
Abstract
Comprehension of adipocyte function has evolved beyond a long-held belief of their inert nature, as simple energy storing and releasing cells. Adipocytes, including white, brown, and beige, are capable mediators of global metabolic health, but their intersection with inflammation is a budding field of exploration. Evidence hints at a reciprocal relationship adipocytes share with immune cells. Adipocyte's capacity to behave in an “immune-like” manner and ability to sense inflammatory cues that subsequently alter core adipocyte function might play an important role in shaping immune responses. Clarifying this intricate relationship could uncover previously underappreciated contribution of adipocytes to inflammation-driven human health and disease. In this review, we highlight the potential of largely underappreciated adipocyte “immune-like” function and how it may contribute to inflammation, immunity, and pathology of various diseases.
Introduction
Dogma has long suggested that adipose tissue (AT), mainly described to consist of white adipose tissue (WAT) and brown adipose tissue (BAT), was a simplistic organ. The common understanding is that WAT plays a central role in energy storage, whereas BAT is tied to energy expenditure through adaptive nonshivering thermogenesis. Although research focused on AT biology and function had stayed relatively dormant, the obesity pandemic has reignited research interests into AT biology and its contribution to disease pathogenesis. In fact, the recently uncovered appreciation of intricate biological processes that govern AT development, function, and contribution to health and disease makes the AT enigmatic function a highly attractive research area.
AT houses a highly rich milieu of cells, including adipocytes, preadipocytes (progenitor stem cells within the stromal vascular fraction), fibroblasts, endothelial cells, and immune cells, which collectively play an important role in the metabolic regulation/whole-body energy homeostasis (Sarantopoulos and others 2018; Ghaben and Scherer 2019). Sensing of environmental homeostatic (eg, energy flux) or disease (eg, obesity, infection, and cancer) cues by AT alter its composition to adapt and contribute, either in beneficial or detrimental means (Desruisseaux and others 2007; Park and others 2014; Wernstedt Asterholm and others 2014; Grant and Dixit 2015; Kajimura and others 2015; Deng and others 2017; Ghaben and Scherer 2019). In fact, recent discoveries revealing that AT is a highly plastic organ with dynamic immune-like characteristics (Ouchi and others 2011; Enerback 2013; Grant and Dixit 2015; Villarroya and others 2018) further support the call for a better functional understanding of AT constituents in homeostasis and disease pathogenesis.
The dogma surrounding AT inflammatory capacity suggests that immune cells infiltrating AT are the primary source of inflammation. Multiple published reviews have previously focused on the contribution of WAT infiltrating immune cells (eg, macrophages, dendritic cells [DC], T cells, B cells, and natural killer [NK] cells) (Ghigliotti and others 2014; Huh and others 2014) and hence will not be discussed in this review. Although underdefined and an active area of investigation, compared with WAT, the immune cell milieu is also present in reduced numbers in BAT and beige AT (eg, macrophages and T and B cells) (Herrero and others 2010; Fitzgibbons and others 2011; Villarroya and others 2018). Traditional approaches to study AT inflammation have almost entirely focused on examination of WAT as a whole or specific WAT infiltrating immune cells. Importantly, however, the intrinsic contribution of adipocytes and/or their potential role in the establishment and modulation of the overall AT inflammatory capacity is underappreciated and has not been well discussed. Hence, this review will summarize the recently established landscape of adipocyte contribution to AT inflammatory capacity (eg, cytokine/chemokine production and immune cell crosstalk) and will invoke critical future directions that should be investigated to broaden our understanding of this topic.
Adipocytes
A fundamental feature of adipocytes, which form the core of AT, is nutrient handling, energy storage and utilization, and secretion of adipokines (eg, leptin, adiponectin, and resistin) that can modify total body metabolic homeostasis (Ouchi and others 2011). Adipocytes are thus appreciated to be highly complex biosynthetic factories. Currently, adipocytes can be divided into multiple subtypes: (a) white adipocytes (energy storing), (b) brown adipocytes (energy expending), (c) beige adipocytes (also called brite adipocyte; inducible, energy expending), and (d) pink adipocytes (lipid-rich, pregnancy/lactation associated) (Fig. 1). Remarkable adipocyte plasticity, which remains an exciting area of investigation, has been extensively discussed elsewhere (Enerback 2013; Sanchez-Gurmaches and others 2016), hence will not be reviewed here.
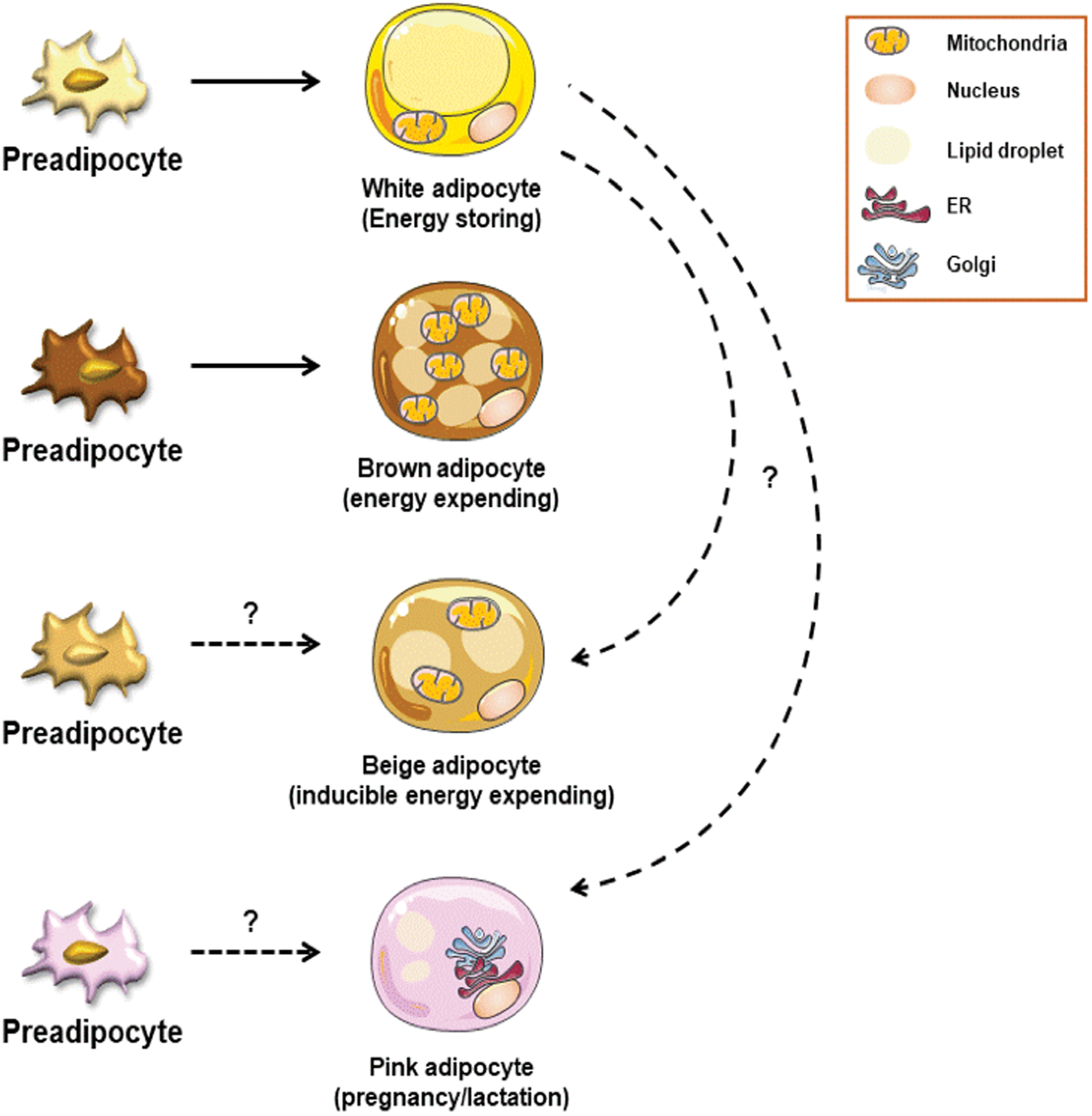
Adipocyte subsets. Adipocytes are divided into various subtypes: white adipocytes (energy storing), brown adipocytes (thermogenesis), beige/brite adipocytes (inducible energy expending), and pink adipocytes (pregnancy/lactation associated). White adipocytes are characterized by large lipid droplets and are known for their energy storing capacity during obesity. Brown adipocytes are characterized by numerous mitochondria. They originate from a separate preadipocyte population. Mitochondrial number and function within brown adipocytes support their thermogenic capacity and maintenance of body temperature. Beige adipocytes arise within WAT and are characterized by unique thermogenic capacity. Induction of beige adipocytes is driven by environmental cues (eg, cold stress, exercise, and β3-adrenergic stimulation). The developmental origins of beige adipocytes are still under investigation. Pink adipocytes are proposed to transdifferentiate from cells within subcutaneous AT. The precise function and developmental origins of pink adipocytes remains underdefined. WAT, white adipose tissue. Color images are available online.
White adipocytes regulate the assimilation of excess energy (eg, circulating dietary lipids) for storage and in part through de novo lipogenesis, resulting in increased lipid droplet size and expansion of the AT (Tan and Vidal-Puig 2008). In contrast, when shortage of nutrients occurs, adipocytes can shrink in size by hydrolyzing stored triglycerides into fatty acids (FA) and glycerol through lipolysis (Lafontan and Langin 2009) to provide other organs/cells of sufficient energy (Frayn 2002).
Brown adipocytes consist of numerous mitochondria and are highly active metabolic cells that have the capacity for adaptive nonshivering thermogenesis, the process of using energy to produce heat necessary for temperature regulation (Ikeda and others 2018). Beige adipocytes constitute inducible thermogenic cells that arise within WAT in response to environmental cues (eg, cold stress and exercise) (Petrovic and others 2010; Ikeda and others 2018). Beige adipocytes are derived from distinct progenitors and potentially from transdifferentiation of white adipocytes. However, technical limitations, including lack of a specific marker for differentiated white adipocytes, prohibit precise clarity of transdifferentiation between white and beige adipocytes and further investigation is needed (Ikeda and others 2018).
The recently discovered pink adipocytes appear during pregnancy and are intricately involved in postpregnancy lactation. Arising from subcutaneous white adipocytes within the mammary glands, pink adipocytes are a lipid rich energy source for the production of milk (Cinti 2018). Given their recent discovery, our understanding of the intersection between inflammation and pink adipocytes is limited (Cinti 2018). Although an exciting and novel avenue of investigation, pink adipocytes will not be further discussed within this review.
The ability to generate and manipulate primary adipocytes, from preadipocytes (Ruiz-Ojeda and others 2016; Sarantopoulos and others 2018; Ghaben and Scherer 2019), has enhanced our understanding of the multifactorial impact adipocytes themselves may play in the establishment and modulation of the overall AT immune capacity. Ex vivo derived adipocyte differentiation, from preadipocytes, is a highly complex process wherein activation of peroxisome proliferator-activated receptor gamma (PPARγ), CCAAT/enhancer-binding proteins (C/EBP) and sterol regulatory element binding protein (SREBP1) drive an adipocyte fate (Ruiz-Ojeda and others 2016; Sarantopoulos and others 2018). Despite the ability to generate ex vivo adipocytes from preadipocytes, their direct comparison with AT isolated mature adipocytes remains an area of intense investigation (Ruiz-Ojeda and others 2016).
Representing the specific focus of this review, functional potential for adipocytes to behave like immune cells has recently been invoked. These provocative studies suggest that the breadth of the adipocyte “immune-like” capacity ranges from their ability to express innate immune receptors (Lin and others 2000; Pietsch and others 2006; Batra and others 2007; Kopp and others 2009; Ballak and others 2015), produce proinflammatory cytokines (Sewter and others 1999; Pietsch and others 2006; Alexaki and others 2009; Kopp and others 2009; Muller and others 2014), express chemokines (Frasca and others 2018), and present antigens (Poggi and others 2009; Deng and others 2013, 2017; Guo and others 2013; Huh and others 2013; Xiao and others 2016) (Fig. 2). Such findings highlight a newfound intricacy and potential for adipocytes to robustly modulate AT and systemic inflammation that in turn impact global immune responsiveness under homeostatic and disease state conditions.
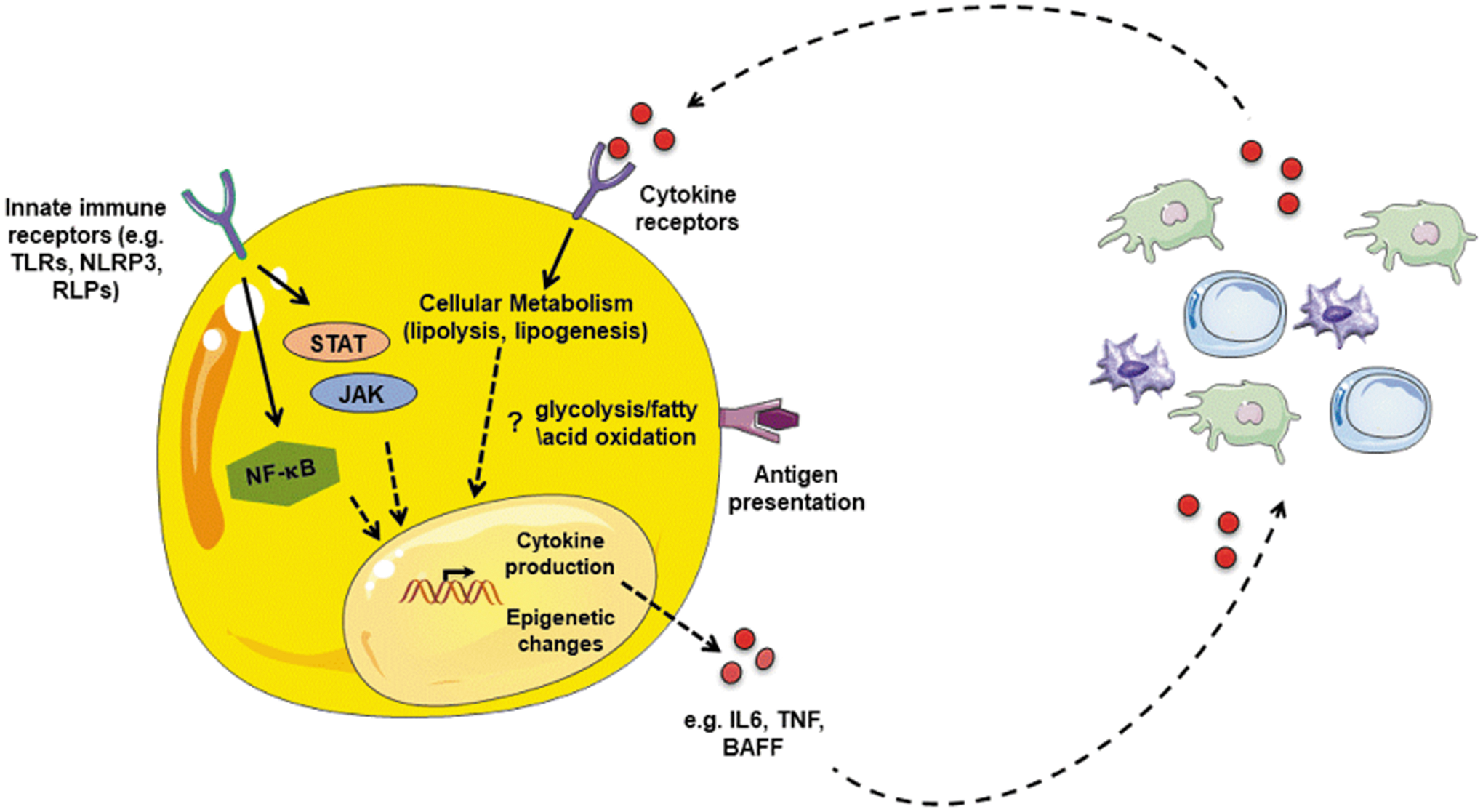
Adipocyte-centric inflammation and crosstalk with immune cells. Adipocytes possess an “immune-like” potential, including (a) expression of innate immune receptors (eg, TLRs, NLRs, and RLRs), (b) cytokine (eg, TNF, IL1β, IL-6, BAFF, and APRIL) and chemokine (CCL2, CXCL8, CXCL1, CXCL10) production, and (c) antigen presentation (eg, MHC-II, CD80, CD40, CD1d, and CIITA). Adipocyte sensing of cytokines alter their function (eg, lipolysis, lipogenesis, and UCP-1 thermogenesis). Cumulatively, these adipocyte inflammatory features suggest potential crosstalk with immune cells (in AT or circulation) that may directly impact adipocyte function, immune cell function, and/or disease pathogenesis. TLR, toll-like receptor; NLR, NOD-like receptor; RLR, RIG-I receptor; BAFF, B cell activating factor; APRIL, a proliferation-inducing ligand; MHC, major histocompatibility complex. Color images are available online.
Adipocyte-Intrinsic “Immune-Like” Potential
Investigations, primarily utilizing mature or preadipocyte-derived primary adipocytes, have uncovered that adipocytes express various innate immune receptors, including toll-like receptors (TLRs), NOD-like receptors (NLRs), and RIG-I-like receptors (RLRs). Screening of TLRs within mouse or human white adipocytes has revealed their expansive presence within these cells. As with immune cells (O'Neill and others 2013), human white adipocytes express all 10 known TLRs with TLR3, TLR4, TLR5, and TLR9 being the most robustly expressed (Ballak and others 2015). In contrast, of 12 known TLRs (O'Neill and others 2013), mouse white adipocytes express TLR1, TLR2, TLR3, TLR4, TLR5, TLR6, TLR7, and TLR9 (Lin and others 2000; Pietsch and others 2006; Batra and others 2007). Furthermore, expression of TLR2 and TLR4 in mouse brown adipocytes has been similarly detected (Bae and others 2014).
In addition, of the 34 NLRs in mice (Kanneganti 2010), white adipocytes are known to express NOD-1, NOD-2, and NLRP3 (Vandanmagsar and others 2011; Zhou and others 2012, 2015), whereas only NOD-1 is detected in brown adipocytes (Bae and others 2014). NOD-1, but not NOD-2, suppresses white adipocyte differentiation and induces the NF-κB pathway to activate innate responses (Zhou and others 2012, 2015). Despite detectable NLRP3 expression in white adipocytes, a critical component in the inflammasome that leads to caspase-1 activation (Vandanmagsar and others 2011), NLRP3 expression is low compared to macrophages thereby raising questions of its role and contribution in adipocytes. The finite knowledge surrounding adipocyte NLR expression and contribution obviously warrants future studies formally interrogating this axis. White adipocytes also express RIG-I (Yu and others 2014), a sensor of double-stranded RNA and viruses, but their functionality remains inconclusive and formal examination on adipocyte “immune-like” function and relevance to viral infections is needed. In addition, given their relevance to shaping immune responses, studies focused on the expression and role of C-type lectin receptors (CLR) and AIM2-like receptors (ALR) in adipocyte (white, brown, and beige) “immune-like potential” are warranted.
To date, expression and contribution of TLRs to adipocyte inflammatory potential has been best studied. Adipocyte sensing of well-established TLR-recognized pathogen-associated molecular patterns (PAMPs) (eg, LPS, zymosan, poly I:C, flagellin, loxoribine, and CpG) by both mature and preadipocytes-derived white adipocytes results in production of multiple proinflammatory mediators (Sewter and others 1999; Pietsch and others 2006; Kopp and others 2009). As obesity is associated with increased systemic endotoxemia (Cani and others 2007), circulating DNA (Nishimoto and others 2016), and likely other PAMPs/danger-associated molecular patterns (DAMPs), these findings suggest that adipocyte-intrinsic innate immune receptor activation may also contribute to the overall tissue and systemic inflammatory environment in obesity or other diseases associated with similar disturbances (eg, inflammatory bowel disease, autoimmunity, and sepsis). Brown adipocytes can likewise produce inflammatory cytokines, through activation of their thermogenic programming; however, this area is nascent and existing literature remains limited (Mauer and others 2014).
In addition to classically quantified inflammatory mediators (eg, TNF and IL-6) adipocytes, like macrophages and DCs, express other inflammatory mediators that are not traditionally reported outside of the field of immunology, including B cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL) (Alexaki and others 2009; Muller and others 2014). Both BAFF and APRIL modify B cell maturation and survival (Schneider and others 1999; Mackay and others 2003) and have been correlated with promoting inflammation in multiple autoimmune diseases (Mackay and others 2003). B cells are well established to infiltrate WAT in the context of obesity (Winer and others 2011) and to modulate obesity pathogenesis (DeFuria and others 2013). Thus, whether adipocytes, through BAFF and APRIL production, can modify B cell status within the WAT and impact disease pathogenesis remains unknown. Adipocytes also express all 3 known receptors of BAFF and APRIL, including BAFF receptor (BAFF-R), transmembrane activator and CAML interactor (TACI), and B cell maturation antigen (BCMA) (Alexaki and others 2009). Whether BAFF and APRIL production by AT infiltrating macrophages or DCs and subsequent sensing by adipocytes can alter adipocyte function and/or the crosstalk among myeloid cells, adipocytes, and adaptive immune cells in AT is an intriguing inquiry.
The importance of adipocyte and immune cell crosstalk likely extends beyond cytokines, as white adipocytes produce multiple chemokines (eg, CCL2, CCL5, CXCL8, and CXCL10) (Kopp and others 2009; Frasca and others 2018) and brown adipocytes can produce CXCL14 (Cereijo and others 2018). Whether this crosstalk is more expansive and includes adipocyte production of other well-established immune cell recruiting chemokines (eg, CCL3, CXCL1, and CXCL9) or immune cell skewing cytokines (eg, TGF-β, IFN-γ, IL-1β, IL-4, IL-10, and IL-23) is understudied. The impact of such knowledge is robust as it might shed light on how adipocytes affect immune cell polarization (eg, M1/M2 macrophages, Th1, Th2, Th17, regulatory T cells [Tregs], and B-1 and B-2 cells) and subsequent disease pathogenesis. Conversely, whether adipocytes can sense pro- and anti-inflammatory mediators and subsequently are themselves skewed/polarized toward a “pro- or anti-inflammatory” phenotype is unknown.
The underpinning mechanisms underlying the capacity to regulate and uncover adipocyte-intrinsic inflammatory potential are underdefined. Epigenetic modifications alter cytokine production in immune cells (Phan and others 2017). Adipocyte epigenome is an established contributor to their thermogenic programming (Peng and others 2018). Hence, the epigenome may similarly impact adipocyte inflammatory programming. Furthermore, adipocytes are highly metabolic cells and cellular metabolic programming in immune cells is intricately tied to their inflammatory vigor (O'Neill and others 2016; Pearce and Pearce 2018). As proinflammatory cytokines (eg, TNF, IL-6, Type I IFNs, and IL-2) are known to modulate core immune cell metabolism, if they similarly regulate adipocyte metabolic capacity remains underdefined. Conversely, if alteration of adipocyte metabolic capacity is tied to adipocyte “immune-like” potential is unknown.
Adipocyte expression of innate immune receptors and production of proinflammatory mediators insinuate adipocytes converse with adaptive immune cells. Recent studies have shed light into direct crosstalk between adipocytes and adaptive immunity. Initial microarray analyses of primary adipocytes uncovered robust expression of major histocompatibility complex II (MHCII) and genes involved with antigen processing and presentation (eg, CIITA, CD80, CD86, and CD40) (Deng and others 2013; Zhong and others 2014; Xiao and others 2016). Closer examination revealed that antigen-specific presenting adipocytes activated CD4+ T cells, in a direct manner, demonstrating the functionality of this antigen processing machinery. This capacity to present antigen and activate CD4+ T cells was largely attributed to large adipocytes (Xiao and others 2016). As adipocyte size is a large determinant of metabolic health (McLaughlin and others 2016; Tandon and others 2018), with larger adipocytes typically an indicator of metabolic derangements, these findings suggest that adipocytes may be an overlooked contributor to MHC-driven processes in metabolic diseases.
Recent examination of adipocyte-specific deletion of MHCII revealed dampened obesity-associated AT-driven IFN-γ production and an enhanced abundance of Tregs within AT (Deng and others 2017). Although lack of adipocyte-specific MHCII preserved insulin sensitivity and glucose metabolism in the context of obesity, in contrast, CD80/86 and CD40 are proposed to play beneficial roles in maintenance of insulin resistance (Guo and others 2013; Zhong and others 2014). Hence studies to formally parse apart the interplay between MHC, costimulation, and their underpinning mechanisms, in adipocytes are needed. In addition, broader understanding of whether adipocytes express MHC class I and can activate CD8+ T cells is unknown and should be studied. Adipocytes also highly express CD1d and directly present lipid antigens to activate invariant NKT (iNKT) cells (Huh and others 2013). AT iNKT numbers and activity have been reported to decline in obesity (Schipper and others 2012). iNKT-deficient mice exhibit insulin resistance and adipocyte hypertrophy, suggesting an interplay between adipocytes and iNKT cells (Huh and others 2013). Future investigations focused on the interplay between adipocytes and iNKT cells in disease pathogenesis are warranted.
Collectively, published findings suggest that adipocytes may contribute to the crosstalk with immune cells in a manner that is potentially either beneficial or detrimental to disease pathogenesis—the outcome being dependent on disease setting. Thus, it is conceivable that adipocyte function and inflammatory capacity may modulate pathogenesis of metabolic, cancer, autoimmune, and infectious diseases. Published evidence of how adipocytes may impact various diseases is discussed below.
Inflammation and Alteration of Adipocyte Function
Core observations that patients with increased circulating proinflammatory cytokines (eg, TNF and IL-6) display augmented energy expenditure and mobilization of lipids suggest a connection between inflammation and AT function (Spitzer and others 1988; Kamimura and others 2007). Additional characterizations have demonstrated that inflammatory cytokines TNF (Feingold and others 1992; Gasic and others 1999; Zhang and others 2002; Green and others 2004; Laurencikiene and others 2007), IL-1β (Price and others 1986; Feingold and others 1992), IL-6 (Ji and others 2011; Zhang and others 2014), IL-4 (Tsao and others 2014), IL-17a (Shin and others 2009), IFNα (Feingold and others 1992), and IFN-γ (Feingold and others 1992) are capable of inducing lipolysis in 3T3-L1 cells, a traditionally utilized adipocyte-like cell line (Ruiz-Ojeda and others 2016). Although adipocyte-specific deletion of a rate limiting enzyme of lipolysis, adipose triglyceride lipase, impacted obesity-driven hepatic immune cell infiltration and activation, it was insufficient to dampen AT inflammation (Schoiswohl and others 2015). In contrast, deletion of a separate rate limiting enzyme of lipolysis, hormone sensitive lipase, resulted in enhanced AT inflammation (Xia and others 2017). Hence more extensive investigations are necessary to define the relevance of cytokine-mediated lipolysis in vivo toward systemic inflammation and disease pathology. In addition, the source of the cytokines (adipocytes and/or immune cells) and whether these proinflammatory cytokines drive lipolysis in all types of primary adipocytes (eg, white, brown, beige, and pink) or if they regulate contrasting functions is unknown.
Adipocyte sensing of cytokines modifies their energy storage machinery. TNF inhibits FA uptake, through downregulation of FA transport protein and fatty acid translocase and lipogenesis in vitro (Memon and others 1998; Ruan and others 2003). Although these collective findings suggest proinflammatory cytokines may drive detrimental effects in WAT, recent evidence has emerged to suggest that inflammation may also play vital beneficial roles in adipocyte health. Specifically, lack of AT TNF sensing in vivo prohibits accumulation of lipids into WAT, leading to exacerbated metabolic dysfunction and ectopic lipid accumulation (Wernstedt Asterholm and others 2014). Mirroring these findings, lack of adipocyte-specific TLR4 sensing dysregulates WAT expansion and exacerbates metabolic disease (Tao and others 2017). Despite this, the contribution of other TLRs and inflammatory mediators (eg, IL-6, IL-17, IL-1β, BAFF, and APRIL) in adipocytes to obesity-associated inflammation remains underdefined with further investigations required to mechanistically comprehend adipocyte-specific contributions to WAT remodeling.
Well-studied signaling pathways downstream of PAMP and cytokine recognition in immune cells, including NF-κB (Berg and others 2004; Weidemann and others 2016) and Jak/STATs (eg, Jak1/2/3, Tyk2, and STAT1/3/5A/5B) (Xu and others 2013), appear to play a similar role in adipocytes. Ability of these pathways to modulate adipocyte lipolysis and adipogenesis has been previously reviewed (Xu and others 2013). In addition, NF-κB and Jak2 may also play a role in modulation of UCP-1-dependent thermogenesis (Chiang and others 2009; Mowers and others 2013; Shi and others 2016). However, tissue-specific genetic investigations are necessary to provide greater clarity on its impact on brown adipocytes.
One potential unifying immune mediator is the NF-κB-regulated transcription factor interferon regulatory factor 4 (IRF4), an established modulator of lymphocyte development and function (Hagman 2017) that can beneficially modify both white adipocyte lipolysis and brown adipocyte adaptive thermogenesis (Eguchi and others 2011). Jak/STAT are critical mediators of interferon α/β receptor (IFNAR) signaling, a ubiquitously expressed receptor that regulates both innate and adaptive inflammatory potential. Interferon (IFN) β impacts thermogenic capacity of brown adipocytes (Kissig and others 2017). Whether type I IFN is sensed by white adipocytes and if this uncovers their inflammatory potential, akin to immune cell counterparts (Thomas and others 2006; Cappelletti and others 2017a, 2017b), is unknown and should be formally examined. Given that adipocytes have an “immune-like” capacity and express similar signaling pathways to immune cells, it is plausible that the regulatory mechanisms (eg, epigenetic, transcriptional, and metabolic) likewise overlap between these cells.
The existence of beige adipocytes was hinted at >3 decades ago, wherein brown adipocytes were observed in WAT (Young and others 1984), something that was enhanced under cold stress (Loncar and others 1986). Although mechanisms regulating development of the beige adipocyte lineage and modulation of their function is still heavily investigated, links have emerged suggesting immune mediators and inflammation may modulate beige adipocyte function. Sympathetic stimulation, through catecholamines, drives beige adipogenesis (Kajimura and others 2015). A suggestion that sensing of IL-4 by AT alternatively activated macrophages upregulates tyrosine hydroxylase (TH; rate limiting enzyme of norepinephrine [NE] synthesis) and contributes to a local NE production and subsequent adipocyte beiging was proposed (Qiu and others 2014; Lee and others 2015) and elicited significant interest in the field. However, a detailed follow-up study revealed that bone-marrow-derived macrophages do not produce NE in response to IL-4 nor do AT resident macrophages express TH (Fischer and others 2017). Furthermore, neither hematopoietic-specific TH deletion nor chronic exposure to IL-4 in WT or IL-4ra−/− mice was sufficient to modify energy expenditure (Fischer and others 2017). These latter findings clearly suggest existence of alternate mechanisms linking IL-4-driven macrophage contribution to beiging—one plausibility being the capacity for macrophages to capture and release catecholamines in an IL-4-regulated manner.
In contrast to IL-4, autocrine and paracrine TGFβ signaling has been demonstrated to constitutively suppress beige adipogenesis (Babaei and others 2018). Activation of Jak/STAT3 pathway, through IL-6 and IL-11, inhibits TGFβ suppression of beiging (Babaei and others 2018). It is posited that sympathetic stimulation augments adipocyte lipolysis, which drives a transient elevation in IL-6/IL-11 to promote beiging of adipocyte progenitors in a paracrine manner (Babaei and others 2018). However, whether the source of these cytokines is from adipocytes or resident AT immune cells remains undefined. Furthermore, in states of acute (eg, sepsis/infection) or chronic inflammation (eg, obesity and autoimmunity), whether this pathway of beiging becomes tolerized or altered is not well understood.
Although cytokines and inflammation may have indirect means to drive adipocyte beiging/browning, recent evidence points to the possibility of immune cells directly regulating adipocyte beiging. Mature adipocytes isolated directly from AT express vascular adhesion protein 1 (VCAM-1) (Chung and others 2017). Expression of VCAM-1, which is enhanced in obesity, facilitates direct interaction with α4-integrin on macrophages driving a reciprocal modulation of macrophage activation and downregulation of UCP-1 expression in adipocytes (Chung and others 2017). Lack of α4-integrin promoted beiging and improved obesity-associated metabolic derangements (Chung and others 2017) suggest that this direct interaction between macrophages and adipocytes facilitates a key component to beiging. However, whether UCP-1-independent thermogenesis is likewise modulated by this interaction remains unknown. Combined existing literature suggests that adipocyte (white, brown, and beige) sensing of inflammatory mediators and interaction with immune cells can modify adipocyte function. If adipocyte to adipocyte communication, through inflammatory mediators or released lipids, can also alter adipocyte “immune-like” potential and function is poorly understood and should be investigated.
Adipocyte-Inflammation Axis in Disease Pathogenesis
Among various diseases, AT contribution to obesity pathogenesis and obesity-associated inflammation has naturally been the most well examined (Ghigliotti and others 2014; Grant and Dixit 2015; Tandon and others 2018). Obesity-associated chronic low-grade inflammation is central to virtually all downstream derangements, including type 2 diabetes, atherosclerotic cardiovascular disease (CVD), nonalcoholic fatty liver disease (NAFLD), Alzheimer's disease, diverse cancers, and increased complications from infectious sequelae (Haslam and James 2005; Falagas and Kompoti 2006). Obesity-driven enhancement of inflammatory mediators, including TNF, IL-6, and IL-17, are well-defined critical mediators of obesity-associated NAFLD (Braunersreuther and others 2012; Harley and others 2014; Giles and others 2016a, 2017; Mukherjee and others 2018) and CVD (Mehra and others 2005; Giles and others 2016b; Robert and Miossec 2017) pathogenesis (Fig. 3).
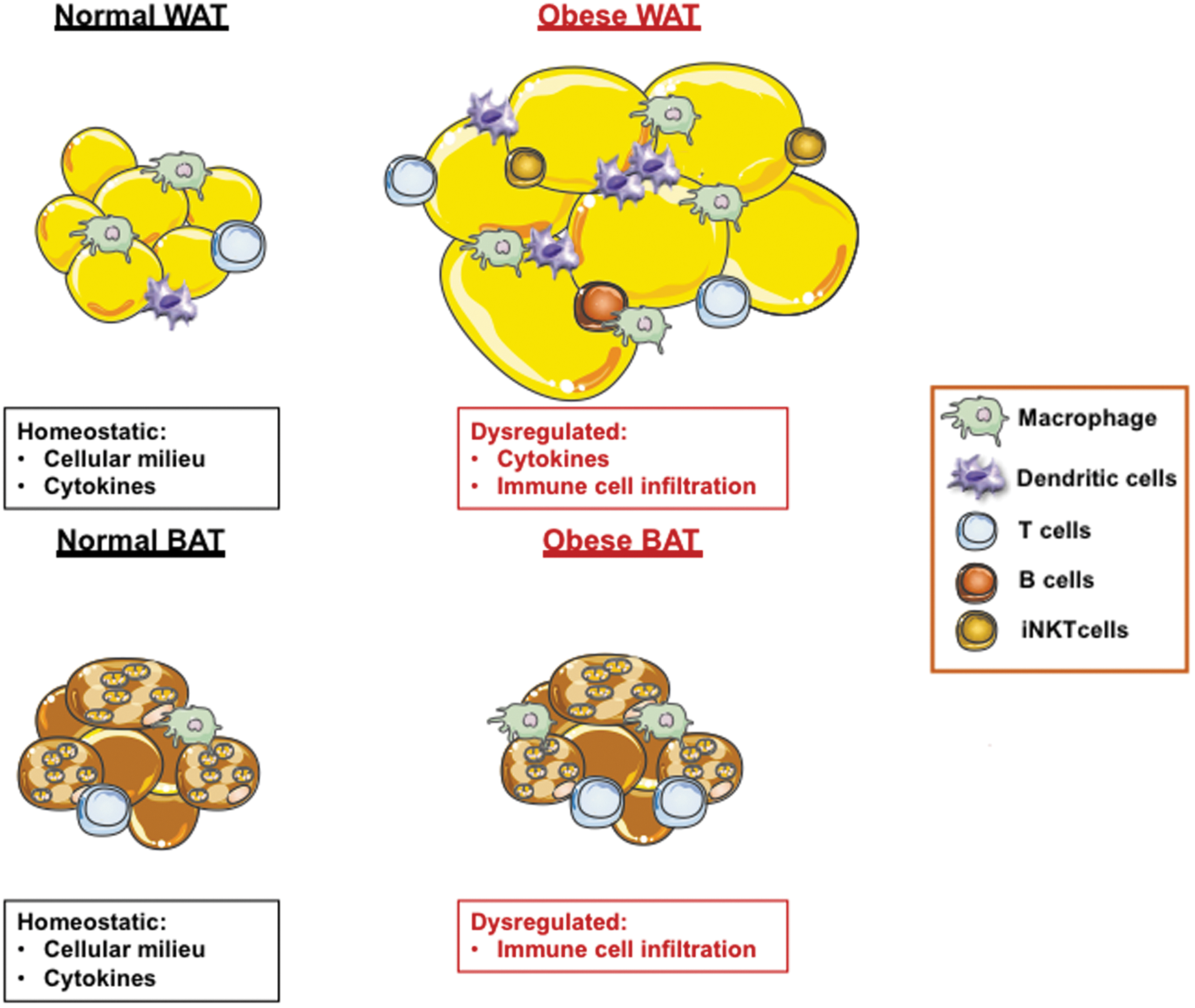
Obesity-driven AT inflammatory plasticity. White and brown AT in a lean state consists of a diverse cellular milieu (eg, adipocytes, macrophages, T cells, and B cells) and capacity to produce cytokines. Pathological expansion of AT in obesity promotes dysregulation of cytokine production (e.g., TNF, IL-6, IL-1, and IFN-γ), enhances immune cell infiltration/polarization/activation and exacerbates the AT inflammatory milieu. Although contribution of AT to obesity-associated inflammation is well investigated, adipocyte-specific contribution to inflammation is understudied and remains a critical gap in knowledge. Color images are available online.
Despite the intuitive sense that adipocytes may contribute to obesity-associated inflammation and downstream derangements, given their “immune-like” potential and sensing of inflammatory mediators to modify functional capacity, whether and how obesity exacerbates adipocyte inflammatory capacity, and whether adipocyte-intrinsic production of these proinflammatory mediators drives disease pathogenesis in obesity is not well understood. One hint interlinking obesity with adipocyte inflammation stems from evidence that lack of adipocyte-specific TLR4 sensing, in the context of obesogenic-diet feeding, exhibits both acute (eg, protection from insulin resistance) and chronic-term effects (eg, lack of AT remodeling, ectopic lipid deposition, and exacerbation metabolic disease). Adipocyte-specific TLR4 deletion led to decreased TLR4 expression in other tissues, including the liver and peritoneal macrophages (Tao and others 2017). These findings reaffirm that adipocytes are critical and complex players in global metabolic health. Thus, additional mechanistic interrogations focused on the contribution of adipocyte-specific inflammation to endocrine regulation of systemic tissues are warranted.
Although the mechanisms that uncover adipocyte inflammatory capacity remain unknown, undifferentiated 3T3-L1 cells can produce type I IFN (McCall and others 2010), a regulator of innate and adaptive immune responses. Whether primary adipocytes can produce type I IFNs and if sensing of type I IFNs can modulate adipocyte inflammatory potential should be elucidated and further investigated. Robust literature underscores the contribution of the type I IFN/IFNAR axis as a promoter of metabolic dysfunction (eg, regulation of intrahepatic CD8+ T cell pathogenicity (Ghazarian and others 2017), interferon regulatory factors (Wang and others 2013, 2014; Kumari and others 2016), suppression of thermogenesis (Kissig and others 2017), and exacerbation of insulin resistance and hepatic dysfunction (Koivisto and others 1989; Tremlett and others 2004; Franceschini and others 2011; Bhattacharya and others 2015)). In contrast, a recent study demonstrated that adipocyte deletion of IFNAR, through use of fatty acid binding protein 4 (FABP4)-driven deletion, augmented obesogenic-diet-induced metabolic disease (Wieser and others 2018). However, FABP4 expression is not restricted to adipocytes (gene expression in macrophages, endothelial cells, osteogenic cells, ganglion, adrenal medulla, and liver could be affected (Martens and others 2010; Lee and others 2013). Thus, conclusions driving the specific contribution of adipocyte IFNAR signaling to metabolic disease require additional interrogations—something that could be addressed using a more specific adipocyte-targeted in vivo deletion using Adipoqcre mice (Lee and others 2013).
Studies of AT/adipocyte-specific contribution in other pathologies including cancer, autoimmunity, and infections remain limited. Obesity is a major risk factor for the development and pathogenesis of 13 different forms of cancer, including breast, endometrial, liver, pancreatic, colorectal, and ovarian (Park and others 2014). Increased systemic FAs and inflammatory cytokines are posited mechanisms fueling cancer pathogenesis. AT milieu, including adipocyte paracrine and endocrine effects (Park and others 2011), has been linked to tumor pathogenesis (Coelho and others 2016). Adipocyte release of FA, through lipolysis, may represent a potential fuel source to tumorigenic cells. In turn, FA can enhance β-oxidation, a modulator of immune cell inflammatory vigor (Moreno-Fernandez and others 2018), within oncogenic cells and promote their migration and invasiveness (Balaban and others 2015; Yang and others 2018). Thus, whether cytokine-driven adipocyte lipolysis plays a role in cancer pathogenesis should be examined. Furthermore, if adipocytes-intrinsic production of proinflammatory cytokines (eg, TNF and IL-6) and/or crosstalk with immune cells (eg, Tregs) directly impact proliferation, survival, invasiveness, and metastasis of tumors should be examined.
Like obesity, there is a sharp rise in development of autoimmune diseases in developed countries. Autoimmune diseases (eg, psoriasis and rheumatoid arthritis) are associated with AT inflammation and increased immune cell influx into the AT (Guzik and others 2017; Hjuler and others 2017). B cells within human AT can produce autoimmune antibodies (Frasca and others 2018). These collective events in autoimmunity are energy exhaustive processes. Cytokines that are augmented in autoimmunity can likewise facilitate adipocyte lipid handling (eg, TNF) and potentially provide a source of fuel to the autoimmune inflammatory flame. Given that adipocytes can produce inflammatory mediators known to modulate pathology of autoimmune diseases (eg. TNF, IL-6, BAFF, APRIL) the contribution of adipocyte-specific inflammation to autoimmunity should be defined. Hence, the direct or indirect impact of adipocyte-specific contribution to T- and B cell-driven autoimmunity should be examined.
Accumulation of AT, through obesity, is linked with greater complications, morbidity, and mortality to infections (Falagas and Kompoti 2006). Notably, these studies have exclusively examined WAT/white adipocytes. For instance, a close relationship between AT/adipocytes and HIV has been proposed. HIV-infected individuals exhibit T cell infiltration into AT, lipodystrophy, and metabolic dysfunction (Desruisseaux and others 2007). Coculturing of HIV-infected T cells with adipocytes enhanced detection of HIV replication subunits (Couturier and others 2015). Adipocytes express the necessary receptors for HIV entry (eg, CD4, CCR5, and CXCR4) (Hazan and others 2002) and thus it is plausible that adipocytes are an important long-term reservoir of HIV that mediates relapse of infection (Couturier and others 2015). Whether HIV itself can trigger changes in adipocyte-intrinsic function needs to be further elucidated.
In addition to HIV, various bacterial (eg, Mycobacterium tuberculosis [Mtb]), viral (eg, cytomegalovirus, influenza A, respiratory syncytial virus, and adenovirus), and parasitic (eg, Trypanosoma cruzi) pathogens can infect adipocytes (Desruisseaux and others 2007; Bouwman and others 2008; Beigier-Bompadre and others 2017). Transfer of WAT from Mtb-infected mice to an uninfected host contributed to Mtb dissemination (Beigier-Bompadre and others 2017). One intriguing conjecture, given that adipocytes are a rich energy source, is that evolutionarily it would have been energetically advantageous for pathogens to develop the ability to reside within adipocytes. Whether pathogens can likewise infect other subsets of adipocytes (brown, beige, and pink) and can trigger an adipocyte's “immune-like” potential is unknown and should be explored. Furthermore, if adipocyte-specific inflammation contributes beneficially (eg, immune cell polarization and pathogen clearance) or detrimentally (eg, bystander host damage and dampened immune cell function) in the context of infections should be formally defined. Lastly, as an extension to apparent correlations, examination of adipocyte contribution to other obesity-impacted health complications (eg, neurological diseases, aging, and reproductive fitness) is lacking and would be of significant future interest.
Conclusions
Overall, this review highlights the necessity to consider and evaluate the contribution of adipocyte-specific inflammation within the enclave of AT inflammatory capacity. The reciprocal nature between adipocytes and inflammation is grossly underappreciated. The expansive knowledge gain of mechanisms regulating immune cell inflammation could be harnessed and in turn explored in adipocytes. In addition to greater definition of crosstalk between adipocytes and immune cells, whether adipocyte to adipocyte communication occurs (eg, white adipocyte induction of brown/beige adipocyte thermogenesis) is unknown and would be an exciting area to explore. Definition of these mechanisms regulating adipocyte-intrinsic inflammatory capabilities and contribution to disease pathogenesis (Fig. 4) could uncover previously unappreciated therapeutic avenues. Notably, existing therapeutics targeting inflammatory pathways in immune cells could be readily repurposed and utilized to target adipocytes and adipocyte inflammation.
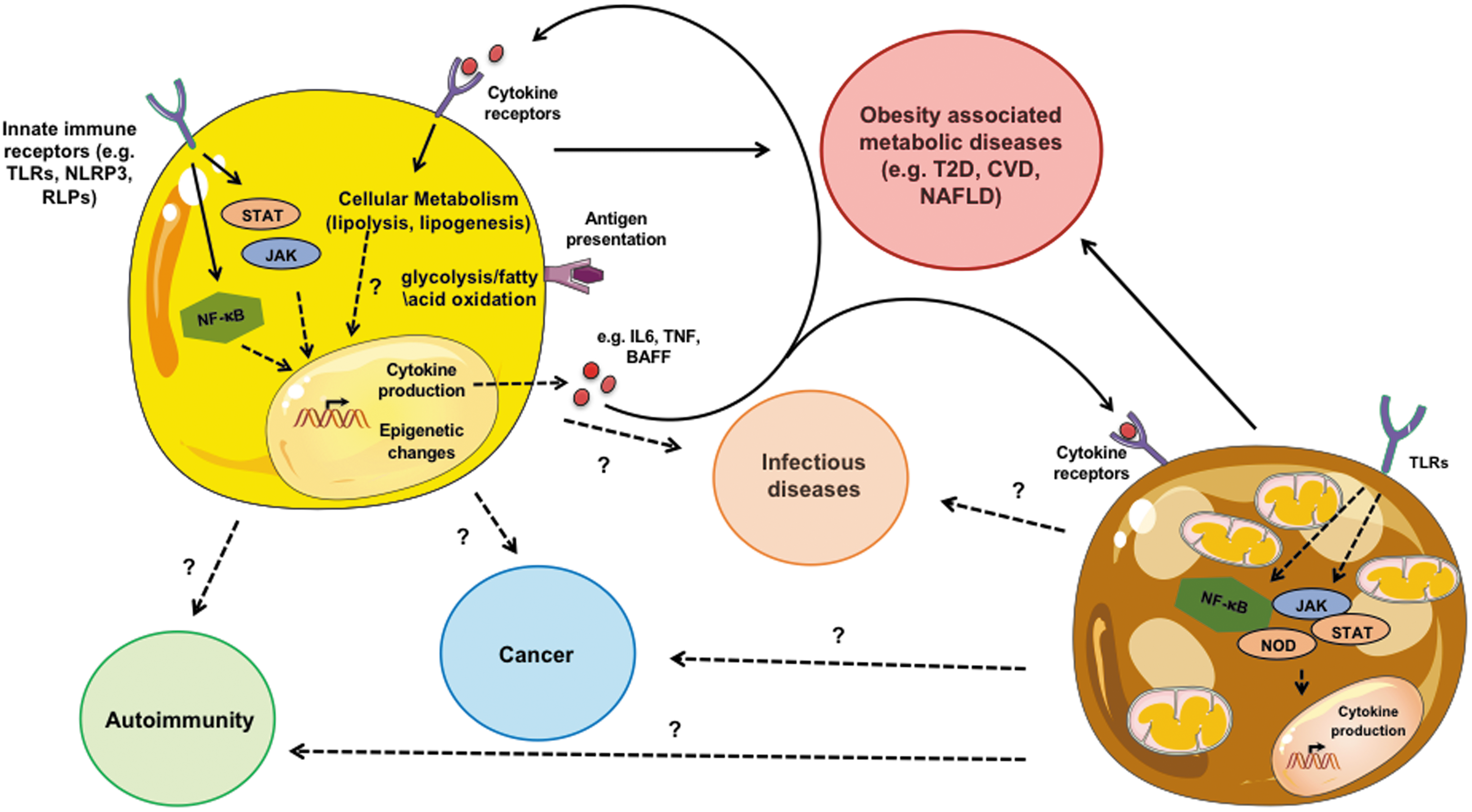
Adipocyte contribution to disease. Adipocytes have “immune-like” capabilities that, in part, overlap with traditionally investigated immune cells. White adipocyte-specific inflammatory capacity can impact obesity-associated metabolic diseases. The contributions of other subsets of adipocytes (eg, brown) to disease should be formally examined. Relevance of the interconnection in adipocytes between signaling pathways (eg, NF-κB and Jak/STAT), core metabolism (e.g., glycolysis and β-oxidation), and epigenetic programming to promote adipocyte-intrinsic inflammatory capacity and contribute to disease pathogenesis warrants future investigation. In addition, if adipocyte crosstalk with immune cells likewise contributes to diseases, including autoimmunity, cancer and infections are a natural extension to current investigations. Color images are available online.
Footnotes
Acknowledgments
This study was supported, in part, by CCHMC Pediatric Diabetes and Obesity Center (to S.D.), NIH R01DK099222 (to S.D); NIH T32AI118697 and T32GM063483-14 (associated with C.C.C.).
Authors' Contributions
C.C.C., M.S.M.A.D., P.C.A., J.S-G., and S.D. participated in the conception of this review and wrote the article.
Author Disclosure Statement
The authors declare no competing financial interests.