
Abstract
Enhanced endogenous interferon (IFN) stimulated gene (ISG) signature has been associated with nonresponsiveness to hepatitis C treatment using pegylated-IFNα (pegIFNα) and ribavirin (RBV) in human immunodeficiency virus/hepatitis C virus (HIV/HCV) coinfected patients. Using a proteomic approach, we identified high levels of IFNα receptor 2a (IFNαR2a) in the serum of null responders to pegIFNα/RBV. IFNαR2a inhibited antiviral activity of all formulations of IFNα in JFH/Huh7.5 cells. Furthermore, serum from null responders, but not from those who achieved sustained virologic response, suppressed IFN-signaling and ISG expression in IFNα-stimulated PBMCs of healthy donors in an IFNαR2a specific fashion. An IFNαR2a transgenic mice model (C57BL/6) was generated that had significantly higher levels of IFNαR2a in the serum than the controls (P=0.001). Total ISG expression in the lymph nodes was significantly higher compared to wild-type mice (P value=0.0016). In addition, IFITM1 and SP110 had significantly increased expression in the liver, IFITM1 and ISG15 in the lymph node, and ISG15 and PLSCR1 in the spleen (P value<0.05). The underlying mechanism of resistance to hepatitis C treatment may involve transsignaling of the JAK/STAT pathway by the sIFNαR2a-IFNα/β complex and result in the enhanced ISG signature observed in null responders. In this regard, the transgenic mice model simulated nonresponders to IFNα therapy and provides valuable insights into the role of sIFNαR2a-IFNα interactions in vivo.
Introduction
H
The current standard of treatment for HCV infection involves pegylated IFNα (pegIFNα) and ribavirin (RBV). This regimen may achieve viral clearance or sustained virologic response (SVR) in up to 30% of patients infected with HCV genotype 1, but is significantly lower for those of African descent or coinfected with HIV (Fried and others 2002; Chung and others 2004; Muir and others 2004).
IFNs have remained the backbone of HCV treatment since the 1990s (Poordad and Dieterich 2012). IFNs and their receptors (IFNαR1 and 2 heterodimer) are important in antiviral and antiproliferative activities through the JAK-STAT signaling paradigm (Schindler and others 2007). Modulated by the IFNα receptor 1 (IFNαR1) and IFNα receptor 2 (IFNαR2), the type I IFN regulates transcription of IFN stimulated genes (ISGs) (Liu and others 2011). Several mechanisms have been suggested as reasons for poor HCV clearance rates among HIV/HCV coinfected subjects. Recently we showed an enhanced endogenous ISG signature that was associated with lack of response to pegIFNα and RBV treatment in HIV/HCV infected subjects (Lempicki and others 2006). However, only limited data are available regarding the host and viral factors that result in this enhanced ISG response in some but not all HIV/HCV infected subjects. Furthermore, why these patients do not respond to exogenous IFN is unclear.
In this study, we performed a proteomic approach to identify soluble factors associated with the lack of response to IFN-based HCV treatment. We further tested the ability of candidate proteins to interfere with IFN signaling in vitro and in vivo using a transgenic mice model.
Materials and Methods
Study subjects
HIV/HCV coinfected study subjects were selected based on previous treatment with pegIFNα2a (180 mg/week) or pegIFNα2b (1.5 mg/kg/week) with weight based RBV (1.0–1.2 g/day) for 48 weeks. Subjected belonged to 2 groups: (1) null responder (NR) those who did not have more than 2 log HCV RNA decline from baseline by 2 weeks of treatment and (2) SVR: those who had less than detectable levels of HCV RNA 24 weeks after stopping treatment. All patients signed informed consent, which was approved by the National Institute of Allergy and Infectious Diseases Institutional Review Board.
Proteomic profiling of pooled serum by antibody array
Pooled serum was used to detect differentially expressed proteins using Cytokine 507 array (RayBiotech, Inc.) where 507 cytokine antibodies were printed on a microscope glass slide. Pooled serum at day 0 and 3 was prepared by mixing equal portions of 14 NR or 14 SVR. The samples were first labeled with Cy3 and applied to the arrays in duplicate per manufacturer's protocol. The arrays were then scanned by Agilent microarray scanner with 10 μm resolution and their spot intensities were analyzed by Agilent Feature Extraction 9.0.
ELISA to quantify serum IFNαR2 levels
Blood was collected by venipuncture longitudinally when subjects were undergoing treatment. Serum was separated from whole blood, spun and stored at −80°C for further use. Serum was thawed and levels of soluble IFNαR2 in subjects were determined using the EIA kit (Mybiosource, Inc.) per the manufacturers' instructions. The range of IFNαR2a detected by EIA was 25–1,600 pg/mL. Serum samples were diluted 1/10 and 1/100 and tested in duplicate.
In vitro assay for biological activity of IFNαR2
Ability of IFNαR2a (R&D Biosystems) to block anti-HCV activity of various formulations of IFNα was determined using JFH/Huh7.5 cells in continuous culture system as previous described (Zhang and others 2009). Freshly isolated PBMCs from healthy donors were incubated with human sera from HIV/HCV coinfected (NR or SVR) and IFNα2b in the presence or absence of IFNαR2a antibody (R&D Biosystems). Total RNA was isolated and ISG expression was determined using the Applied Biosystems Custom TaqMan Expression Assay, a multiplex PCR platform.
Transgenic mice construction
Four founder clones were produced: (1) Mus musculus IFNαR2a (Mmu.IFNαR2a) with aminoterminal Kozak translation initiation sequence, (2) CAG2 promoter with the chicken β-actin promoter with CMV enhancer, with the sequence based on the pCCALL2 series vectors, and containing Gateway attL4 and attR1 sequences for multisite recombination, (3) albumin promoter with the sequence derived from literature sources and cloned from a DNA already at the NCI, Frederick Protein Expression Laboratory with Gateway attL4 and attR1 sequences, and (4) Pol2 promoter with the murine Pol2 promoter. These clones were completely sequenced to verify that they were correct and matched the expected database sequences. The clones were spectinomycin-resistant, enabling growth in any Escherichia coli strain using 50 μg/mL spectinomycin to maintain the plasmids.
Expression clones were produced by using Multisite Gateway recombination to merge promoters with the IFNαR2a gene into constructs utilized to generate transgenic mice. Three expression clones were created, including the CAG-Mmu.IFNαR2a, Alb-Mmu.IFNαR2a, and Pol2-Mmu.IFNαR2a. DNA was prepared using the Sigma GenElute maxiprep kit and verified by agarose gel electrophoresis.
The construct DNA from 3 expression clones were microinjected onto fertilized eggs of C57BL/6 mice to create the first generation of IFNαR2a transgenic mice in the NIH animal facility in Frederick, MD and housed under the NIH animal welfare rules and regulations. All 3 transgenic mice strains were developed and confirmed to have overexpression of sIFNαR2a. The presence of the transgene was confirmed using the Sigma-Aldrich Extract-N-Amp Tissue PCR kit and 2% agarose gel electrophoresis from mice tails. The overexpression of sIFNαR2a was verified by Western blot analysis using mice serum. Two microliters of serum was added to 24 μL water, 10 μL 4× LDS, and 4 μL β-Me and heated for 10 min at 70°C. Samples were run on −12% BT gels with 5% milk in TBS-T (0.05% T in TBS) as blocker. The transgene was detected using 1 μg/mL rat anti-IFNαR2 in 5% milk TBS-T and 1:10,000 anti-rat HRP in 5% milk TBS-T.
The CAG-IFNαR2a transgenic mice with the chicken β-actin promoter were backcrossed to produce the stable second generation of transgenic mice (n=9). C57BL/6 mice were used as wild-type controls (n=4).
ISG expression
RNA was extracted from lymph node, spleen, and liver of 6–12-week-old transgenic and control mice using the Qiagen RNeasy Mini Kit and cDNA was generated using the Applied Biosystems High-Capacity cDNA Archive Kit. Real-time PCR was performed using the 96-well Applied Biosystems Custom TaqMan Expression Assay to analyze ISG expression in each tissue type. The following ISGs were analyzed: IFI27L1, IFITM1, IRF3, ISG15, ISG20, OAS1b, PLSCR1, SP110, and STAT1. Each ISG expression was normalized to that of GAPDH.
Statistical analysis
The F-test to compare variances and unpaired t-test were used to determine statistical significance of high levels of IFNαR2a and mean ISG expression in NR versus SVR. Paired t-test was used to determine statistical significance of inhibitory effects of IFNαR2a on IFNα. A Student's t-test was used to determine statistical significance of ISG expression in transgenic mice compared to control mice.
Results
Null responders to PegIFN and RBV have increased IFNαR2a in serum
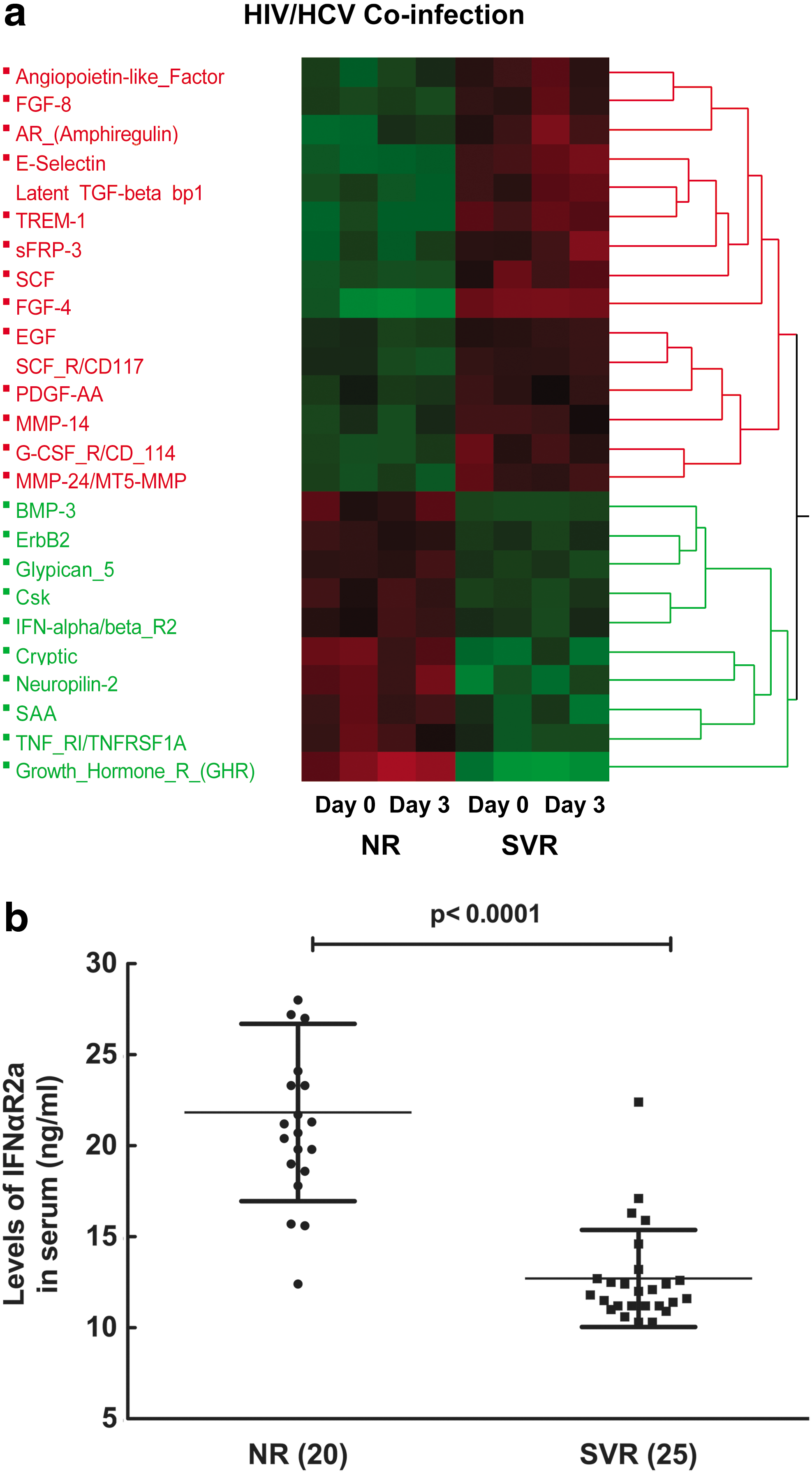
To identify the soluble factors differentially expressed in the serum of NR and SVR subjects, we performed protemic analysis using an antibody array (Raybiotech). Twenty-five proteins were selected based on criteria of P value<0.05 and fold change greater than 2 as shown in Fig. 1a. There were no differences in proteomic levels between day 0 and 3 in both NR and SVR subjects. Fifteen proteins were upregulated in SVR compared to NR, and 10 proteins were upregulated in NR compared to SVR. Among the 10 upregulated proteins in NR, high levels of sIFNAR2a in the serum of NR were validated using a confirmatory ELISA as shown in Fig. 1b by the unpaired t-test (P value<0.0001) and F-test to compare variances (P value=0.005). Due to the potential biological significance of the molecule, IFNαR2a was selected for further validation by EIA.
IFNαR2a inhibits antiviral activity of all formulations of IFNα
We then tested whether the presence of high levels of IFNαR2a could inhibit the biological activity of IFNα. Figure 2 illustrates the inhibitory effect of IFNαR2a on different formulations of IFNα in JFH/Huh7.5 cells. The addition of IFNαR2a reduced suppression of HCV replication by IFNα-2b from 64.7%±13.0% to 20.7%±5.8% (P value=0.0128). In PegIFNα2b, suppression of HCV replication was reduced from 57.3%±11.4% to 9.7%±3.5% (P value=0.0284). In PegIFNα2a, suppression of HCV replication was reduced from 59.7%±10.3% to 7.0%±2.6% (P value=0.0095). In Alb-IFNα, suppression of HCV replication was reduced from 75.3%±4.6% to 6.0%±2.0% (P value=0.0018). Suppression of HCV replication was dramatically reduced with the addition of IFNαR2a in each of the formulations of IFNα.
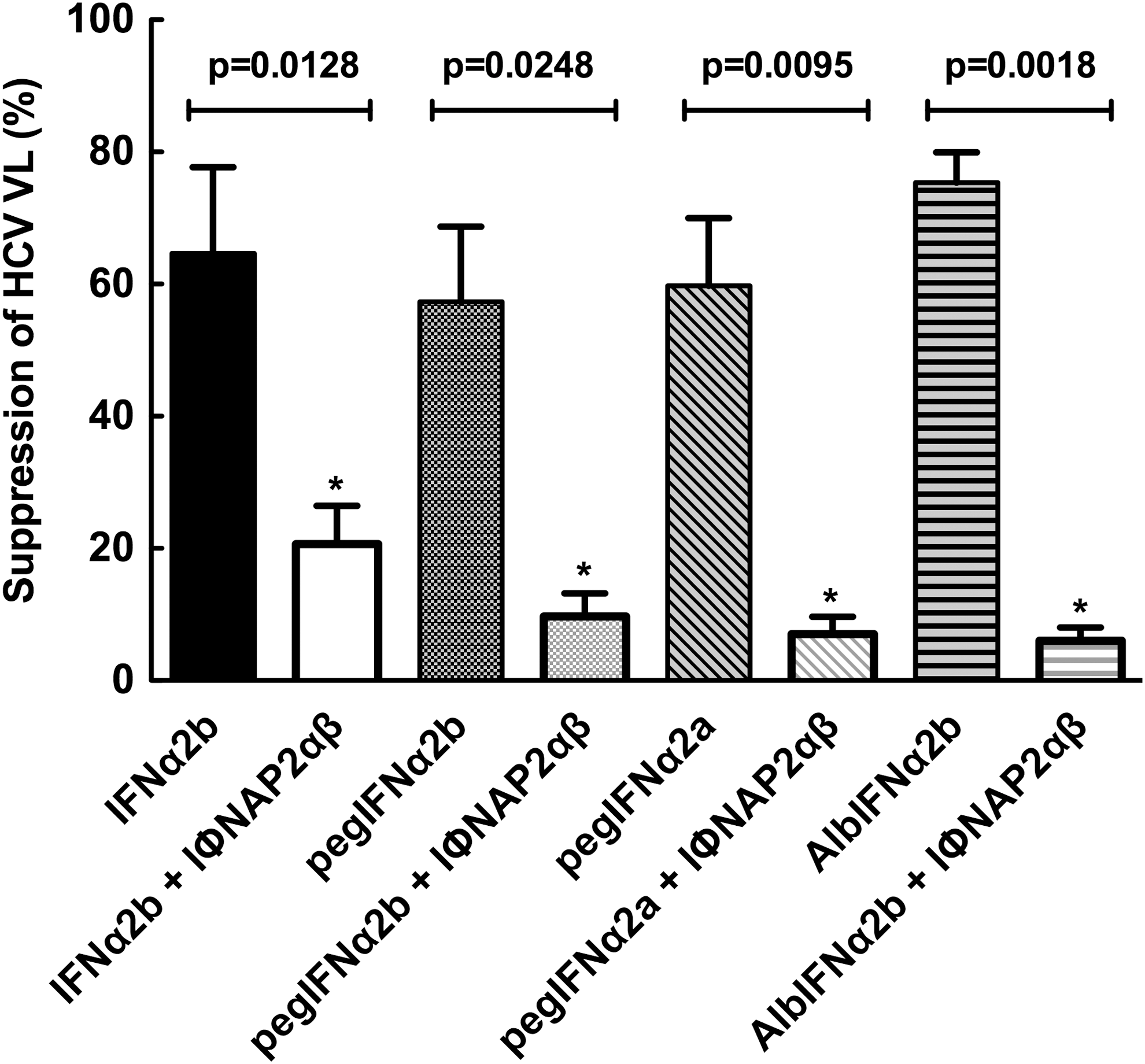
Soluble IFNα/β receptor 2 can inhibit the anti-HCV effects of various IFNα formulations in vitro. Using an in vitro HCV continuous culture system (J6/JFH-1/Huh7.5), copy numbers of J6/JFH-1 were quantitated by real-time PCR in culture supernatants with or without addition of equimolar IFNα formulations (IFNα2b, pegIFNα2a, pegIFNα2b, AlbIFNα2b) in the presence or absence of IFNα/β receptor 2 (5 μg/mL) as described in the Materials and Methods section. As shown in Fig. 3, all IFN formulations exhibited potent suppression of HCV replication and addition of sIFNαR2 abrogated this effect significantly (all P values<0.05).
Serum from NR suppress IFNα-mediated ISG expression
We then determined whether serum from NR, which has high levels of IFNαR2a, would have an effect on the biological activity of IFNα. Freshly isolated PBMCs were stimulated with recombinant IFNα with serum from NR and SVR in the presence or absence of anti-IFNαR2a antibodies. As shown in Fig. 3, serum from NR specifically inhibited ISG expression in PBMCs (P value<0.0001), which was abrogated by the addition of neutralizing anti-IFNαR2a antibodies (P value<0.0001). Sera from SVR had minimal effect on the expression of ISG since they had lower levels of sIFNαR2a.
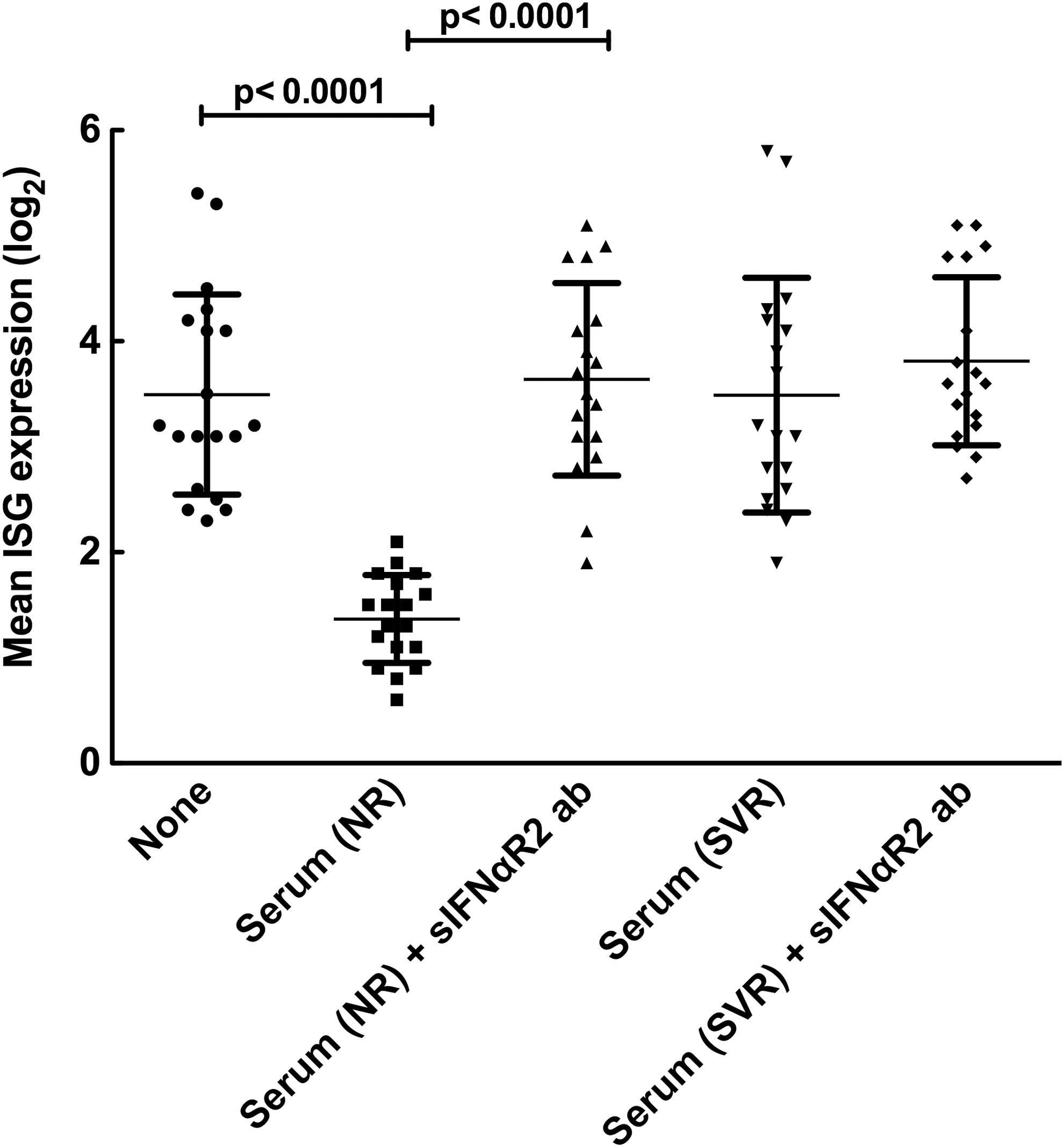
Inhibition of IFNα signaling in peripheral mononuclear cells by serum from null responders is mediated by soluble IFNαR2. Mean IFN-stimulated gene (ISG) expression (n=30) in PBMCs after treatment with 20 IU/mL of IFNα in vitro for 60 min with or without serum from NR and SVR in the presence or absence of anti-IFNαR2 antibody were measured by real-time PCR as described in the Materials and Methods section. Expression of each ISG was normalized with that of GAPDH and averaged for each experimental condition. As shown in Fig. 4, incubation of serum from NR, but not from SVR resulted in the suppression of IFNα mediated induction of ISG (P value<0.0001 and >0.05, respectively). The inhibition of IFN signaling by serum from NR was entirely reversed by addition of antibodies to IFNαR2 (P value<0.0001), which suggest the mechanism of suppression is mostly, if not entirely, mediated by interactions between IFNα and soluble IFNαR2 present at high levels in the serum of NRs. However, no such effect was observed with SVR, as the levels of sIFNαR2 in the serum of SVR are considerably lower (P value>0.05).
IFNαR2a expression in transgenic mice
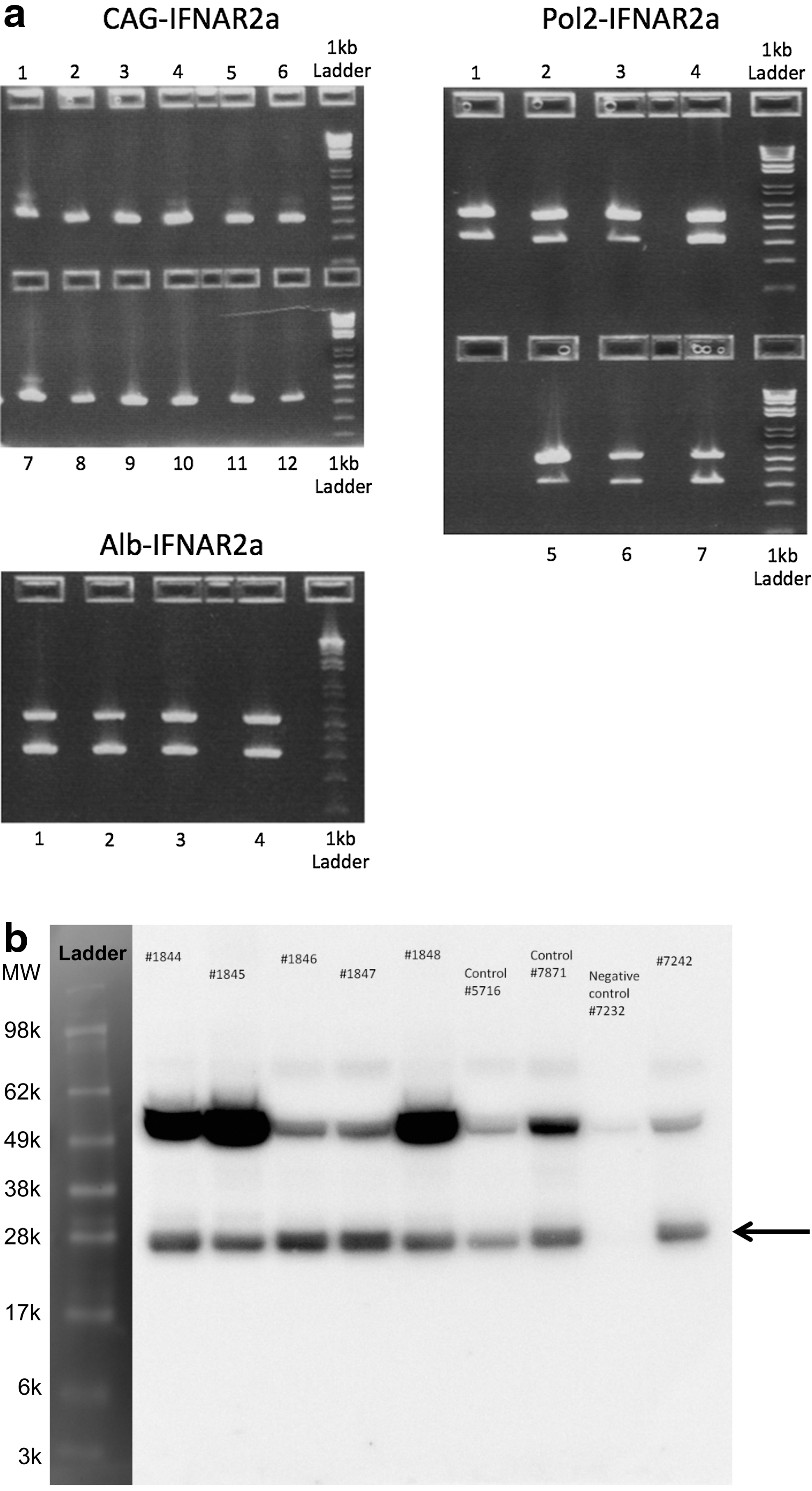
Three lines of mice expressing IFNαR2a transgene were generated as described in the methods. Two of them used ubiquitous promoters, while the third used an albumin promoter, restricting the expression of the transgene to the liver. All mice were tested for presence of transgene as shown in Fig. 4a and expression of transgene via western blot in Fig. 4b.
Increased ISG expression in IFNαR2a transgenic mice
To determine the effect of high expression of IFNαR2a on IFN signaling, we performed multiplex PCR for ISG expression using various tissues from CAG-IFNαR2a transgenic mice as shown in Table 1. Total ISG expression in the lymph node was significantly higher (P value=0.0016) compared to wild-type mice. In addition, IFITM1 (P value=0.0420) and SP110 (P value=0.0190) were identified to have significantly increased expression in the liver, IFITM1 (P value=0.0121) and ISG15 (P value=0.0121) in the lymph node, ISG15 (P value=0.0091) and PLSCR1 (P value=0.0182) in the spleen.
Significantly different (P<0.05) between the transgenic and wild-type samples.
ISG, interferon-stimulated gene; avg. Ct±SD, average cycle threshold numbers ± standard deviation.
Discussion
We describe a novel mechanism mediated by excess levels of soluble IFNαR2 in the serum of patients who subsequently failed to respond to IFNα treatment for hepatitis C. Furthermore, using an in vitro system, we demonstrate that excess levels of IFNαR2a can inhibit IFN-mediated antiviral activity against HCV. In this regard, serum from null responders inhibited IFNα signaling in an IFNαR2a specific manner. Moreover, we developed an in vivo model of transgenic IFNαR2a mice, which demonstrated enhanced endogenous IFN signaling, suggesting transsignaling as a mechanism of interference of soluble IFNαR2a with JAK/STAT signaling.
In hepatitis C treatment with exogenous IFNα, several theories exist to explain the different host responses (NR versus SVR) to IFN treatment. Although PBMCs from NR have increased baseline ISG expression in vivo as demonstrated by DNA microarray and real-time PCR data (Lempicki and others 2006), PBMCs from NR and SVR respond to IFNα similarly in vitro (data not shown). This suggests a role for inhibitory mechanisms in vivo that determines such a response. The higher basal levels of soluble IFNαR2a in NR compared to SVR have been observed to possibly affect this IFN-signaling pathway.
The mechanism by which sIFNAR2a would enhance the endogenous ISG expression is not clearly understood. Recent studies have demonstrated that soluble IFNαR2a is one among 3 isoforms of the IFNαR2, which are generated by alternative splicing in both human and mouse and produces the transmembrane IFNαR2b, as well as the complete receptor IFNαR2c (Owczarek and others 1997; Hardy and others 2001; de Weerd and others 2007). In the murine model, IFNαR2a exhibited antagonistic and agonistic properties (Hardy and others 2001). Recombinant IFNαR2a acted as a competitive inhibitor of type I IFN antiproliferative activity in an in vitro assay. However, in the murine IFNαR2 knockout thymocytes, recombinant IFNαR2a produced an antiproliferative response, suggesting transsignaling capability of the soluble receptor (Hardy and others 2001). The specific mechanism of soluble IFNαR2a is not clear in its interaction in vivo with endogenous and exogenous IFN, especially when administered as therapy. After IFNα treatment in patients with hepatitis C, expression of the IFNα/β receptor in the liver was much stronger in SVR than NR, suggesting host differences in response to IFN (Meng and others 2005). Hence, the soluble form of IFNαR2 receptor soluble may act as important regulators of immune response as in the case on many other such cytokine receptors, including IL-6 and IL-15 (Fernandez-Botran 1991). For example, soluble receptors, IL-6R and IL-15R, have been shown as agonists in enhancing stimulation of target immune cells. Transsignaling is a process by which the soluble component of a heterodimer can interact with the ligand and the membrane bound counterpart resulting in partial activation of the signaling pathway. In this regard, our data demonstrates that sIFNαR2a can also act in such a manner and result in activation of JAK/STAT pathway to enhance endogenous IFN gene signature observed in HIV/HCV coinfected subjects. Transsignaling has been previous described with both IL-6 and IL-15 receptors. It has been shown that IL-6/sIL-6R complex induces the dimerization of gp130 in cells to regulate intracellular signal transduction (Jones and others 2005). Similarly, the IL-15/IL-15Rα complex has been demonstrated to have superior stabilization and increased bioactivity manifesting in selective induction of natural killer and CD8+ T cells (Rubinstein and others 2006; Bergamaschi and others 2008, 2009). Our data demonstrating that IFNAR2a transgenic mice have enhanced endogenous expression of ISG, is consistent with this hypothesis and could contribute to the IFN signature observed in NRs in vivo, most likely by JAK/STAT phosphorylation, resulting in ISGF3–ISRE interactions.
Elevated levels of soluble IFNαR2a (sIFNαR2a) in serum and urine have been associated with hairy cell leukemia and adenocarcinoma, resulting in high resistance to IFN therapy (Novick and others 1992; Ambrus and others 2003). In hepatitis C, where treatment is mainly dependent on IFN therapy, high levels of sIFNαR2a have been associated with poor response rates to pegIFNα and RBV (Mizukoshi and others 1999). In this regard, soluble IFNαR2a may potentially act as a blocking agent in the IFN-signaling pathway. As shown in our experiments, we can block the pathway and inhibit the antiviral activity of IFN. However, it was unclear how the blocking of the pathway led to increased ISG expression. Clinical data suggests that high baseline ISG and ISG15 are negative predictors for treatment response in hepatitis C to IFN treatment (Chen and others 2005; Sarasin-Filipowicz and others 2008). Therefore, we generated the transgenic mice with overexpression of IFNαR2a, which suggests that IFNαR2a leads to high endogenous IFN signaling.
In our proposed model, high levels of soluble IFNαR2 is associated with a lack of response to exogenous IFN. We propose that the excess amount of IFNαR2a is generated by alternative splicing, probably as a result of a yet unidentified SNP in some but not all HCV-infected individuals. An excess of IFNαR2a then combines with membrane bound IFNαR1 and IFN and leads to phosphorylation of the JAK/STAT pathway, resulting in an enhanced ISG expression (transsignaling) in tissues, such as the liver. Finally, this persistent activation most likely leads to further blunting of JAK/STAT signaling, resulting in a lack of response to exogenous IFN.
Overall, these results suggest that high ISG expression may explain most of the IFN signaling abnormalities described in the hepatic and peripheral lymphatics in patients with hepatitis C. Finally, further studies will extend these observations in an IFN-free treatment paradigm to clarify the role of high ISG expression in predicting treatment response.
Footnotes
Acknowledgments
This research was supported in whole by the Intramural Research Program of the NIH (National Institute of Allergy and Infectious Diseases, National Cancer Institute and the Clinical Research Center). We acknowledge Apath LLC for providing us with J6/JFH-1 HCV clone and Huh7.5 cells.
Disclaimer
The content of this publication does not necessarily reflect the views of policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Author Disclosure Statement
None of the authors have any conflicts of interest to report.