
Abstract
Atopic diseases, such as atopic dermatitis (AD), allergic asthma (AA), and allergic rhinitis (AR), are increasingly becoming a worldwide issue. This atopic triad originates at an early age and on a multifactorial basis, causing significant discomfort to susceptible individuals. The global case number is now reaching new highs, so exploring immune system regulation and its components is becoming critical. One cytokine, interleukin-32 (IL-32), is involved in inflammation and regulation of the immune system. It has nine isoforms that show varying degrees of expression, both intracellularly and extracellularly. IL-32 is secreted by immune cells, such as monocytes, macrophages, natural killer cells, and T cells, and by nonimmune cells, including fibroblasts, keratinocytes, and endothelial cells. Its production is regulated and augmented by microorganisms, mitogens, and other cytokines. Early studies demonstrated that IL-32 was an immune regulator that functioned to protect against inflammatory diseases, including AD, AA, and AR, and proposed a proinflammatory role for IL-32 in immune regulation and symptom exacerbation. However, several later reports suggested that IL-32 is downregulated in inflammatory diseases and exerts an anti-inflammatory effect. This review article focuses on recent findings regarding the detrimental and protective roles of IL-32 in development and management of inflammatory diseases. The exact role of IL-32 in AD, AA, and AR still remains to be elucidated. Future research should explore new avenues of IL-32 functionality in human inflammatory diseases.
Background
Allergy and atopy are used interchangeably in inflammatory diseases. Atopy is defined as the tendency of an organism to produce an exaggerated immunoglobulin E (IgE) immune response to common allergens, resulting in development of allergic diseases such as atopic dermatitis (AD), allergic asthma (AA), and allergic rhinitis (AR). Atopy is generally linked with the term, atopic march, which describes the disease progression typically starting in early childhood with development of AD, followed by AA, and finally progressing to AR (Aw and others 2020) (Fig. 1a). Atopic disease prevalence is increasing globally, especially in developing countries. It has been reported that AD affects up to 20% of children and up to 3% of adults, the peak incidence of asthma is higher in children aged <5 years, and AR incidence has been estimated at up to ∼25% in children and >40% in adults (Wu and others 2012; Nutten 2015; Brożek and others 2017; Barbarot and others 2018; Aw and others 2020; Mattiuzzi and Lippi 2020). Atopic individuals with AD are susceptible to bacterial, viral, and fungal infections, further increasing disease severity (Casagrande and others 2006; Grigoryev and others 2010; Tomczak and others 2019). Skin barrier defects and primary infections may cause detrimental effects on AA and AR, worsening individual disease symptoms and significantly burdening patients. Understanding the interplay between risk factors and immunity, which can increase the likelihood of developing atopic diseases, would allow for early diagnosis and treatment.
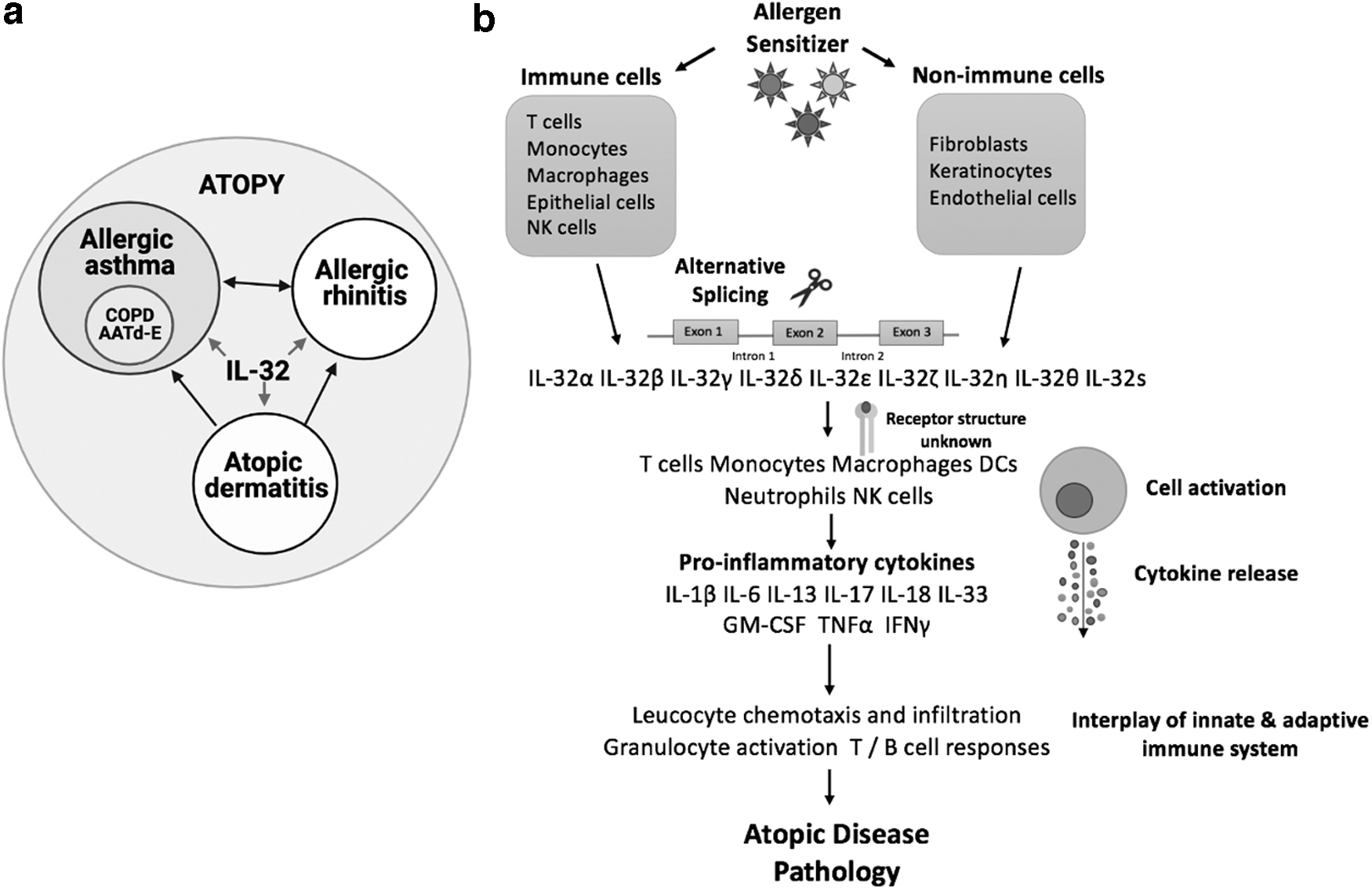
IL-32 pathway leading to the onset of atopic disease pathology.
Autoimmunity is also related to atopy as these are both manifestations of immune system dysfunction involving distinct immunological reactions. It is widely discussed that T helper (Th) cell subsets, Th1 and Th2, are closely related to one another as Th1 cells increase the likelihood of autoimmunity and Th2 cells induce atopy-related cell activities (Magnan and others 2000; Romagnani 2004; D'Angelo and others 2016). The analysis by D'Angelo and others showed a strong positive association between Th2-mediated allergic diseases and Th1-regulated autoimmune disorders. Human studies also show that Th1 cells contribute to hypersensitivity reactions and immunopathology (Woodfolk and Platts-Mills 2001; Romagnani 2004). However, the mechanisms explaining the nexus between atopy and autoimmunity are not fully understood.
The immune system is a complicated network consisting of a variety of cells and molecules that interact to defend the body against bacterial, viral, and fungal pathogens and regulate unwanted immunity responses (Qin and others 2016; Schönrich and Raftery 2019; Chai and others 2020). Cytokine molecules form an important part of immune disease regulation, orchestrating innate and adaptive immune cell responses. Over the past several decades, it has become evident that cytokines contribute to human disease immunopathogenicity, symptom exacerbation, and severity. Understanding cytokine mechanisms would allow us to treat disease pathogenicity and develop therapeutic and prevention strategies.
The search for novel cytokines has been the focus of immune system research for more than two decades. The pleiotropic cytokine, interleukin-32 (IL-32), has recently been associated with inflammatory diseases, including AD, AA, and AR (Calabrese and others 2008; Meyer and others 2010; Kwon and others 2017; Gonnet and others 2018; Hong and others 2018; Lee and others 2020). The IL-32 gene was discovered in 1992, and the biological role of the protein, also known as natural killer cell transcript 4 (NK4), was first described in the scientific literature in 2005 (Dahl and others 1992; Kim and others 2005; Joosten and others 2013). Only recently, exciting new concepts of pivotal roles of IL-32 in atopic diseases have emerged (Meyer and others 2012; Nold-Petry and others 2014; Khawar and others 2015; Kwon and others 2018; Sohn and others 2019; Fadaei and others 2020). Cytokines, including IL-32, orchestrate AD, AA, and AR allergic reactions, and a better understanding of mechanisms of IL-32 in inflammation would allow for earlier diagnosis and more efficient treatment. As autoimmunity and atopy are regulated by a balance of Th1 and Th2 subsets, IL-32 may form part of the additional impetus in the leverage of either T cell subset and lead to increased disease severity and symptom exacerbation. Recent studies suggest that IL-32 serves as both a proinflammatory and anti-inflammatory cytokine, although its exact role remains controversial. In view of the above information, this review article focuses on the old and new data concerning IL-32 in inflammatory atopic diseases and its underlying role as a pro- and anti-inflammatory cytokine.
Characteristics of IL-32
IL-32 was first identified on the human chromosome 16p13.3 and has not been assigned to any cytokine family due to its specific cell surface receptor (Dahl and others 1992). IL-32 is expressed by monocytes, macrophages, natural killer (NK) cells, T cells, and epithelial cells, as well as some nonimmune cells, namely fibroblasts, keratinocytes, and endothelial cells (Fig. 1b) (Akdis and others 2016). IL-32 mRNA undergoes alternative splicing and produces nine isoforms, including IL-32α, IL-32β, IL-32γ, IL-32δ, IL-32ɛ, IL-32ζ, IL-32η, IL-32θ, and IL-32s (El-Far and others 2016; Ribeiro-Dias and others 2017; Palstra and others 2018). Each IL-32 form is unique due to its specific signaling mechanisms, cell targets, cytokine-specific interactions, potency, and pro- and anti-inflammatory properties. IL-32 is found only in humans and has little homology with any other cytokine. IL-32γ is the most active, secreted, and studied isoform, whereas IL-32β, although expressed in various cells, is less potent than IL-32γ (Choi and others 2009; Huang and others 2015; Damen and others 2017). IL-32 targets monocytes, macrophages, dendritic cells (DCs), T cells, neutrophils, and NK cells (Fig. 1b).
The cell surface receptor is yet to be identified due to its unique structure and is believed to mediate its effects through intracellular mechanisms (Joosten and others 2013). According to Heinhuis and others (2012), the structure of IL-32 comprises coils and α-helices. Both IL-32α and IL-32β bind αVβ3 and αVβ6 integrin extracellular domains and activate procaspase-3, leading to apoptosis. Moreover, IL-32α interacts with intracellular proteins associated with integrin signaling and focal adhesion, including paxillin and focal adhesion kinase 1. This indicates that IL-32 may have a role in integrin signaling and modulation of cell death. Postsynthesis IL-32 is found intracellularly and is released upon apoptosis or in exosomes (Kim and others 2005; Goda and others 2006; Zahoor and others 2017). Several studies suggest that minuscule amounts of IL-32γ can be secreted by human rheumatoid arthritis synovial fibroblasts, indicating a potential IL-32 secretion mechanism (Heinhuis and others 2011; Kim and others 2014).
IL-32 has been shown to be important in bacterial, viral, fungal, and protozoan infections and atopic diseases, including AD, AA, and AR (Deplanche and others 2016; Gasiuniene and others 2016; dos Santos and others 2017, 2019; Tian and others 2019; Koeken and others 2020; Liu and others 2020). IL-32 is generally accepted to be involved in ameliorated Th1-related responses. However, the data remain equivocal. Several studies have highlighted the protective response of IL-32 and the dampening of the immune system in inflammatory diseases. Different factors could alter conventional modulation effects of IL-32 and switch its role in the exacerbation of inflammatory diseases, resulting in a different cytokine milieu and increased local and systemic pathogenicity. A number of studies have discussed the proinflammatory effects of IL-32, while others have stressed the importance of its anti-inflammatory effects in the atopic triad; to date, the anti-inflammatory role of IL-32 in patients with AR is unknown.
Proinflammatory Effect of IL-32 in Atopic Diseases
AD is a chronic and relapsing inflammatory skin disease that is genetically complex and usually characterized by pruritus and chronic dermatitis with varying degrees of severity (Silverberg and Simpson 2013). It most often begins in infancy and is frequently followed by other atopic disorders such as asthma. The emergence of these diseases is age dependent and affected by both environmental and genetic factors. Research on the effect of IL-32 on the pathogenesis of these diseases has been limited; however, in recent years, research has suggested that IL-32 may act as a proinflammatory cytokine in AD, AA, and AR.
Keratinocytes play important roles in the immunopathology of AD as both protective barriers and innate immune system mediators (Tokumaru and others 2005; Pietka and others 2019). Meyer and others (2010) showed that IL-32 is expressed in human primary keratinocytes when stimulated with interferon gamma (IFNγ) and tumor necrosis factor alpha (TNFα), both of which have been correlated with higher IL-32 serum levels. Similarly, IL-32 has been observed in patients with chronic plaque psoriasis (CPP); IL-32 was weakly expressed in a healthy human skin keratinocyte nucleus, but increased in individuals with CPP (Hu and others 2007). Transfection of primary keratinocytes with small interfering RNA to IL-32 decreased keratinocyte apoptosis, indicating that IL-32 promotes cell death and contributes to AD pathophysiology. Addition of the immunosuppressive drug, cyclosporine, for treatment of severe AD decreased IL-32 serum levels, further highlighting its significance as a proinflammatory cytokine (Kitayama and others 2017). In discussing the proinflammatory role of IL-32 in AD, (Oh and others 2011) showed that IL-32 indirectly induces anticancer activities by causing apoptosis. Recent research has suggested that IL-32 acts as a molecular link between keratinocytes and Langerhans cells (LCs) in healthy skin (Gonnet and others 2018). It has been proposed by Gonnet and others that IL-32 contributes to LC activation, decreased surface expression of adhesion molecules, and CXCL10 production. Moreover, Gorvel and others (2017) demonstrated that LCs secrete four isoforms of IL-32 (α, β, γ, and δ), with IL-32α being produced the most. Based on these findings, IL-32 levels may be increased during the onset of AD, leading to pathophysiological changes in the skin.
According to a recent report by Kwon and others (2017), IL-32γ exhibits synergistic effects with the NOD1 ligand and dsRNA to induce IL-6 from human bronchial epithelial (BEAS2B) cells. In an in vitro study by Kwon and others, the level of IL-32γ in serum and plasma was higher in asthmatic patients than in healthy controls. Notably, a low dose of NOD1 ligand stimulation with IL-32 exhibited synergistic induction of IL-6 and IL-33 in BEAS2B cells. Meyer and others (2012) observed that the serum IL-32 level was positively correlated with serum TNFα and IFNγ levels in asthmatic patients compared with healthy individuals. Concurrently, IL-13 and IL-17 proinflammatory cytokine levels were also elevated. According to Meyer and others, serum IL-32α and IL-32β were detected more frequently in asthmatics than in healthy controls; conversely, no relationship between eosinophilic inflammation and serum levels of total IgE was observed. TNFα is generally understood to act indirectly on Akt and JNK signaling pathways and promote apoptosis and cell migration (Papa and others 2004; Bieler and others 2007).
Emerging evidence suggests that IL-32 is involved in airway inflammation through induction of TNFα (Li and others 2014). IL-32 and proinflammatory cytokine (TNFα, IFNγ, IL-13, and IL-17) interplay was seen in other diseases, indicating that IL-32 may have a direct or indirect role in promoting inflammation (Moon and others 2012; Nold-Petry and others 2014; Catalán and others 2016). Furthermore, stable asthmatic IL-32γ levels were elevated in induced sputum (Kwon and others 2017). Kwon and others showed that in sputum, IL-32γ negatively correlated with the forced expiratory volume (FEV1) and positively correlated with the annual exacerbation rate. These findings build on the idea that IL-32γ directly and indirectly promotes asthmatic inflammation.
Several studies show that inflammatory diseases such as chronic obstructive pulmonary disease (COPD) and alpha-1 antitrypsin (AAT)-deficient emphysema have links with atopy and provide evidence of a possible proinflammatory role for IL-32 in AA (Fig. 1a) (Eden and others 1997; Fattahi and others 2013; Neves and others 2013; Backer and others 2020; Putcha and others 2020). In patients with COPD, lung tissue macrophages express IL-32 along with TNFα (Calabrese and others 2008; Baraldo and others 2015). AAT is known to inhibit IL-32 (Marcondes and others 2011; Lee and others 2017). Interestingly, COPD patients with AAT-deficient emphysema maintain elevated IL-32 levels, whereas individuals with AAT-deficient emphysema and without COPD show AA symptoms (Hill and others 1999; Van Veen and others 2006; Vidal and others 2006; Eden and others 2007; Eden 2010). Moreover, laboratory studies of human airway epithelial cells have clearly shown that IL-32 expression increases followed by IFNγ-induced IL-32 proinflammatory effects on the airway epithelium (Kudo and others 2012). Ota and others (2015) found evidence that synthetic dsDNA induces IL-32 production in bronchial epithelial cells, whereas siRNA targeting p65 and TAK1 inhibits IL-32 expression. Furthermore, recombinant IL-32 (rIL-32), when subjected to human lung adenocarcinoma A549 cells, induced fibrosis, suggesting that it has a proinflammatory effect on pulmonary alveolar epithelial cells (Gong and others 2020). In line with previous reports, it has been demonstrated that in asthmatic patients, serum IL-32 was negatively correlated with lung function compared with healthy controls (Jia and others 2014). These findings explore the link between AA, AAT deficiency, and IL-32, where increased IL-32 levels may contribute to airway hyper-responsiveness and airflow obstruction, leading to AA.
Supporting this view, IL-32 expression has been shown to correlate with the number of neutrophils infiltrating the alveolar walls (Calabrese and others 2008). Neutrophil-derived proteinase 3 (NP3) binds to IL-32α and mediates proteolysis activities (Novick and others 2006). IL-32α appears to be first inserted into the plasma membrane; NP3 then cleaves IL-32α, thus enhancing its functional activities. Moreover, NP3 cleaves the separate domains of IL-32 differently, further affecting the proinflammatory cytokines (IL-1β, IL-6, IL-8, and TNFα) (Kim and others 2008). In view of the above, further study shows that IL-32 stimulates eosinophils through NP3, resulting in a higher frequency of infiltrating eosinophils in the lungs (Wong and others 2013). In patients with AR nasal mucosa, IL-32 favors the production of proinflammatory cytokines IL-1β, IL-18, and granulocyte–macrophage colony-stimulating factor (GM-CSF) (Jeong and others 2011). Research by Netea and others (2008) revealed that IL-32γ amplifies the effects of GM-CSF and IL-4 on the DC CD83 marker. Together, these findings suggest that effects of IL-32 on the innate immune system are related to increased eosinophilic inflammation due to upregulated chemotactic factors and proinflammatory cytokines. Based on the limited data, it is difficult to clearly define a proinflammatory role of IL-32 in AA; however, recent evidence shows IL-32 as a possible contributor to disease immunopathology.
AR, also known as hay fever, is inflammation in the nostrils triggered by environmental allergens and is usually apparent in individuals with AD and AA. Upon allergen sensitization, the typical AR immune response involves IgE production, mast cell activation, and subsequent histamine release (Pawankar and others 2011; Bernstein 2016). Several lines of evidence have suggested IL-32 as a contributor to AR symptom exacerbation (Jeong and others 2011; Nam and others 2014). Jeong and others (2011) determined that levels of inflammatory cytokines IL-1β, IL-18, and GM-CSF increase upon production of IL-32 from human nasal mucosal samples and EoL-1 cells. Moreover, in the context of in vivo experiments, AR mouse models have demonstrated IL-32 activation and its relationship with higher IgE levels. IL-32 was shown to induce the production of proinflammatory cytokines, which may be affected by different IL-32 isoforms (α, β, γ, and δ) found in nasal mucosal tissues. A report by Nam and others (2014) stated that IL-32 upregulates p38, MAP, NFkβ, and caspase 1, resulting in subsequent production of thymic stromal lymphopoietin (TSLP). Assays with a human THP-1 cell line showed that IL-32 regulates the production of IL-8, NO, and TNFα. Experiments with human EoL-1 cells indicated that IL-32 controls IL-8 and GM-CSF levels, whereas assays with THP-1 cells showed that IL-32 promotes IL-1β and IL-6 production through activating caspase-1. These findings are congruent with other studies investigating IL-32, IL-1β, IL-6, and caspase-1 (Kim and others 2005; Netea and others 2005; Nold-Petry and others 2009; Lin and others 2017). Of interest, treating THP-1 cells with bamboo salt inhibited IL-32 and downregulated proinflammatory cytokines (IL-1β, IL-8, TNFα, IFNγ, and TSLP). In these studies, it is reasonable to assume that IL-32 is responsible for regulating inflammatory responses at both post-transcriptional and cellular levels. Further investigations into the role of IL-32 are warranted and will lead to a more complete understanding of proinflammatory mechanisms that affect immunity, specifically in patients with AR.
Proinflammatory effects of IL-32 are summarized in Table 1, together with other studies that have established a possible proinflammatory role of IL-32 in AA.
Proinflammatory Effect of Interleukin-32 on Atopic Diseases
AR, allergic rhinitis; BALB/c, Bagg and Albino mice strain c; GM-CSF, granulocyte–macrophage colony-stimulating factor; HUVEC, primary human umbilical vein endothelial cells; IFNγ, interferon gamma; IgE, immunoglobulin E; IL-32, interleukin-32; NHBE, normal human bronchial epithelial cells; PBMCs; TNFα, tumor necrosis factor alpha; TSLP, thymic stromal lymphopoietin.
Anti-Inflammatory Effect of IL-32 in Atopic Diseases
The anti-inflammatory role of IL-32 in AD is weakly understood; however, a recent study by Lee and others pointed out several key aspects. Administering rIL-32 to a PA-induced AD model significantly decreased AD progression by reducing AD-related cytokines and inflammatory cell recruitment. In an in vitro study, IL-32γ overexpression decreased TNFα and IFNγ. Lee and others showed that IL-32 blocks IL-31 and IL-33 expression, reducing skin lesion inflammation. Furthermore, IL-32 suppresses miR-205 and nuclear factor kappa B (NF-kB) expression, diminishing chronic skin inflammation and dampening the proinflammatory cytokine response. Studies suggest that IL-32 may have a protective role in AD; however, the sparsity of data emphasizes the need for additional human studies investigating the role of IL-32.
In contrast to the proinflammatory effects of IL-32 on AA, Bang and others (2014) evaluated the therapeutic effect of rIL-32 on allergic inflammation by performing bronchoalveolar lavage fluid analysis. The study showed that IL-32γ transgenic mice had significantly reduced airway inflammation compared with wild-type (WT) mice. IL-32γ production decreased neutrophil and basophil recruitment and reduced the chemotactic leukocyte factor, eotaxin, in the lungs. Th1 (TNFα and IFNγ) and Th2 (IL-4, IL-5, and IL-13) cytokine levels were decreased in the lungs of IL-32γ transgenic mice. Moreover, in the lungs of ovalbumin-sensitized IL-32γ transgenic mice, IL-10 levels were markedly increased by CD11b+ monocytic cells. Remarkably, further analysis proved that the lungs of IL-32γ transgenic mice possessed elevated levels of CD11b+Ly-6C+Ly-6G− monocytic cells, which are known for suppressing T cell activation and proliferation (Zhu and others 2007; Haile and others 2008; Deshane and others 2011). Vascular endothelial growth factor (VEGF) induces angiogenesis in the airways of asthmatic patients and is an established biomarker in asthma (Lee and others 2004; Bucher and others 2018; Liu and others 2018). Meyer and others (2012) observed that IL-32 inhibition increases VEGF levels in human bronchial epithelial cells. A similar trend was seen with platelet-derived growth factor (PDGF), further highlighting the anti-inflammatory role of IL-32. Furthermore, the results from the study by Hong and others provide convincing evidence that rIL-32 attenuates collagen deposition and Alpha Smooth Muscle Actin production in mouse models and inhibits fibronectin in MRC-5 cells stimulated with transforming growth factor beta (TGF-β). Taken together, these experiments define the role of IL-32 in preventing fibrosis in asthmatic patients and exemplify involvement of IL-32 in maintaining immunological homeostasis during AA. IL-32 clearly possesses a protective role in AA; however, with limited data, the definitive mechanism of IL-32 remains to be determined.
Anti-inflammatory effects of IL-32 are summarized in Table 2.
Anti-Inflammatory Effect of Interleukin-32 on Atopic Diseases
AD, atopic dermatitis; NF-kB, nuclear factor kappa B; PDGF, platelet-derived growth factor; VEGF, vascular endothelial growth factor.
Conclusions
IL-32 may play a significant role in the pathogenesis of inflammatory diseases by regulating different pathways of the immune system. IL-32 demonstrates proinflammatory activities in AD, AA, and AR through upregulation of TNFα, IL-1β, IL-6, IL-8, IL-17, IL-33, GM-CSF, CXCL10, and PR3. Accumulating evidence now indicates that IL-32 also exhibits anti-inflammatory properties in atopic diseases such as AD and AA by increasing IL-10 levels and downregulating chemotactic factors, including TNFα, IL-1β, IL-6, IL-31, IL-33, VEGF, PDGF, and miR-205. Therefore, IL-32 acts as a pleiotropic cytokine on a multifactorial basis depending on its secreted form, the surrounding cytokine milieu, the disease state, and genetic factors. Our emerging understanding of the functional role of IL-32 in human inflammatory diseases and the factors regulating the balance between pathogenic and protective disease states might aid in the development of cytokine-directed therapeutics. However, additional research on human samples must be conducted to clarify further details regarding the role of IL-32.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
B.S. received a 2020–2022 grant from the Mutual Funds of the Lithuania–Latvia–Taiwan Cooperation Project (registration no. P-LLT-20-4, Research Council of Lithuania).