
Abstract
Influenza A viruses (IAVs) cause mild to severe infections in humans with considerable socioeconomic and global health consequences. The host interferon (IFN)-α/β response, critical as the first line of defense against foreign pathogens, is induced upon detection of IAV genomic RNA in infected cells by host innate pattern recognition receptors. IFN-α/β production and subsequent activation of cell signaling result in the expression of antiviral IFN-stimulated genes whose products target various stages of the IAV life cycle to inhibit viral replication and the spread of infection and establish an antiviral state. IAVs, however, encode a multifunctional virulence factor, nonstructural protein 1 (NS1), that directly antagonizes the host IFN-α/β response to support viral replication. In this review, we highlight the mechanisms by which NS1 suppresses IFN-α/β production and subsequent cell signaling, and consider, therefore, the potential for recombinant IAVs lacking NS1 to be used as live-attenuated vaccines.
Introduction
I
Interferon-α/β Induction by IAVs
IAVs spread via aerosol droplets, nasal secretions, saliva, and virus-contaminated surfaces (Bean and others 1982; Noti and others 2012). In wild aquatic birds and poultry, IAVs also spread via oral–fecal transmission (Rohani and others 2009). In all cases, IAVs enter the host through the oral or nasal cavities, crossing the layer of mucus coating the upper respiratory tract before infecting lung epithelial cells and resident immune cells, namely macrophages and dendritic cells (DCs) (Bhardwaj and others 1994; Tate and others 2010). IAVs typically induce a rapid and robust innate immune response, which in turn contributes to the activation of an adaptive immune response, resulting in viral clearance and immunological memory (Fig. 1). However, highly pathogenic strains can cause severe pathology and mortality.
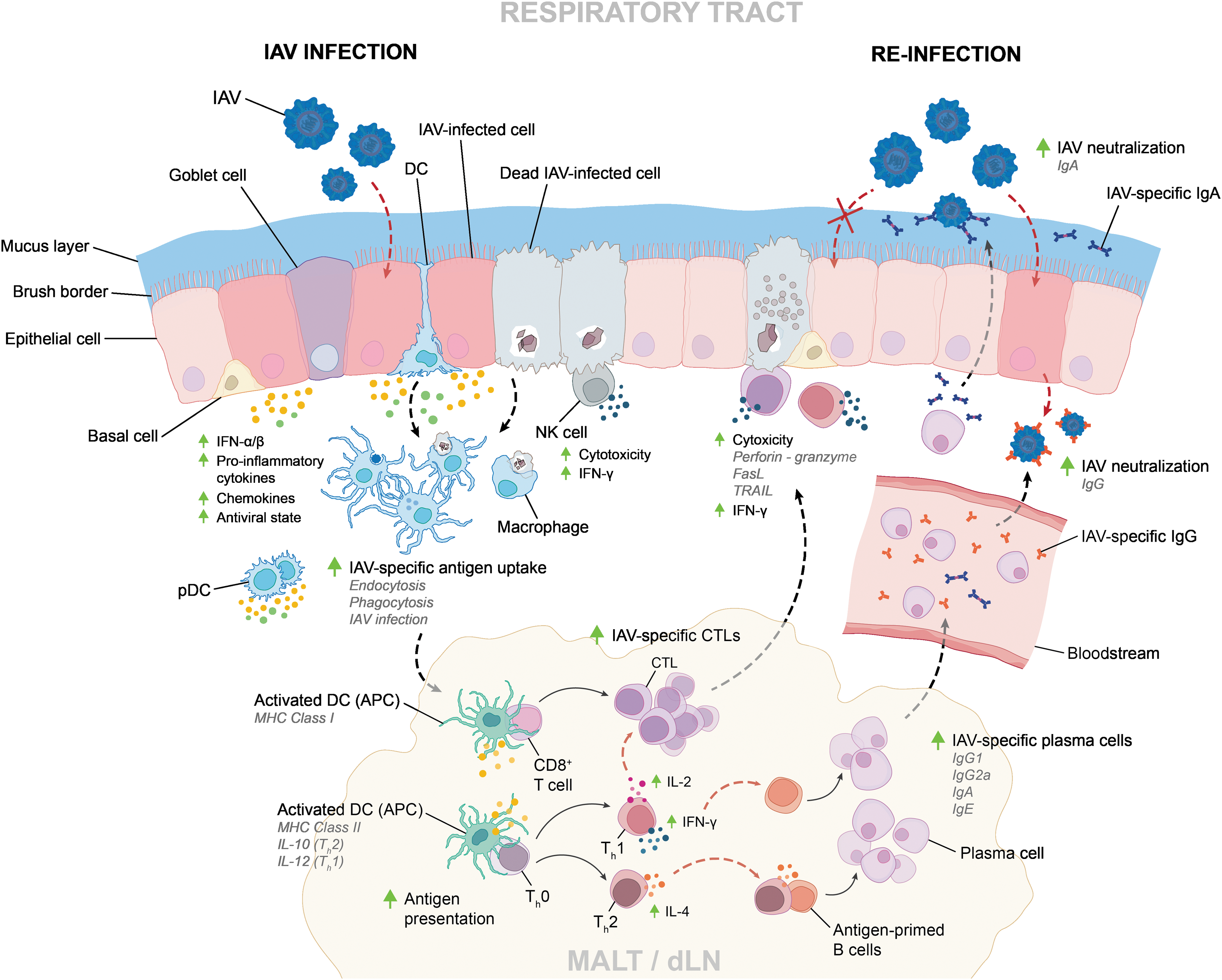
Innate and adaptive immune responses to IAV infection. IAVs encounter innate immune cells in the respiratory tract, triggering innate and adaptive immune responses that lead to viral clearance and the development of a strain-specific memory response. Infection by highly pathogenic strains or an immunocompromised host can result in severe pathology and mortality. CTL, cytotoxic T lymphocyte; dLN, draining lymph node; IAV, influenza A virus; MALT, mucosa-associated lymphoid tissue.
Multiple innate pattern recognition receptors (PRRs) are essential for recognizing pathogen-associated molecular patterns by damage/danger-associated molecular patterns (DAMPs) to induce the expression of interferons (IFNs)-α/β, proinflammatory cytokines, and chemokines in response to IAV infection: toll-like receptor (TLR) 3, TLR7, TLR8, retinoic acid-inducible gene I (RIG-I), melanoma differentiation-associated protein 5 (MDA5), nucleotide-binding oligomerization domain-containing protein 2 (NOD2), and NACHT, LRR and PYD domains-containing protein 3 (NLRP3) (Fig. 2) (Diebold and others 2004; Le Goffic and others 2007; Loo and others 2008; Wang and others 2008; Allen and others 2009; Sabbah and others 2009; Ichinohe and others 2010). Dependent on where they are expressed within the infected host cell, PRRs detect viral RNAs either during the IAV life cycle when viruses fail to undergo fusion and are degraded in the endosome or during viral genome replication when viral RNAs are translocated into the cytosol. In select innate immune cells such as macrophages and neutrophils, PRRs can also detect dsRNAs (DAMPs) released by dying IAV-infected cells that have been phagocytosed.

PRR activation during IAV infections. TLRs, RLRs, and NLRs recognize ssRNAs (TLR7, RIG-I, and NOD2), dsRNAs (TLR3 and MDA5), and protons (NLRP3 inflammasome) produced during viral replication and by dying IAV-infected cells. PRR activation results in the expression of IFN-α/β and proinflammatory cytokines as part of the innate immune response to IAV infection. IFN, interferon; MDA5, melanoma differentiation-associated protein 5; NLRP3, NACHT, LRR, and PYD domains-containing protein 3; NOD2, nucleotide-binding oligomerization domain-containing protein 2; PRR, pattern recognition receptor; RIG-I, retinoic acid-inducible gene I; TLR, toll-like receptor.
TLR3, TLR7, and TLR8 are expressed in the endosomes of multiple cell types present within the respiratory mucosa. TLR3 recognizes dsRNA and is expressed in respiratory epithelial cells, alveolar macrophages, and DCs (Guillot and others 2005). In addition to inducing the expression of IFN-α/β and proinflammatory cytokines in response to IAV infection (Le Goffic and others 2007), TLR3 activation has also been linked with tissue damage and lung pathology (Le Goffic and others 2006). When infected with a lethal dose of IAV, TLR3 −/− mice have higher lung viral titers but survive longer than wild-type (wt) mice and mount a normal T cell response (Le Goffic and others 2006).
TLR7 and TLR8 recognize ssRNAs. TLR7 and TLR8 are expressed in DCs, monocytes, macrophages, and neutrophils (Diebold and others 2004; Wang and others 2008; Ablasser and others 2009). In pDCs, TLR7 activation by viral RNAs results in IFN regulatory factor (IRF) 7-dependent IFN-α/β production, whereas in granulocyte–macrophage colony-stimulating factor-primed neutrophils, TLR7 activation primarily induces proinflammatory interleukin (IL)-8 production (Jewell and others 2007; Wang and others 2008). Studies have shown that TLR7 expression is important for inducing an IAV-specific antibody response in the context of sublethal IAV infections, while TLR7 signaling is not required for an antiviral T cell response (Heer and others 2007; Koyama and others 2007).
RIG-I, MDA5, NOD2, and NLRP3 are cytosolic PRRs that detect various components of the replicating IAV. Localized to stress granules, formed in part by the IFN-inducible antiviral protein kinase RNA-activated (PKR) (Onomoto and others 2012), RIG-I is able to detect IAV 5′ triphosphate ssRNAs and 5′ disphosphate dsRNAs, which are synthesized during viral replication (Kato and others 2006; Pichlmair and others 2006). Recognition and binding to viral RNA induce conformational changes within RIG-I that allow it to associate with mitochondrial antiviral-signaling protein (MAVS) and induce IFN-α/β and proinflammatory cytokine production in lung epithelial cells and alveolar macrophages (Le Goffic and others 2007; Wang and others 2012). While RIG-I is essential for IFNB and IFNA gene expression in infected cells, knockout studies have also shown that the RIG-I-like receptor, MDA5, which binds dsRNA, contributes to IRF7 expression and the induction of antiviral IFN-stimulated genes (ISGs): OAS, ISG15, IFIT1, and STAT1 (Benitez and others 2015).
The NOD-like receptors, NOD2 and NLRP3, detect ssRNAs and are also critical for limiting IAV infection. In response to IAV infection, NOD2 −/− mice produce less IFN-β, recruit fewer activated DCs and IAV-specific CD8+ T cells to the lungs, and have higher morbidity in comparison to wt mice (Lupfer and others 2014). The NLRP3 inflammasome is expressed in monocytes, macrophages, DCs, and neutrophils and is activated by IAV ssRNA and cytosolic acidification caused by newly synthesized IAV matrix 2 (M2) protein within the trans-Golgi network (Allen and others 2009; Ichinohe and others 2010). NLRP3 activation results in the recruitment and autocatalytic cleavage of procaspase-1 into caspase-1 (Yang and others 1998). Caspase-1 dimerization enables the cleavage of pro-IL-1β and pro-IL-18 into IL-1β and IL-18 proinflammatory cytokines, respectively (Pirhonen and others 1999).
Effects of IFN-α/β on IAV Replication
IFN-α/β upregulate the expression of more than 3000 ISGs * encoding proteins with a variety of functions, including innate host defense, growth inhibition, antigen processing and presentation, metabolism, cell adhesion, immune modulation, and transcriptional activation (Rusinova and others 2013). During an IAV infection, IFN-α/β produced by infected cells and innate immune cells induce the expression of antiviral ISGs, which encode proteins that target specific stages of viral replication and establish an antiviral state in both infected and neighboring uninfected cells. IFN-inducible antiviral proteins that target various stages of IAV replication include myxovirus resistance gene A (MxA), ISG15, PKR, 2′-5′-oligoadenylate synthetase (OAS), IFN-induced transmembrane protein (IFITM), and viperin.
In brief, MxA and its mouse homolog Mx1 form oligomeric ring structures that bind IAV nucleoprotein (NP) and inhibit viral ribonucleoprotein (vRNP) translocation (Kochs and others 2005; Verhelst and others 2012; Nigg and Pavlovic 2015). Feral mice, that express Mx1, are much less susceptible to IAV infection compared with laboratory strains, which lack the Mx1 gene (Staeheli and others 1988; Tumpey and others 2007; Shin and others 2015).
ISG15, an ubiquitin-like protein, binds to lysine residues within viral and host proteins through a process similar to ubiquitination, called ISGylation (Loeb and Haas 1992; Shi and others 2010; Zhao and others 2010). Many cellular proteins are ISGylated following IFN-α/β treatment, which may affect their antiviral functions (Malakhova and others 2003; Shi and others 2010). ISG15 −/− mice are more susceptible to IAV infections than wt mice (Lenschow and others 2007; Hsiang and others 2009); likewise, A549 lung epithelial cells lacking ubiquitin-like modifier-activating enzyme 7 (UBA7), which is required to catalyze ISGylation (Yuan and Krug 2001), are attenuated in their ability to inhibit IAV nonstructural protein 1 (NS1), hemagglutinin (HA), NP, and matrix 1 (M1) protein expression when infected with IAV (Hsiang and others 2009).
PKR as an intracellular sensor of dsRNA recognizes IAV mRNA panhandle structures during viral replication (Hatada and others 1999). PKR is also a target for inhibition by NS1, as it phosphorylates and inhibits the eukaryotic translation initiation factor 2-α subunit (EIF2α) to block cellular and viral mRNA translation (Min and others 2007). PKR −/− mice are extremely susceptible to infection by recombinant IAVs (rIAVs) that do not express NS1 (Bergmann and others 2000).
Similar to PKR, 2′-5′-OASs also bind dsRNAs (Chebath and others 1987; Hovanessian and others 1988; Donovan and others 2015). dsRNA binding activates OAS, which synthesizes 2′-5′ olygoadenylates (Chebath and others 1987; Hovanessian and others 1988; Rusch and others 2000; Li and others 2004). These 2′-5′ olygoadenylates act as substrates for ribonuclease L (RNAseL) (Floyd-Smith and others 1981; Zhou and others 1993), which cleaves cellular and viral RNA to block IAV replication and induce apoptosis (Wreschner and others 1981; Cooper and others 2015). RNA fragments generated by RNAseL in the cytoplasm can also activate the NLRP3 inflammasome (Chakrabarti and others 2015) and are able to activate RIG-I and MDA5 to induce IFN-α/β production (Malathi and others 2007).
IFITMs (IFITM1, IFITM2, and IFITM3) are expressed on the cell surface and inhibit viral entry into yet uninfected cells, which have been primed by IFN-α/β signaling (Brass and others 2009; Huang and others 2011). The exact mechanism of inhibition by IFITMs needs further characterization, but recent single-virion imaging studies by Desai and others (2014) have demonstrated that IFITM3 blocks the formation of fusion pores in the late endosome, which is required for IAVs to release vRNPs into the cytosol.
Viperin, on the other hand, is expressed in the endoplasmic reticulum and restricts IAV budding by blocking the activity of farnesyl diphosphate synthase—an enzyme that is involved in the synthesis of isoprenoids (Wang and others 2007). Reduced isoprenoid synthesis results in changes to the composition and fluidity of the host cell membrane (Wang and others 2007).
In addition to the induction of antiviral ISGs, IFN-α/β signaling is also critical for establishing an antiviral immune response by activating innate and adaptive immune cells (Fig. 3). In comparison with wt mice, mice lacking intact IFN-α/β receptors (IFNARs) exhibit significantly more weight loss and a more rapid time to death when infected with IAVs, including H5N1 and H1N1 subtypes (Garcia-Sastre and others 1998a; Koerner and others 2007; Mordstein and others 2008; Szretter and others 2009; Seo and others 2011; Arimori and others 2013). Using a mouse-adapted H1N1 IAV, Arimori and others (2013) showed that, in addition to increased mortality, IAV-infected IFNAR −/− mice also experienced greater levels of proinflammatory IL-1β and IL-6 expression in their lungs compared with wt mice, characteristics of the elevated cytokine signature seen in infections with highly pathogenic IAV strains (de Jong and others 2006; Teijaro and others 2011). Increased IL-1β and IL-6 production in IFNAR −/− mice also correlated with severe lung tissue damage and excessive neutrophil infiltration. Moreover, IFNAR −/− mice had much lower levels of anti-inflammatory IL-10 expression in their lungs compared with wt mice, and treatment with exogenous IL-10 was able to reduce mortality (Arimori and others 2013).
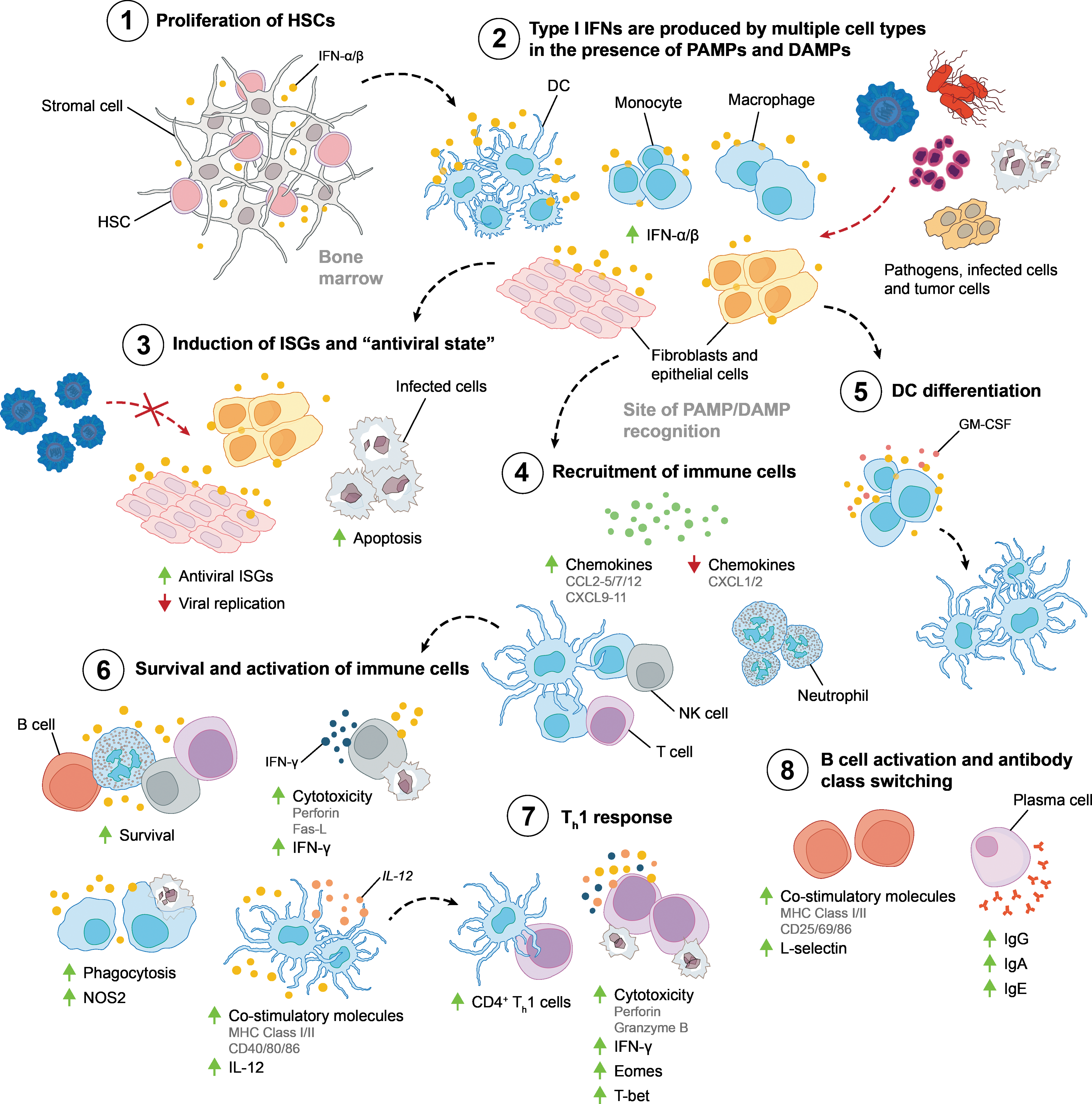
Effects of IFN-α/β on the immune system. IFN-α/β have a critical role throughout the development of innate and adaptive immune responses by affecting the recruitment, proliferation, activation, and function of multiple cell types.
Influenza A Virus NS1
The IAV NS1 is encoded by the NS gene (segment 8). NS1 is one of the first viral proteins to be expressed in the infected host cell (Shapiro and others 1987; Manicassamy and others 2010). NS1 exhibits strain-specific distribution in the cytoplasm and the nucleus (Melen and others 2007; Ayllon and others 2012), where it is able to interact with RNA and host proteins to suppress host cell functions, most notably the host IFN-α/β response while promoting viral replication (Fig. 4). NS1 is able to regulate viral RNA synthesis, viral mRNA splicing and translation, inhibit host mRNA processing, and prevent the host from mounting an effective IFN-α/β response. Hence, rIAVs that do not express NS1 are attenuated in hosts that have a normal IFN-α/β response (Garcia-Sastre and others 1998b) and, accordingly, have also been identified as vaccine candidates.

NS1-host protein interactions. IAV NS1 is expressed in the cytoplasm and nucleus of infected cells. NS1 interactions with host proteins influence multiple cellular processes, resulting in enhanced IAV replication. NS1, nonstructural protein 1.
NS1 Inhibits Antiviral ISGs: PKR and OAS
NS1 inhibits both PKR and OAS, which detect viral RNA and block IAV replication. The NS1 RNA-binding domain (RBD; residues 1–73) is not required for PKR inhibition (Li and others 2006a; Min and others 2007). Instead, NS1 has been shown to bind a linker region within PKR, between its dsRNA-binding domain and kinase domain, to prevent the conformational changes from its inactive to active state upon binding to dsRNA or protein activator of IFN-induced protein kinase EIF2AK2 (PACT) (Li and others 2006a, 2006b). Residues 123–127 within the NS1-binding effector domain (ED; residues 74-230/237) have also been shown to be important for binding to PKR (Min and others 2007). In comparison with rA/Udorn/1972 [H3N2] (rUd) encoding wt NS1, rUd encoding NS1 expressing 123A and 124A or 126A and 127A is unable to inhibit PKR-mediated phosphorylation of eIF2α (Min and others 2007). In regard to inhibition of OAS activity, NS1 sequesters IAV-associated dsRNA to prevent detection by 2′5′-OAS/RNaseL (Min and Krug 2006). IAV rUd expressing a truncated NS1 lacking an RBD or an NS1 encoding an R38A mutation in the RBD to abrogate dsRNA binding is unable to prevent 2′5′-OAS/RNaseL-mediated RNA degradation and this results in up to 1,000-fold lower viral titers than wt rUd (Min and Krug 2006).
NS1 Inhibits IFN-α/β Production
The ability for NS1 to inhibit IFN-α/β production is strain specific and occurs at both the pretranscriptional and posttranscriptional stages of IFN induction. Pretranscriptional inhibition of IFN-α/β production occurs when NS1 binds 5′ triphosphate viral RNAs produced during IAV replication, to form a complex with the host PRRs, RIG-I, and MAVS, preventing the innate detection of viral genomic material within the infected cytoplasm (Ferko and others 2004; Pichlmair and others 2006; Mibayashi and others 2007). NS1 is able to inhibit IFN-β production in cells expressing constitutively activated RIG-I (Mibayashi and others 2007). Binding of NS1 to viral RNA and RIG-I has been shown to prevent the activation of NF-κB, IRF3, and the c-Jun: activating transcription factor 2 heterodimer, suppressing IFN-α/β production by IAV-infected cells (Wang and others 2000; Ludwig and others 2002; Mibayashi and others 2007; Ruckle and others 2012). Compared with wt rA/Puerto Rico/8/1934 [H1N1] (rPR8), infection with rPR8 encoding NS1 expressing R38A and K41A mutations, which abrogate NS1-mediated RNA and RIG-I binding, induces more IFN-α/β production (Ramos and others 2013). Consequently, rIAVs encoding RNA-binding mutant NS1s replicate to lower titers in cells and in mice that have an intact IFN-α/β response.
Recently, NS1 has been shown to interact directly with RIG-I, independent of RNA binding (Jureka and others 2015). Based on solved protein structures, the pandemic 1918 IAV [H1N1] NS1 RBD is able to interact with 1 of 2 RIG-I caspase activation and recruitment domains (CARDs)—CARD2 (Jureka and others 2015). This NS1-RIG-I interaction may inhibit the conformational changes required for RIG-I-mediated MAVS activation and IFN-α/β expression (Jureka and others 2015). Binding of NS1 to RIG-I CARD2 may also mask residue K172 in RIG-I from undergoing tripartite motif-containing 25 (TRIM25)- and ring finger protein 135 (RNF135)-dependent ubiquitination, which is necessary for MAVS activation (Gack and others 2009; Rajsbaum and others 2012; Jureka and others 2015). Notably, NS1s isolated from multiple strains of IAV are able to block RIG-I ubiquitination by TRIM25 and RNF135 to inhibit IFN induction (Gack and others 2009; Rajsbaum and others 2012). The MDA5 pathway is another target of pretranscriptional inhibition of IFN-α/β production by NS1 (Liniger and others 2012; Wei and others 2014). Cotransfection studies in duck embryonic fibroblasts show that H5N1 NS1 expression significantly reduces the activity of MDA5 CARD, which by itself induces both IFN-β and IRF7 expression (Wei and others 2014).
In contrast, posttranscriptional inhibition of IFN-α/β production is largely due to NS1-dependent regulation of the host mRNA processing machinery—namely cleavage and polyadenylation specific factor 4 (CPSF4). Posttranscriptional inhibition of IFN-α/β expression may also occur as a result of 5′ mRNA cap-snatching, NS1 binding to host poly(A)-binding protein II (PABPII), and NS1-mediated inhibition of the nuclear export of host mRNAs. IFN-β pre-mRNA accumulates in wt rUd-infected cells, but not in the nucleus of cells infected with rUd encoding NS1-G184R, which fails to bind CPSF4 (Das and others 2008). Additional mutagenesis studies by Kochs and others (2007) have shown that A/Texas/36/1991 (Tx) [H1N1] NS1, which binds CPSF4, inhibits IFN-α expression and that F103S and M106I mutations in the Tx NS1 ED abrogates NS1-CPSF4 binding and NS1-mediated inhibition of IFN-α expression. On the contrary, mutations that restore CPSF4 binding in PR8 NS1, S103F, and I106M also restore NS1-mediated antagonism of IFN-α expression (Kochs and others 2007). Similarly, Ramos and others (2013) showed in human DCs that the Tx NS1 CPSF4-binding region is critical for suppressing IFN-α production, whereas the RBD is critical for PR8 NS1-mediated inhibition of IFN-α expression and production. Constitutive expression of the CPSF4 F2F3 zinc finger domains, as a tagged peptide, in the nucleus of IAV-infected cells inhibits viral replication, correlating with an increase in IFN-β mRNA expression (Twu and others 2006). Therefore, the NS1 CPSF4-binding domain may present as a potential antiviral target.
NS1 Inhibits IFN-α/β Signaling
Not only is NS1 able to inhibit IFN-α/β production, but it may also be involved in suppressing IFN-α/β signaling responses. For instance, NS1 has been shown to regulate the activity of phosphoinositide 3-kinase (PI3K) and Crk-like protein (CRKL), which are effectors in distinct signaling cascades activated by IFN-α/β. The current research suggests a strain-specific mechanism for IAVs and their NS1s to downregulate IFN-inducible JAK-STAT signaling (Pauli and others 2008; Uetani and others 2008; Jia and others 2010).
In a 2008 study, Uetani and others (2008) found that A/Aichi/2/1968 [H3N2] infection significantly downregulates both IFN-α/β and IFN-γ-inducible tyrosine phosphorylation of STAT1 in A549 cells. In the context of IFN-γ, IAV infection reduced JAK1 and IFN-γ receptor 1 (IFNGR1) protein expression, but not JAK2 or IFNGR2 expression (Uetani and others 2008). NS1 expression alone in A549 cells was not sufficient to inhibit IFN-α/β- or IFN-γ-inducible STAT1 phosphorylation. In contrast, Pauli and others (2008) did not observe any downregulation in IFN-γ-inducible tyrosine phosphorylation of STAT1 in A549 cells infected with PR8 [H1N1] IAV. Instead, PR8 inhibited IFN-β-inducible tyrosine phosphorylation of STAT1 and STAT2 after 6 h of infection, owing to upregulated SOCS3 expression (Pauli and others 2008).
In addition to these findings, we have shown (Jia and others 2010) that A/duck/Hubei/L-1/2004 [H5N1] NS1, which encodes the 5 amino acid deletion at residues 80–84, is able to inhibit IFN-β-inducible phosphorylation of STAT1, STAT2, and STAT3 in HeLa cells. In addition, cells expressing H5N1 NS1 exhibited a reduction in the level of IFN-inducible STAT1-STAT1, STAT1-STAT3, and STAT3-STAT3 binding to sis-inducible elements (SIEs). These H5N1 NS1-expressing cells also had lower levels of surface IFNAR1, but not IFNAR2 expression. Notably, treatment with IFN-α before challenge with IAV resulted in increased ISG expression and decreased viral M gene expression in IAV-infected primary human lung explants, suggesting that viral inhibition of the IFN response can be overcome with exogenous IFN treatment (Jia and others 2010).
IFN-α/β as a Treatment for IAV Infection
Current treatments for IAV infections in humans include inactivated and live-attenuated vaccines delivered via intramuscular injection or via intranasal spray, and direct-acting antivirals, which target specific IAV proteins to inhibit viral replication. IFN-α has been tested in clinical trials as a prophylactic for IAV infections, with varying degrees of success. Studies in mice and ferrets have shown that IFN-α/β treatment inhibits IAV replication, significantly reducing viral titers (Kugel and others 2009; Yoo and others 2010). A low-dose rIFN-α nasal spray was shown to be an effective and safe prophylactic against IAV infection in a randomized controlled trial among military personnel (Gao and others 2010). In this study, 6.4% of subjects (30 of 472) who received rIFN-α treatment were positive for IAV-specific antibodies in comparison with 22.6% of subjects (104 of 460) from the control group—an absolute risk reduction of 16.3% (Gao and others 2010). Conversely, in a double-blind controlled trial conducted by Bennet and others (2013), low-dose oral rIFN-α was ineffective in reducing overall incidence of acute respiratory illness. Moreover, rIFN-α was effective in reducing the incidence and severity of acute respiratory illness in male subjects older than 50 years who had received the seasonal IAV vaccine. IFN-α/β may therefore be promising prophylactics for IAV in high-risk populations, while providing the added benefit of being a broad-spectrum antiviral (Gao and others 2010).
NS1Δ Viruses as Candidate Vaccines
rIAVs encoding NS genes lacking an NS1 open-reading frame (rIAV-NS1Δ) (Garcia-Sastre and others 1998b) or expressing truncated NS1 (Falcon and others 2005; Haye and others 2009) can be generated by reverse genetics. Multiple studies have shown that despite being highly attenuated in IFN-α/β-competent hosts (Garcia-Sastre and others 1998b; Kochs and others 2009), rIAVs-NS1Δ are able to induce robust strain-specific—and in some cases cross-protective—humoral responses (Talon and others 2000; Falcon and others 2005; Wacheck and others 2010). In early animal studies, mice immunized with rPR8 expressing truncated NS1-99 or NS1Δ were protected from subsequent challenge with up to 5,000-times the lethal dose (LD50) of wt rPR8 (Talon and others 2000). Immunization with rPR8-NS1-99 and rPR8-NS1Δ induced PR8-specific antibodies and comparable levels of PR8 NP-specific CD8+ T cells in their spleens, in comparison with mice infected with wt rPR8 (Talon and others 2000). In view of these findings, NS1Δ viruses are considered promising candidates for live-attenuated IAV vaccine (LAIV) development.
In 2010, Wacheck and others (2010) published a seminal article describing the safety and efficacy of a rIAV-NS1Δ (A/New Caledonia/20/1999; H1N1) in a small randomized, double-blind, placebo-controlled, dose-escalation trial. In brief, 58% of healthy subjects vaccinated with a high-dose of rIAV-NS1Δ had a greater than 2-fold increase in vaccine-specific serum IgG. The same cohort also experienced a greater than 4-fold increase in HA-inhibiting antibody titers and a 2-fold increase in nasal IgA. Interestingly, ∼50% of the sera collected from this cohort were able to further neutralize a number of other IAV strains: A/Solomon Islands/3/2006 and A/Brisbane/59/2009, in addition to the wt A/New Caledonia/20/1999, suggesting that LAIVs using rIAV-NS1Δ can confer some degree of cross-protection.
Given its multifunctional role in promoting viral replication and suppressing host immune responses, NS1 presents as a conserved virulence factor that could be a target beyond rIAV-NS1Δ vaccines for limiting infections by a broad range of IAV strains. Indeed, novel strategies to suppress NS1 activity can be developed by identifying critical structural features (Darapaneni and others 2009) and understanding the underlying mechanisms of NS1-mediated host antagonism.
Summary
The IFN-α/β response is important for limiting viral replication, through the induction of an antiviral state, and, as a result, many viruses have evolved mechanisms to suppress IFN production and signaling. IAVs not only induce IFN-α/β via multiple PRRs but also encode a multifunctional protein, NS1, which directly antagonizes IFN-α/β production and signaling. IAV NS1 may therefore be a potential target for the development of both novel LAIV and direct-acting antivirals.
Footnotes
Author Disclosure Statement
No competing financial interests exist.