protein kinase R (PKR) is one of the first and best characterized of these effector molecules. PKR mediates responses via phosphorylation of protein substrates and promotes signal transduction pathways to maintain homeostasis, mediate immune responses, and, upon sustained activation, promote apoptosis. As a number of reviews have dealt with PKR-dependent resistance to virus, this review will cover broader roles ascribed to PKR and the mechanism(s) by which PKR exerts its effects.
Introduction
P
PKR's Induction and Response to IFN
The eif2ak2 gene, which encodes PKR, is constitutively expressed in all differentiated tissues at low levels, and then further induced by a variety of stress-associated responses (Ank and others 2006). Expression of eif2ak2 is coordinately regulated by an IFN-stimulated response element (ISRE), a kinase-conserved sequence (KCS), as well as the transcription factors Sp-1 and -3, and the tumor suppressor protein p53 (Fig. 1) (Kuhen and Samuel 1997, 1999; Ward and Samuel 2002, 2003; Das and others 2004, 2006; Yoon and others 2009). Putative roles for Ets, Myb, myogenic differentiation antigen (MyoD), E2F, and the nuclear factor-κB (NF-κB) have also been proposed based upon identification of regulatory elements for these transcription factors in the gene promoter (Tanaka and Samuel 1994). In addition, an IFN-γ activation site-like element has been identified in the promoters of the human and mouse eif2ak2 gene, although this element does not appear to be functional (Tanaka and Samuel 1994, 2000; Xu and Williams 1998).
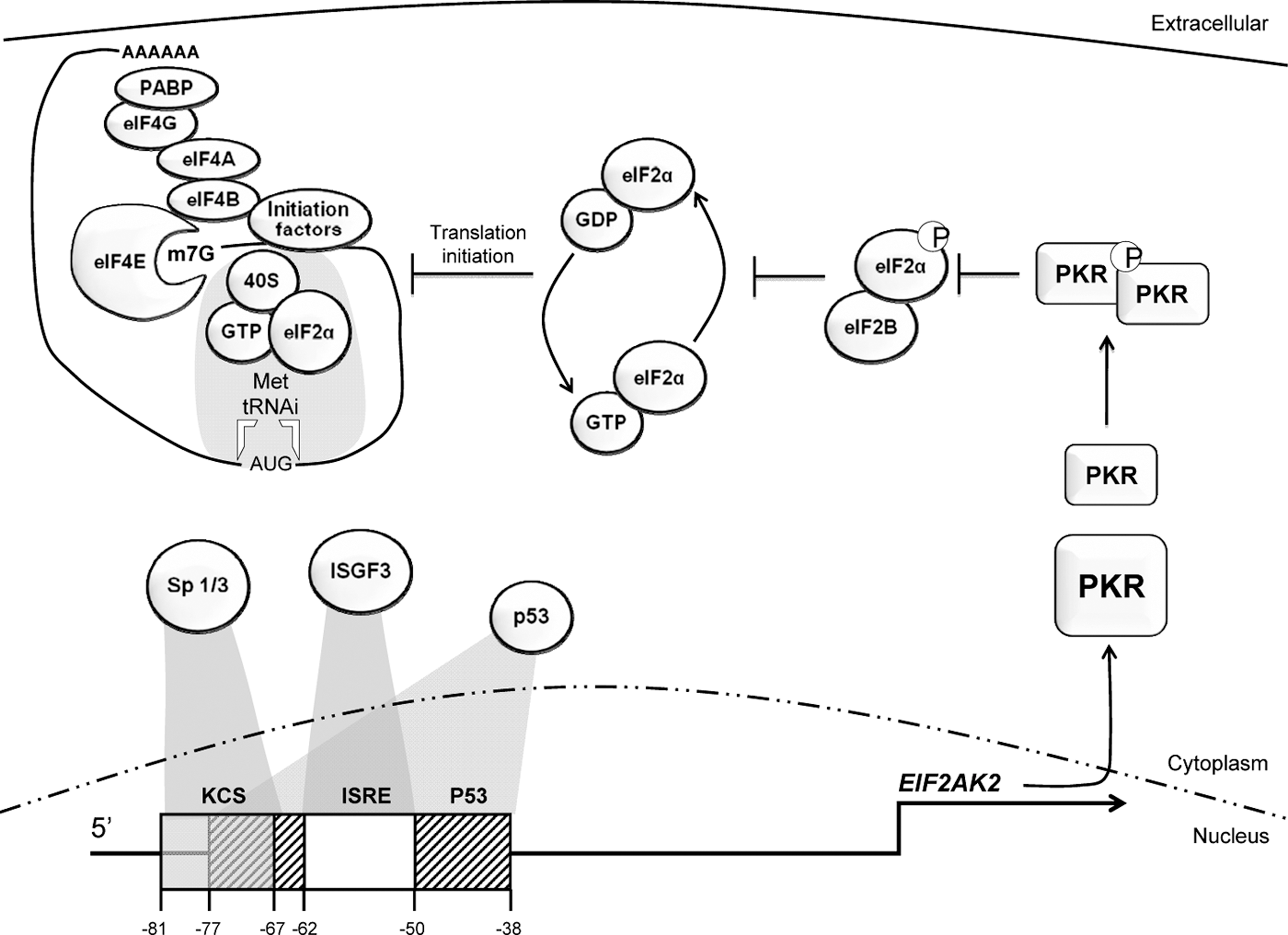
Transcriptional regulation of the human eif2ak2 gene and subsequent translational control. Transcription factor-binding sites in the 5′-untranslated regions of the eif2ak2 gene include the KCS (shown as a gray box), ISRE (white box), and p53 (patterned box) responsive elements. The p53 responsive element is split by the ISRE, and overlaps with the KCS element. The Sp-1 and -3, ISGF-3, and p53 transcription factors bind to the KCS, ISRE, and p53, respectively, for constitutive expression and subsequent induction of the eif2ak2 gene. Induced PKR must then be activated before it phosphorylates eIF2α to impose global translation regulation. Phosphorylated eIF2α sequesters the guanine nucleotide exchange factor, eIF2B. This inhibits replacement of the inactive eIF2-GDP with eIF2-GTP to enable formation of an active 43S preinitiation complex and subsequent recruitment to the mRNA via eIF4 factors that are attached to the mRNA cap structure (m7G). eIF2α, eukaryotic initiation factor 2α; IFN, interferon; ISRE, IFN-stimulated response element; ISGF-3, IFN-stimulated gene factor 3; KCS, kinase conserved sequence; PKR, protein kinase R.
PKR expression is also regulated post-transcriptionally. At heightened protein levels, activated PKR autoregulates expression of its own mRNA via the inhibition of global translational initiation (Thomis and Samuel 1992). As was identified by Garaigorta and Chisari (2009) this mechanism is applicable to other IFN-stimulated genes (ISGs) (Garaigorta and Chisari 2009). This apparent paradox, in which IFN-induced PKR represses ISG translation, occurs in cells with activated PKR, while cells in which PKR is inactive remain permissive to translation of ISGs. In this way, PKR tailors the IFN response, so that it can be partitioned to elevate immune surveillance in uninfected cells but is constrained in cells already infected. The significance of this PKR-dependent adaptation of the IFN response is largely unexplored but is likely to have considerable significance. In light of this conjecture, it would be interesting to know if any of the ISGs contain structures in their 5′-untranslated regions (UTRs) that permit escape from PKR-dependent translational control.
The identification of PKR as a p53-regulated gene was significant for our understanding of PKR expression (Yoon and others 2009). It was demonstrated that p53 directly induces the eif2ak2 promoter via a response element that is split by the ISRE (Fig. 1). Accordingly, PKR was shown to play a role in p53-mediated inhibition of translation, apoptosis, and tumor growth by phosphorylation of eIF2α in response to DNA damage (Yoon and others 2009). The IFN response has subsequently been demonstrated to be modulated by p53 regulation of the IFN-regulated factor-9 (IRF-9) (Munoz-Fontela and others 2008). Moreover, p53 itself is an ISG (Takaoka and others 2003). This supports previous studies that demonstrate a role for IFNs in growth control and apoptosis. Not only does p53 induce eif2ak2 expression, but it has also been shown to associate with PKR. Pull-down experiments using glutathione S-transferase fusion proteins demonstrated that p53 associates with PKR through its C-terminal domain. Additionally, it is proposed that PKR phosphorylates p53 at serine residue 392 (Cuddihy and others 1999). Together, these observations describe partly redundant functions for p53, PKR, and the IFN response that may account for the previous inability to confirm an anticipated role of PKR as a tumor suppressor, as will be discussed later.
Activation of PKR by dsRNA
As stated, PKR is constitutively expressed but is not functional until activated. A variety of activating stimuli have been characterized. The classical activator of PKR is dsRNA. The mechanism by which dsRNA activates PKR has been extensively covered by others, so it will not be discussed here in detail. Briefly, dsRNA is cooperatively bound by tandem RNA-binding motifs (RBMs) at the N-terminus of PKR. Binding of RNA relieves steric inhibition of the kinase domain. Although there is excellent data supporting regulation by autoinhibition, this is challenged by some who contend the sole role of the RBMs is to colocalize PKR monomers to a dsRNA molecule (reviewed by Cole 2007). Colocalization of PKR monomers enables transautophosphorylation to stabilize the active dimeric enzyme (Cosentino and others 1995; Nanduri and others 2000; Vattem and others 2001; Sadler and Williams 2007). In keeping with this model, it had been established that activating RNA must be double stranded and of a minimum length (30 bp) to accommodate 2 PKR monomers (Manche and others 1992). This convention was overturned by Nallagatla and others (2007), who showed that RNAs with limited duplexed regions could activate PKR if these molecules contained a 5′-triphosphate moiety. This feature distinguishes host-capped mRNAs from viral uncapped RNAs. Significantly, PKR could only be activated by 5′-triphosphate-RNAs when induced by IFNs. This may be due to elevated levels of PKR obviating the requirement for colocalization. Alternatively, PKR activated in this manner may be functionally different, as has been shown for PKR activated by select other processes (as will be discussed below). This modification of the faculty of PKR to detect alternative forms of viral RNA is a remarkable feature of the IFN response.
In addition to activation by viral RNAs, endogenous transcripts can activate PKR. The best characterized instance of this is the IFN-γ mRNA (Osman and others 1999; Ben-Asouli and others 2002). It has been demonstrated that PKR binds to a pseudoknot structure in the gene's 5′UTR, which subsequently activates PKR to then inhibit translation of the transcript via phosphorylation of eIF2α. PKR only binds to the pseudoknot structure at induced levels due to competitive exclusion by the ribosome. Hence, this delineates a mechanism by which type I IFNs regulate the production of toxic gene products. Also, this mechanism may provide an explanation for the observed suppression of IFN-γ by IFN-α/-β (Rayamajhi and others 2010).
Activation of PKR by Polyanionic Molecules
PKR is activated by the polyanionic molecules dextran sulfate, chondroitin sulfate, poly
Activation of PKR by heparin is intriguing. The distribution of heparin in the body is restricted largely to secretory basophil and mast cells. Consequently, it is believed to function in wound and antimicrobial responses. Therefore, heparin-mediated effects would seem to indicate a PKR-dependent immune function in the skin, mucosa of the lungs, gut, mouth, nose, and eyes. Little has been done to gauge the function of PKR in mucosal immunity. The closely related proteoglycan heparin sulfate has a broader expression than heparin, occurring on the surface of most, if not all, cells as well as extracellularly. Accordingly, heparin sulfate has been purported to have a wide variety of biological activities. It has been identified as a receptor for some viruses (Spear and others 1992; Hallak and others 2000a, 2000b). Intracytoplasmic heparin sulfate could indicate viral entry into a cell, and might present an early warning to the innate immune system. Therefore, it would be interesting to test if PKR is also activated by heparin sulfate and if this is relevant to PKR's established role in the antiviral response.
Activation of PKR by Caspases
Activation of PKR by caspases involves proteolytic cleavage of PKR at asparagine residue 251 by the apoptotic initiator caspase-8 and effectors caspase-3 and -7 (Saelens and others 2001). Removal of the autoinhibitory N-terminus of PKR by caspase cleavage generates a constitutively active kinase. As with heparin activation, the cleaved kinase is described to function as a monomer (Wu and Kaufman 2004). However, it is not clear what would prevent the truncated PKR from dimerizing. PKR monomers associate via residues in the kinase domain, which are present in the truncated protein. Similar truncated kinase constructs have been demonstrated to dimerize (Dey and others 2005). Presumably, the removal of the RBM prevents colocalization of PKR monomers. It seems possible that the truncated PKR protein might dimerize at higher concentrations during, for instance, induction with IFNs.
The promoter of the gene encoding caspase-8 has been shown to contain an ISRE and so is induced as part of the type I IFN response (Casciano and others 2004; De Ambrosis and others 2007). Caspase-8 has been demonstrated to be part of the innate immune response to virus and dsRNA. Moreover, mouse embryonic fibroblasts that are null for caspase-8 have an attenuated inflammatory response (Takahashi and others 2006). Hence, the IFN regulation of caspase-8 parallels the nucleotide-binding oligomerization domain-like receptor induction of caspase-1 and -5 and assembly of inflammasomes.
PKR's Activity
Once activated PKR has 2 roles: first, to directly affect protein function by phosphorylation; second, to activate transcription factors by modulating cellular signaling networks. It has been established that, at least in the instance of NF-κB activation, PKR functions in signaling pathways as an adaptor protein, without phosphorylating substrates.
As a kinase, PKR phosphorylates serine and threonine residues in protein substrates (Table 1). It has also been reported that PKR phosphorylates tyrosine residues (Su and others 2006). Moreover, it is claimed that autophosphorylation of tyrosines is essential for RNA-binding and subsequent kinase activity (Su and others 2006). Structures of PKR's kinase domain may provide support for this (Dar and others 2005). The structure of the kinase domain shows that the sub-element of the activation segment, termed the p + 1 loop, adopts a conformation that is intermediary between that expected of serine/threonine and tyrosine kinases. However, the autophosphorylated tyrosine residues identified as being essential for PKR's activity are either in regions that are not previously associated with activity (Y101, Y162), or that would appear to disrupt catalytic function (Y293). Also, autophosphorylation is triggered after binding of dsRNA, and so the claimed requirement for phosphorylation of tyrosine residues within the N-terminus of PKR to enable dsRNA binding seems unlikely. Rather, phosphorylation within the RBMs is more consistent with ejection of dsRNA after activation (Jammi and Beal 2001).
Reported serine (S), threonine (T), and tyrosine (Y) residues phosphorylated by PKR in protein substrates (indicated by bold and underlined text).
Conditions for an in vitro kinase assay for PKR were established that recommended the inclusion of manganese in the reaction buffer (Galabru and Hovanessian 1987). As we have observed a consequence of manganese for kinase specificity, we have indicated PKR substrates identified without manganese in the buffer.
The serine residue 307 within IRS-1 was not validated as a PKR phosphorylation site but is assumed from Western blot analysis of mouse cell lysates.
Citations for PKR's peptide substrates are Romano and others 1998, Taylor and others 1996, Zhang and others 2001, MKK6 Silva and others 2004 and HIV TAT Endo-Munoz and others 2005. Citations for others substrates are listed in the text.
eIF2α, eukaryotic initiation factor 2α; ILF-3, interleukin factor-3; IRS-1, insulin receptor substrate-1; MKK6, mitogen-activated protein kinase kinase 6; PKR, protein kinase R; RHA, RNA helicase A.
No substrate motif has been recognized that dictates PKR phosphorylation specificity. Although no definitive sequence is apparent, there may be a bias in the residues encoded at phosphorylated sites (Table 1). This speculative bias shows a preference for lysine and arginine residues proximal to phosphorylated amino acids. Analysis of kinase substrate preference, using short randomly assorted peptides from yeast, showed that another eIF2α kinase, GCN2, has low specificity but clustered, by weak association, with kinases that had a preference for arginine residues proximal to the phosphophorylated serine/threonine (Mok and others 2010). A qualification to the low specificity for short peptide substrates demonstrated for GCN2, and extrapolated for PKR, is that a crystal structure of PKR's kinase domain in association with eIF2α shows specificity is mediated by protein contacts outside of the catalytically active pocket of the enzyme. Hence, although there may be little specificity, for short peptides, substrate specificity is increased by restricting access to the catalytic pocket of PKR (Dar and others 2005).
Phosphorylational Control of eIF2α
The best-characterized substrate of PKR is eIF2α. Through this activity PKR controls translation. The mechanism of this translational control is well established, so this will be only briefly covered here (Fig. 1). However, many mRNAs have evolved mechanisms that allow them to escape this translational repression through features encoded in their 5′UTR regions, such as internal-ribosomal entry sites and multiple upstream open reading frames (Petryshyn and others 1996; Wek and others 2006). Transcripts regulated in this manner assuage stress responses. In addition, a number of transcripts that are resistant to this translational repression, such as the CCAAT/enhancer binding protein-α and -β, mediate cellular differentiation (Calkhoven and others 2000). PKR has been ascribed a role in cellular differentiation (Salzberg and others 1995). Appropriately, PKR is not expressed in undifferentiated cells, but its expression increases as cells differentiate (Wang and others 2002). However, as PKR-null mice do not have developmental defects, there cannot be a strong dependence on PKR for cellular differentiation.
It had previously been demonstrated that defects in the regulation of eIF2α by phosphorylation caused malignant transformation (Donze and others 1995; Perkins and Barber 2004). Hence it was presumed that PKR would act as a tumor suppressor. Although this was supported by data showing that expression of kinase-dead PKR in NIH-3T3 cells results in malignant transformation, mice lacking PKR do not develop tumors at an increased rate (Chong and others 1992; Koromilas and others 1992; Meurs and others 1993; Yang and others 1995; Abraham and others 1999). An explanation for this variance in PKR-dependent effects, in cell lines compared to in vivo, has been attributed to IFN-priming in murine models. The regulatory network identified between IFN, p53, and PKR suggests that each gene may partially compensate for each other in this network. Correspondingly, Yoon and others (2009) demonstrated, using a tumor xenograft model in nude mice, that deletion of p53 with simultaneous knockdown of PKR promoted tumor development.
PKR has subsequently been shown to participate in the activity of one of the most commonly mutated tumor suppressive genes, the phosphatase and tensin homolog deleted from chromosome 10 (PTEN). Mutations of PTEN have been identified in a large fraction of tumors, notably prostate cancers and glioblastomas. Mounir and others (2009) demonstrated that cell lines expressing PTEN showed enhanced PKR and eIF2α phosphorylation and impaired colony formation. Consequently, the antiproliferative and pro-apototic effects of PTEN were reduced in cells ablated for PKR.
Phosphorylation of B56α
PKR was shown to physically interact with the B56α regulatory subunit of the phosphatase 2A (PP2A) proteins. PP2A consists of a family of heterotrimeric enzymes, composed of a structural and catalytic subunit, which associates with one of several regulatory B subunits. PP2A is the major serine and threonine phosphatase in the cell and as such regulates a wide array of processes. PKR was shown to phosphorylate B56α at serine residue 28 (Xu and Williams 2000). As a result, PKR was shown to block B56α-mediated inhibition of PP2A and, thus, enhances PP2A activity. PKR was demonstrated to regulate dephosphorylation and, consequentially, the activity of the eIF4E via activity of PP2A (Joshi and others 1995; Xu and Williams 2000). In addition, PKR was shown to regulate PP2A-mediated dephosphorylation of BCL2 to mediate apoptotic processes (Ruvolo and others 2008).
PP2A regulates the activity of a range of potent kinases, such as the mammalian target of rapamycin (mTOR). The mTOR kinase pathway transduces stress signals, from mitogens, growth factors, sensors of cellular energy stasis, and the redox state of cells. This is particularly interesting in light of the recent identification of a role for PKR in obesity-related conditions (Nakamura and others 2010). This study proposed a role for PKR in regulating a diet-induced inflammatory response, predominantly through the c-jun N-terminal kinase (JNK), and by phosphorylation of the insulin receptor substrate-1. Relevant to these responses, mTOR has been demonstrated to regulate the nutrient response via insulin and has been demonstrated to phosphorylate insulin receptor substrate-1 (Ueno and others 2005; Tzatsos and Kandror 2006). A recent analysis of the protein kinase networks in yeast demonstrated interactions between the yeast PP2A regulatory subunit RTS1 and the GCN2 regulator GCN1. Further, GCN2 was linked into the TORC1 nutrient-sensing kinase network (Breitkreutz and others 2010).
Coordinate Regulation of Proteins That Encode the RBM
PKR has also been demonstrated to phosphorylate a number of proteins that share the RBM, including the RNA helicase A and interleukin factor-3 (ILF-3) (Parker and others 2001; Sadler and others 2009). A number of additional proteins with RBMs interact with PKR, but have not been shown to be substrates. These include the spermatid perinuclear RNA and transactivating (SPNR) and (TAR)-RNA binding (TRBP2) proteins, adenosine deaminase acting on RNA-1 (ADAR-1), the protein coded DRBP-120, and dihydrouridine synthase 2-like proteins (DUS2L) (Park and others 1994; Coolidge and Patton 2000; Toth and others 2006; Mittelstadt and others 2008; Wang and Samuel 2009). As is evident from this catalog, the RBM mediates protein–protein interactions. Through these interactions the activity of different proteins that encode the RBM are coordinated, predominantly by suppression of their function. In this way, the RBM acts like the caspase recruitment domain and toll/IL-1 receptor domain to mediate protein interactions to coordinate enzyme activity in protein networks. As a number of these proteins have been associated with virus replication, the interaction with PKR has a consequence for the host antiviral response (Fujii and others 2001; Isken and others 2003; Hartman and others 2006; Jeang and Yedavalli 2006; Toth and others 2006; Li and others 2010).
Activation of PKR in Cell Signaling Networks
In addition to the direct binding of activating ligands that triggers phosphorylation of protein substrates, PKR is also activated indirectly and subsequently modulates cellular signaling networks (Fig. 2).
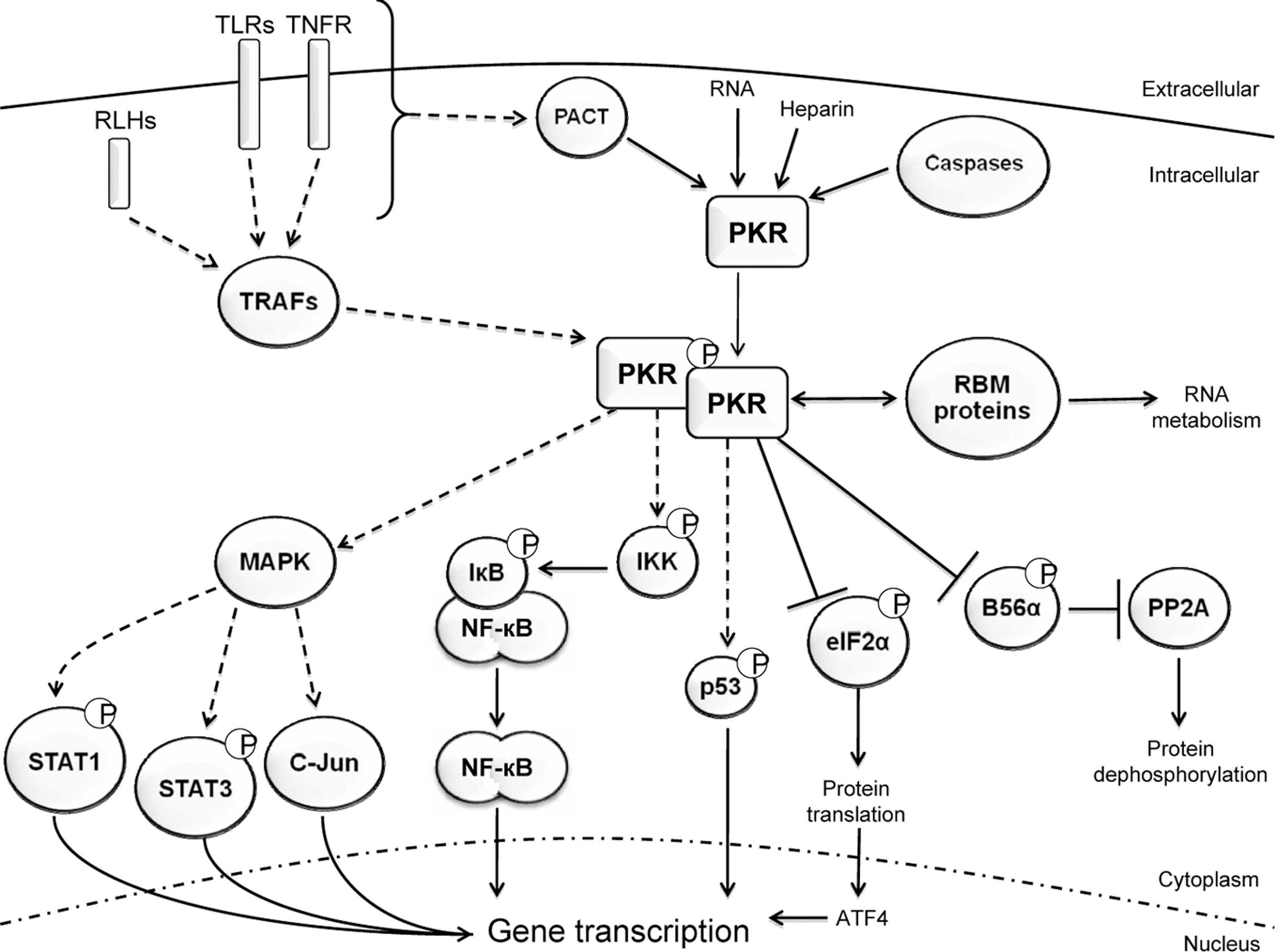
Cellular activities of PKR. PKR is activated by ligand-induced dimerization and transautophosphorylation. Although it is also reported that heparin and caspase generate an active monomeric PKR. Once activated, PKR functions directly by protein phosphorylation, or indirectly by integrating signaling networks to promote the activation of transcription factors and ensuing gene induction. In this figure, direct interactions are indicated as solid lines, whereas indirect actions, or speculative interactions, are drawn with dashed lines. Although PKR has been shown to directly interact with STAT-1 and -3, the kinase does not phosphorylate these proteins. Instead, this is predicted to be mediated by the MAPK family. Therefore, PKR's effect upon STAT-1 and -3 is drawn as being via this signaling cascade. This figure is representative of PKR function and does not include all protein substrates and pathways affected by the kinase. MAPK, mitogen-activated protein kinase; STAT-1, signal transducers and activators of transcription.
PKR responds indirectly to IL-1, IFN-γ, tumor necrosis factor (TNF)-α, platelet-derived growth factor, as well as a variety of pathogen-associated molecules and general stress stimuli (Mundschau and Faller 1995; Yang and others 1995; Kumar and others 1997; Cheshire and others 1999; Goh and others 2000; Ramana and others 2000; Deb and others 2001; Ichikawa and others 2002; Tam and others 2007; Zheng and others 2008). Responses to these molecules are initiated via the cognate receptors: IL-1 receptor, IFN-γ receptor-1 with -2, TNF receptor (TNFR) -1 and -2, and either platelet-derived growth factor receptor-α or -β, and the innate immune toll-like receptors and retinoic acid-inducible gene (RIG)-like helicases, and by undefined pathways. Association between each ligand and the cognate receptors triggers signal transduction pathways that activate latent cytoplasmic transcription factors. The transcription factors that have been demonstrated to be regulated by PKR are NF-κB, c-jun, the signal transducers and activators of transcription (STAT) -1 and -3, IRF-1, and the activating transcription factors (ATF) -3 and -4 (Wong and others 1997; Gil and others 2000; Deb and others 2001; Guerra and others 2006; Lee and others 2007). Of these transcription factors, only regulation of the ATFs by PKR has a clearly established mechanism. ATF-3 and -4 mRNA contain multiple upstream translational start sites. This feature of mRNAs promotes translation upon phosphorylation of eIF2α. Consequently, PKR-dependent induction of ATF-3 and -4 is via control of eIF2α. Consistent with this mechanism, the other eIF2α kinases also induce ATFs (Guerra and others 2006).
PKR-dependent activation of the other transcription factors listed above is independent of phosphorylation of eIF2α. At least in the instance of NF-κB, PKR-dependent activation appears to be independent of phosphorylation of any protein substrate. It has been shown that a kinase-dead mutant of PKR was competent to activate NF-κB (Chu and others 1999; Bonnet and others 2000; Gil and others 2000; Zamanian-Daryoush and others 2000). Activation of NF-κB requires phosphorylation and ubiquitin-dependent proteolysis of the inhibitor-κB (IκB). Under normal circumstances NF-κB exists in a latent state in the cytoplasm bound to its inhibitor IκB. Phosphorylation of IκB renders it susceptible to proteolysis via the ubiquitin-proteosome pathway, which results in the release and translocation of NF-κB to the nucleus, where it binds to DNA and induces gene transcription (Brown and others 1993; Kumar and others 1994). PKR affects NF-κB activity by association with the IκB kinase complex to regulate levels of IκB (Kumar and others 1994). A plausible mechanism for PKR's activation of NF-κB, and likely other transcription factors, has emerged that involves the protein activator of the IFN-inducible protein kinase (PACT) and TNFR-associated factors (TRAFs).
PKR's Association with TRAFs
Gil and others (2004) identified a mechanism by which PKR might mediate indirect phosphorylation and ubiquination of IκB. Human PKR was shown to encode 2 functional binding motifs for TRAF proteins. TRAFs dock with activated cell signaling complexes to then regulate the subcellular location and, as E3-ubiquitin ligases, promote the degradation of key signaling components. Seven TRAF proteins have been identified (TRAF-1 to -7). The relevant residues in PKR, within the N-terminus (TKQE) and kinase domain (PEQIS) of human PKR, appear most similar to consensus motifs that bind TRAF-2 and -3. Correspondingly, PKR was shown to associate with TRAF-2 and also with TRAF-5 and -6. The consensus binding motif for TRAF-6 (KXXPXE) is different from those identified in PKR. However, the various TRAF proteins associate with each other through their TRAF domains. TRAF-2 has been demonstrated to interact with TRAF-6 (Davies and others 2005). Similarly, TRAF-3 interacts with TRAF-5 (Pullen and others 1998). Hence the observed interactions with TRAF-5 and -6 may be indirect. Characterization of truncated constructs of PKR showed that the kinase domain binds TRAF proteins to a greater extent than the N-terminus of PKR. However, a challenge to the consequence of the C-terminal TRAF-binding motif in human PKR is that this motif is not conserved in the mouse kinase. As the same signaling pathways function in mice, the altered motif (PEQLF) may still bind TRAFs, a different TRAF-binding may exist (none is apparent), or this association is mediated by the motif at the N-terminus.
Contrary to the mouse PKR, the C-terminal TRAF-binding motif is otherwise generally preserved (occurring in PKR from rat, pig, cow, and hamster). Indeed, the residues within this region of PKR are highly conserved as they encode part of the activation segment of the kinase domain, which regulates catalytic function of the kinase. The consensus residue in the TRAF-binding motif overlaps with residues that define a feature termed the p + 1 loop within the activating segment. This feature acts as a phospho-acceptor during substrate phosphorylation. Despite the required functional conservation of this feature, the TRAF-binding motif in PKR's kinase domain does not occur in the other eIF2α kinases.
The TRAF-binding motif at PKR's N-terminus occurs at the C-terminal end of the second RBM. The equivalent amino acids are conserved within PKR from different species (including human, mouse, rat, hamster, cat, pig, and cow). However, the motif does not occur in PKR's first RBM, or on RBMs from other human proteins (including TRBP2, PACT, DiGeorge syndrome critical region 8, Dicer, ADAR-1, -2, -3, ILF-3, and SPNR). Amino acid residues on either side of the TRAF-binding motif in RBM-2 (96 to 118 and 162 to 171) interact with the kinase domain (within residues 328 to 335) to instigate autoinhibition (Gelev and others 2006; Li and others 2006). Binding of dsRNA relieves autoinhibition. Indeed, the equivalent location in RBMs from other proteins that constitute the RBM-2 TRAF-binding motif in PKR have been demonstrated to mediate hydrogen bonding to nucleotides in the major groove of dsRNA (Ryter and Schultz 1998). As the association between RBM-2 and the kinase domain is mediated by residues close to both TRAF-binding motifs, it predicts that autoinhibition would prevent an association with TRAFs. Accordingly, it was shown that PKR interacted with TRAFs only after activation.
There is an interesting consequence to this conjecture. The kinase-dead PKR, with a point mutant at the lysine residue 296, is still able to activate NF-κB. This would appear to contradict data that showed PKR had to be activated to interact with TRAFs. However, this mutation (K296R) disrupts the interaction between RBM-2 and the kinase domain. Hence, the TRAF-binding motifs in the kinase-dead PKR would be exposed and able to constitutively interact with TRAFs. Therefore, the functionality of kinase-dead PKR as a signaling molecule may be an artefact of the protein's altered shape.
TRAFs play a major role in signal transduction downstream of pattern recognition receptors. Significantly, the consensus TRAF motif recognized in PKR has been associated with signals triggered by toll-like receptors and RIG-like helicases that then recruit TRAFs to activate NF-κB, via TRAF-2 and -6 (Takada and others 2007; Yamashita and others 2008; Sondarva and others 2010). Consequently, the association between PKR and TRAFs correlates with the observed responsiveness of PKR (Horng and others 2001; Hsu and others 2004; Kalali and others 2008; Zhang and Samuel 2008; Zhang and others 2009). Experiments reported by Takada and others (2007) exploring TNF-α activation of NF-κB corroborated this association with TRAFs in PKR-dependent signaling. A defect in NF-κB activation in PKR-null cells could be rescued by expressing p65, IκB kinase-β, or the NF-κB-inducing kinase, but not TRAF-2, TNFR-1-associated protein, or TNF-α. This delineates the point within the network at which PKR functions as the recruiter of TRAFs to the signaling complexes.
TRAF-2, -3, and -6 have also been shown to trigger activation of JNK and p38 (Dadgostar and others 2003). Hence, PKR's association with TRAFs could also be evoked as a mechanism by which the kinase regulates these 2 mitogen-activated protein kinases (MAPK) (Goh and others 2000; Iordanov and others 2000; Silva and others 2004).
Perturbation of the MAPK cascade also partly accounts for the PKR-dependent effects toward STAT proteins. There is a defect in activation of STAT-1 and -3 in PKR-null cells (Deb and others 2001; Lee and others 2005). Dimerization of STATs is partially instigated through tyrosine phosphorylation of the proteins by the janus kinases. Full transcriptional activity also requires serine phosphorylation at residue 727, at least for human STAT-1 and -3. Although, significantly, serine phosphorylation is not required for transcriptional activity of the ISG factor-3. The identification of the serine kinase has not been confirmed, but the phosphorylated motif (PMSP) in STAT-1 is characteristic of a phosphorylation site for MAPKs, particularly JNK, p38, and extracellular signal-regulated kinase.
Activation of PKR by PACT
It is generally accepted that the PKR-dependent response to stress stimuli that include the sphingolipid ceramide, arsenite, thapsigargin, and hydrogen peroxide is mediated by PACT (also called RAX in mice) (Ito and others 1999; Ruvolo and others 2001). As well as responding to general stress, it is also claimed that PACT mediates many other PKR-dependent activities. PKR-mediated activation of NF-κB, IRF-1, and STAT-1 are reportedly all attenuated in cells in which PACT is knocked down by RNA interference, or that express a mutant PACT that does not activate PKR (Bennett and others 2006). A requirement for PACT in the activation of these transcription factors may not be at odds with signaling mediated by TRAFs. PACT could provide the activating signal that then enables PKR to associate with TRAFs to trigger subsequent activation of the various transcription factors.
PACT encodes 3 RBMs that mediate the interaction with PKR (Patel and Sen 1998; Peters and others 2001; Huang and others 2002). Although PACT encodes 3 RBMs and binds dsRNA, it can activate PKR independently of dsRNA (Patel and Sen 1998; Peters and others 2001). Stress signals result in phosphorylation of PACT (at serine residue 18), followed by its interaction with PKR (Bennett and others 2004). The kinase that phosphorylates PACT has not been identified. Residues around the phosphorylated serine (REDSGTF) form a putative phosphorylation motif for the MAPK family (Blom and others 1999; Xue and others 2008).
The biological significance of PACT-mediated regulation of PKR is not entirely clear, as different transgenic mutant PACT mice have been reported that have markedly different phenotypes (Rowe and others 2006; Bennett and others 2008; Peters and others 2009). In one instance a PACT-null mouse is viable but has developmental defects (Rowe and others 2006). Subsequently, a second heterozygous PACT mutant was reported that was embryonically lethal in homozygous PACT-null mice (Bennett and others 2008). This same group knocked out the equivalent gene in Drosophila (dRAX). Flies homozygous for the mutant allele were viable but died prematurely and had a number of additional defects. These mutant flies demonstrated neurological defects and had impaired locomotion. This phenotype mimics the dystonia and gait abnormalities observed in humans with mutations in the gene encoding PACT (Camargos and others 2008). In any case, these observed phenotypes are not seen in the PKR-null mice, so it raises the question of how these effects are mediated. Generation of PACT and PKR double-knockout mice is required to elucidate the role of each protein in this interaction.
Future Perspectives
PKR is unique among the eIF2α kinases as it is induced by IFNs and plays a more significant role in immune responses as a cell signaling molecule. In this capacity PKR has been categorized to promote inflammation. However, the emphasis to date has been on deciphering PKR's role as an antiviral protein. Viral infection models induce an acute stress event where viral RNAs directly activate PKR. It is now clear that PKR can be activated by other more general stress stimuli. Hence, PKR is likely to play a role in diseases that have chronic stress stimuli. Correspondingly, a recent study has shown that PKR influences obesity-related conditions (Nakamura and others 2010). This study suggests that PKR promotes inflammation caused by diet, thereby compounding metabolic disease. This is consistent with considerable in vitro cell-based studies but far more limited in vivo experiments that have assessed PKR-dependent responses to pathogen-associated molecules and inflammatory cytokines. Pertinently, the transcription factors activated by PKR do not exclusively promote inflammation. It has been demonstrated that PKR induces the anti-inflammatory IL-10 via activation of NF-κB (Cheung and others 2005; Chakrabarti and others 2008). Further, it has been demonstrated that, at least in vitro, expression of the potent inflammatory cytokines TNF-α and IFN-γ is limited by PKR. Also, other functions of PKR are inconsistent with a narrow role in advancing inflammation. PKR-dependent phosphorylation of eIF2α is generally perceived to be protective, rather than pro-inflammatory. In relation to obesity-mediated etiologies, the other eIF2α kinases and phosphorylation control of eIF2α itself have been demonstrated to be protective (Harding and others 2001; Guo and Cavener 2007; Oyadomari and others 2008). Therefore, it will be important to assess the in vivo role of PKR in different diseases that have varying inflammatory states. Similarly, the role of PKR in different cells and tissue types has not been well addressed. PKR activity is generally assessed in vitro using mouse embryonic fibroblast or macrophage cells. Investigation of PKR's role in other immune cells, such as mast cells, or different specialist cells or tissues requires additional analysis. The function of PKR in neurons, for instance, is of relevance given the proposition that PKR regulates diseases associated with the brain to affect the development of dementia (Hugon and others 2009). This is particularly intriguing as the brain is an immune privileged organ and so does not elicit the usual inflammatory responses.
The recent demonstration of PKR as a p53-induced gene supports a previous presumed involvement of PKR in stress-induced growth arrest. The recognition that deletion of PKR in combination with loss of the tumor suppressor p53 advanced tumorigenesis is exciting. A double-knockout murine model, in which both p53 and PKR are ablated, would be informative to test the significance of PKR in tumorigenesis, and the consequences for therapy. Similarly, the dependence of TRAFs for PKR-mediated cell signaling should be addressed with suitable knockout animal models. As the association between the different TRAF proteins complicates these experiments, this could be addressed with animal models that have targeted mutations of PKR. However, unequivocal evidence of precisely which residues are essential to mediate the different functions of PKR is still lacking. Related to this, transgenic mice that have ablated kinase activity are essential to distinguish kinase from merely scaffold functions of PKR. Similarly, a better understanding of the consequence of phosphorylation of PKR's substrates is necessary. Given the broad activity of the PP2A family of phosphatases, it would be anticipated that phosphorylation of B56α by PKR will be shown to have significant effects on the cell. This seemingly counterproductive induction of a phosphatase by a kinase requires a better understanding of the function of the PP2A proteins. Finally, although it was initially thought that much of PKR's effects were mediated through the induction of IFN, the signaling networks that PKR affects have more recently been shown to induce IFN relatively modestly. Studies that assess the activity of PKR in the IFNAR-null mouse are required to gauge the reliance upon IFN in PKR-dependent responses.
Footnotes
Acknowledgments
The authors are grateful to Drs. Frances Cribbin and Claire McCoy for their editorial assistance in the preparation of this article.
Author Disclosure Statement
No competing financial interests exist.