
Abstract
Interleukin-17A is a pro-inflammatory cytokine that plays a key role in the immune response to many pathogens and implicated in autoimmune diseases. This molecule is also involved in providing protection to many bacterial and fungal infections of gastro-intestinal tract and respiratory mucosa. Although molecular aspect of IL-17A has been studied in few species, no data are available for buffalo, which is one of the major sources of milk production in India. Therefore, in the present study, IL-17A gene of Indian Murrah Buffalo origin was cloned, expressed, and analyzed using bioinformatic tools. The coding sequence of buffalo IL-17A gene was cloned in prokaryotic expression vector (pET-28a) followed by its expression, purification, and characterization. A computational analysis was performed to understand the sequence, structure, and evolutionary relationship of buIL-17A. It revealed that the length of buIL-17A sequence without signal peptide is 132 amino acids as in cattle. However, sequence identity is found to be 99% due to one amino substitution difference between buffalo and cattle. After analysis, it can be concluded that buIL-17A recombinant protein can be used as a potential immunobiological reagent for diagnostic and therapeutic purpose.
Introduction
Interleukin-17
Ruminants have higher proportion of γδ-T lymphocytes than human and mice and have role in skin and chronic mucosal infections. Compared to humans having <5% γδ-T lymphocytes of the total circulating lymphocytes, ruminants possess about 15%–60% and play a major role in T cell regulation. These cells continuously secrete IL-10 and proliferate in response to TGF-β, IL-10, and after contacting Antigen-Presenting Cells (APCs) (Guzman et al., 2014). Studies on molecular signaling of IL-17A as cytokine have led various therapeutic approaches toward diseases like rheumatoid arthritis, multiple sclerosis, and psoriasis (Van den Berg and McInnes, 2013). IL-17 and Th-17 cells have been characterized in selective species of veterinary importance, including livestock and companion animals, along with the development of antibodies and diagnostic kits (Flynn and Marshall, 2011; Jing et al., 2012; Loos et al., 2012). Its expression has been shown in specific animal diseases like recurrent airway obstruction and recurrent uveitis in horses, skin inflammation in sheep, bacteremia in both rabbits and chicken, pyometra in bitches, and middle ear inflammation in rabbits (Mensikova et al., 2013). Besides this, the signal produced for the host defense mechanism by IL-17A and its receptor IL-17RA against gut microbiota is well documented (Ivanov et al., 2009; Kumar et al., 2016). Host protection effects against various bacterial and fungal pathogens like Mycoplasma pneumonia, Klebsiella pneumoniae, Candida albicans, Histoplasma capsulatum, Blastomyces dermatitidis, and Coccidioides posadasii have also been reported (Chen and Kolls, 2013).
Limited availability of reagent antibodies and other immune molecules of buffalo have hampered the progress in buffalo immunology. There is need to understand buffalo immune system features and functions with application of modern tools of investigation in the era of postgenomics computational and systems immunology. Since IL-17A has not been characterized in buffaloes, this is the first report to characterize the molecular and structural features of buffalo-IL17A.
Materials and Methods
Isolation of peripheral blood mononuclear cell
Peripheral blood mononuclear cells (PBMCs) were used as a source of IL-17A gene from Indian Murrah Buffalo. The institutional animal ethics committee (IAEC) permission had been taken for blood sample collection.
PBMCs were isolated using Histopaque-1077 (Sigma). Briefly, 4 mL of histopaque was taken in 15 mL centrifuge tube, and equal volume of blood was overlayed gradually with utmost care, avoiding mixing of two layers. Thereupon tubes were centrifuged at 500 rpm for 30 min at room temperature, after which three distinct layers can be seen. PBMCs were present in thin milky middle layer. PBMCs were separated and washed twice with 10 mL of Dulbecco's Phosphate buffered saline (Sigma). Finally, the pellet was resuspended in tissue culture media prepared as follows: 90 mL Iscove's Modified Dulbecco's Medium (Gibco), 10 mL Fetal bovine serum (Sigma), 1 mL of 100 × antibiotic antimycotic solution (Gibco), and 100 μL 50 mM β-mercaptoethanol at a concentration of 106cells/mL. The cells were transferred in 25 cm2 tissue culture flask and incubated in carbon dioxide incubator at 37°C. After 2 h, cells were stimulated with 10 μg/mL Concavalin A (Sigma) to activate IL-17A genes and were further incubated for 60 h. Cells were harvested using centrifuge and stored at −80°C for further use.
Amplification of the buffalo IL-17A gene
RNA was isolated from the stored cells using RNeasy Kit (Qiagen) following manufacturer's instructions, and the cDNA was synthesized using RevertAid cDNA Synthesis Kit (K1621; Thermo scientific) following manufacturer's instructions and stored at −80°C for further use. To amplify the buffalo IL-17A (buIL-17A) gene, primers were designed using National Center for Biotechnology Information (NCBI) Genbank Accession No. XM_006056757.3. Primers used to amplify the genes were: IL17_Fwd: 5′-TCTATCTAGAATGGCTTCTATGAGAAC TTC-3′ and IL17_Rev: 5′-AATACTCGAGAGCCAAATGGCGGACAAT-3′.
The purified DNA amplicon was cloned into cloning vector pJET1.2/blunt. Randomly selected clones were screened by polymerase chain reaction (PCR) and sequenced subsequently.
Subcloning of buIL-17A gene
The buIL-17A gene was cloned without signal peptide sequence using forward and reverse primers as follows: FwdNcoI_WLS: 5′-CTCT
Expression and western blot of recombinant buIL-17A protein
Positive clones were transformed into E. coli BL-21 strain for expression of recombinant buIL-17A. A single colony was inoculated in 10 mL of fresh Luria-Bertani (LB) broth containing kanamycin (50 μg/mL) and grown at 37°C for overnight on shaking incubator at 200 rpm. Next day 100 mL LB broth was inoculated with 1% primary culture and grown at 37°C to reach an optical density of 0.4. Culture was induced with 0.5 mM isopropyl-β-D-thiogalactoside (IPTG) and incubated at 37°C for 4 h on shaker incubator. Cells were harvested by centrifugation, and the pellet was resuspended in 5 mL of lysis buffer B (100 mM NaH2PO4, 10 Mm TrisHcl, 8 M urea, pH-8) followed by sonication with 10 s on-off cycles. The total cell lysate was clarified by centrifugation at 8,000 rpm for 20 min at 4°C. Collected samples were mixed with 2 × Laemmli's buffer and run on 15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE).
For western blotting proteins were separated on 15% SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membrane. After transfer, membrane was blocked in 5% nonfat milk for 1 h and subsequently incubated with the anti-His-horseradish peroxidase (HRP) antibody (1:5,000) overnight at 4°C (not more than 15 h). The membrane was washed four times with TBST for 10 mins each, and subsequently, the membrane was developed using 3,3′-diaminobenzidine (DAB) with 30% H2O2. Finally, reaction was stopped by washing the membrane with water.
Purification of recombinant buIL-17A protein
Recombinant buIL-17A protein was further purified using Ni-NTA column affinity chromatography. Qiagen nickel-nitrilotriacetic acid (Ni-NTA) superflow column was equilibrated with buffer B, and then the cleared lysate was added at a constant rate of 0.5 mL/min using peristaltic pump. The column was washed with Buffer C (100 mM NaH2PO4, 10 Mm TrisHcl, 8 M urea, pH-6.3), and the recombinant protein was eluted with Buffer E (100 mM NaH2PO4, 10 Mm TrisHcl, 8 M urea, pH-4.5). Fractions containing buIL-17A protein were identified by SDS-PAGE. The purified protein was stored in aliquots of 500 μL at −20°C. IL-17A protein purified using Ni-NTA column chromatography was in phosphate buffer containing 8 M urea. The purified protein was dialyzed using 3.5 kDa cutoff dialysis membrane (Repligen) against decreasing concentration of urea. Finally, the protein was in 200 mM NaH2PO4, 10 Mm TrisHcl, pH 7.2.
Evolutionary relationship
A Phylogenetic tree was built for the available IL-17A protein sequences to understand their evolutionary relationship through MEGA X software tool (Kumar et al., 2018). The evolutionary history was inferred using the Neighbor-Joining method (Saitou and Nei, 1987). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) was shown next to the branches (Felsenstein, 1985). The tree was drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Poisson correction method (Zuckerkandl and Pauling, 1965) and are in the units of the number of amino acid substitutions per site.
The evolutionary relationship was inferred using the Maximum Likelihood method and Jones-Taylor-Thornton (JTT) matrix-based model (Jones et al., 1992). The tree with the highest log likelihood (−3,713.34) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained with Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the JTT model and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 101 amino acid sequences. There were a total of 137 positions in the final dataset. Evolutionary analyses were conducted in MEGA X (Kumar et al., 2018).
Sequence analysis
The buIL-17A gene sequence obtained was translated using Expasy server (Gasteiger et al., 2005). The buIL-17A amino acid sequence was compared with the corresponding sequence in human to understand their homology. The hsIL-17A protein sequence (Q16552) was extracted from UniProtKB/Swiss-Prot database (The UniProt Consortium, 2023). The sequences were compared using Clustal × 2.1 (Larkin and others 2007). The target sequence, buIL-17A, was examined for secondary structural region, as well as for their fold through prediction servers (Buchan and Jones, 2019; Kelley et al., 2015; Lobley et al., 2009). A multiple sequence alignment (MSA) was built for a set of representative protein sequences of various species to understand the amino acids conserved for structure and function across the species using Clustal × 2.1 (Larkin et al., 2007). Mapping of the predicted secondary structural regions of IL-17A was performed on MSA.
In silico structure analysis
Neither crystal nor nuclear magnetic resonance structure available for buIL-17A protein therefore modeling a three-dimensional structure was done. It was designed to find out a template structure through two different approaches such as (a) using comparative search algorithm, Basic Local Alignment Search Tool (Camacho et al., 2009) against Protein Data Bank (PDB) database at NCBI (Rose et al., 2012) and (b) using Hidden Markov Model search algorithm through HHpred server (Zimmermann et al., 2018). However, the results obtained by both of the approaches were found to be different; hence, structural details were examined further to pick up the templates. Finally, the coordinates of the top listed proteins were extracted from protein databank (Burley et al., 2021) and were examined for their method of determination, resolution, and molecular features such as oligomerization. Since a single template was not suitable to multiple criteria, modeling was performed through multitemplate approach using MODELLER algorithm (Webb and Sali, 2016).
Twenty numbers of models were generated with an energy minimization, and based on the score, a best energy minimized model was selected and the structure was evaluated for its quality through Ramachandran plot using PROCHECK algorithm (Laskowski et al., 1996). The Ramachandran plot was examined for any deviations from the fully allowed, additionally allowed, and generously allowed regions. The residues were further refined with loop refinement algorithm under MODELLER package. The modeled structure superimposed on the template structure was evaluated.
The biological form of IL-17A exists as dimer. Hence, a dimer structure of the buIL-17A was modeled by superimposing the predicted structural coordinates on to the template structure. The dimer form of the target structure is achieved; then they were examined for their interacting residues. The dimer was superimposed on the structure that is interacting with its receptors such as IL-17RA and IL-17RC and the interacting residues between the IL-17A and its receptors.
Physiochemical properties of buIL-17A
Expasy's ProtParam prediction (Gasteiger et al., 2005) server was used to estimate various physiochemical properties of the protein such as molecular weight, theoretical isoelectric point (pI), total number of positive and negative residues, extinction coefficient, instability index, aliphatic index, and grand average hydropathy (GRAVY).
Results
Amplification of buIL17-A gene
BuIL-17A gene was amplified to give expected fragment length amplicon of 462 bp (Fig. 1). Amplified fragment was sequenced and submitted to gene bank with Accession No. OM524556. BuIL-17A ORF sequence derived from Concavalin A stimulated PBMCs was identical to the Predicted: Bubalus bubalis IL-17A mRNA sequence, GenBank ID: XM_006056757.3.
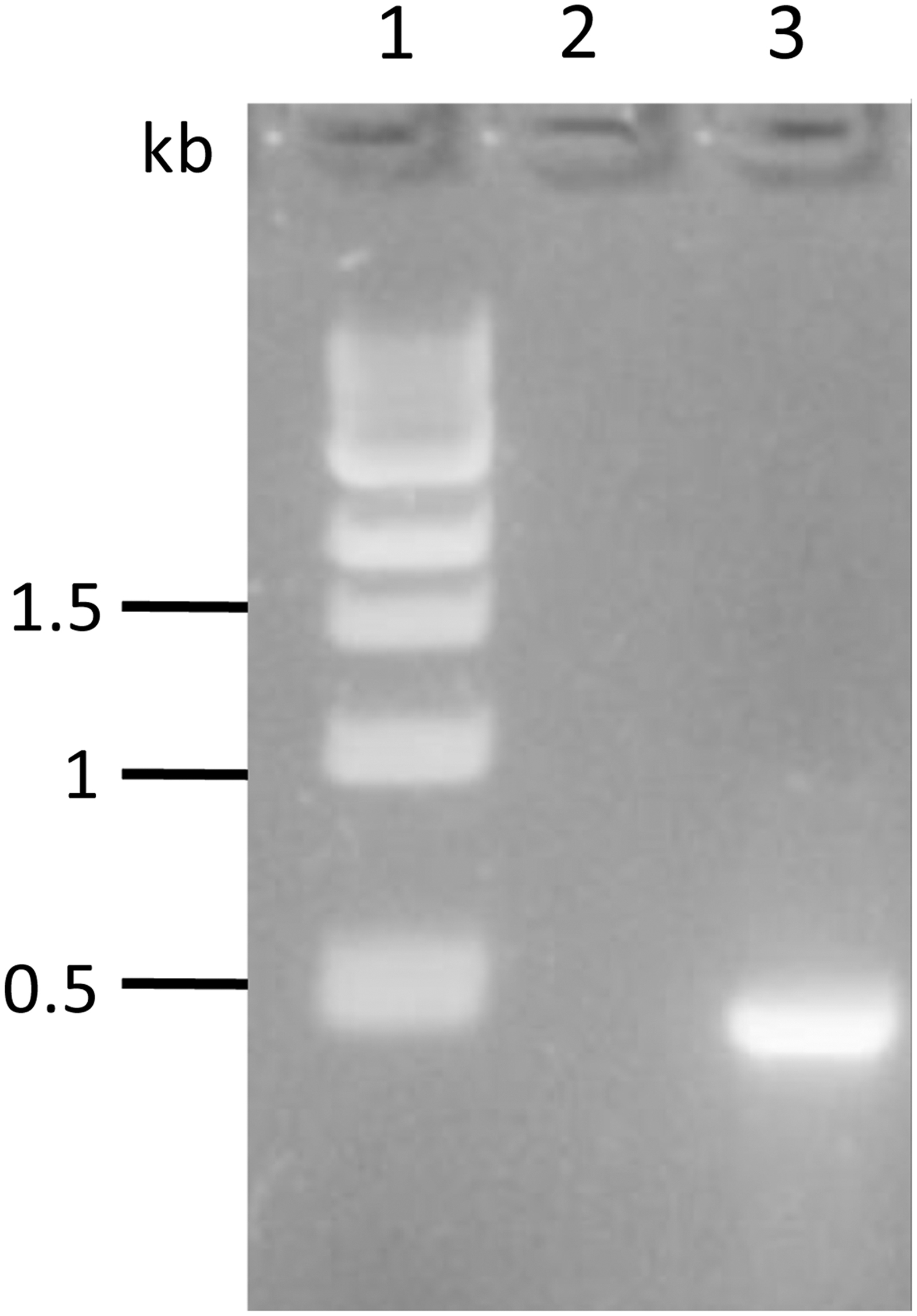
PCR amplification of buIL-17A gene. Lane1: 1kb DNA ladder, Lane 2: negative control, Lane 3: 462 bp buIL-17A gene fragment amplified product.
Expression and purification of buIL-17A
Induction with IPTG showed a protein band at 16 kDa corresponding to calculated recombinant buIL-17A (Fig. 2). The expressed protein was further checked whether it is soluble or not and it was found that protein was coming in the insoluble fraction, indicating that expressed protein was in inclusion bodies. So, the protein was efficiently purified in denaturing conditions (Fig. 2).
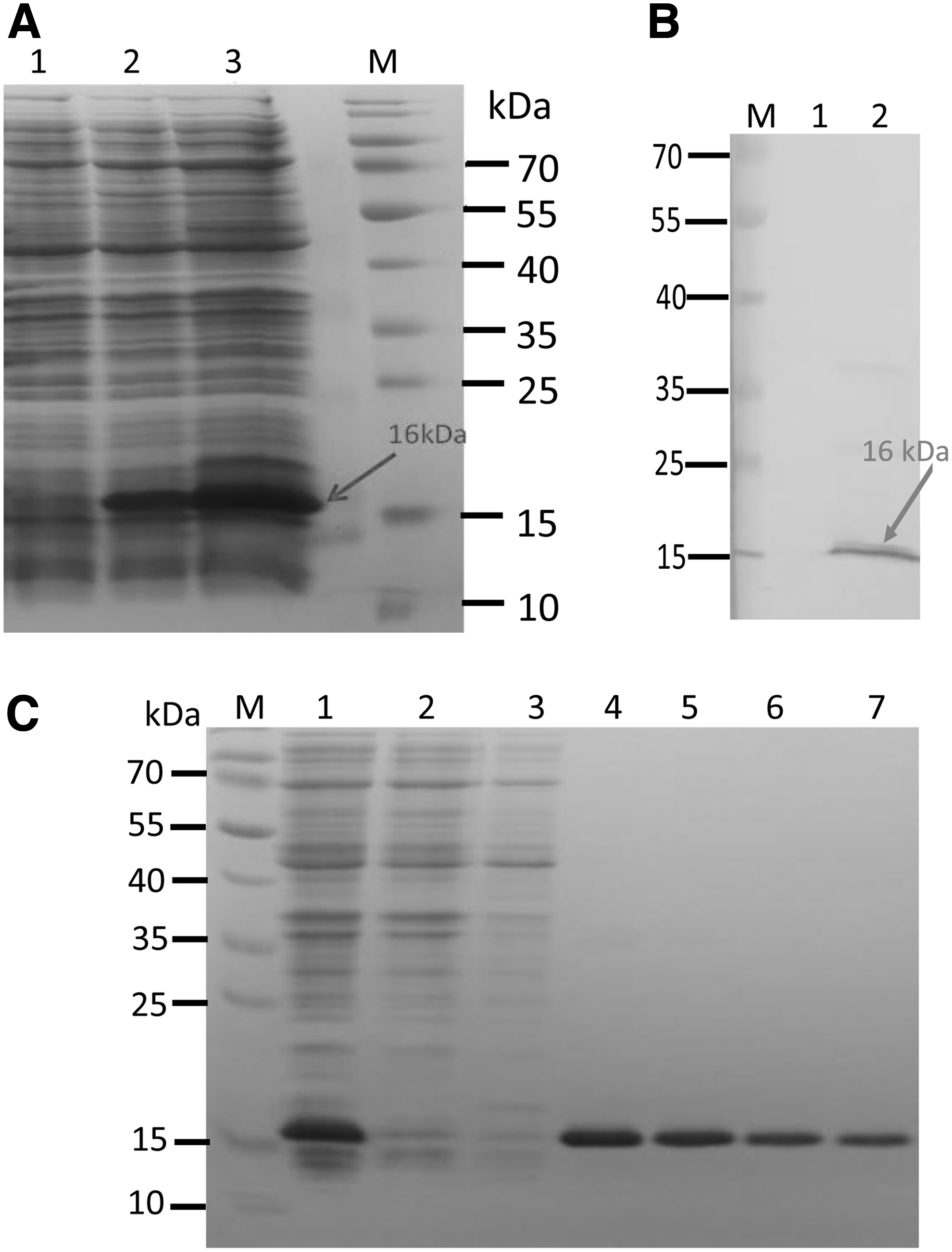
Expression and purification of recombinant buIL-17A.
Sequence analysis
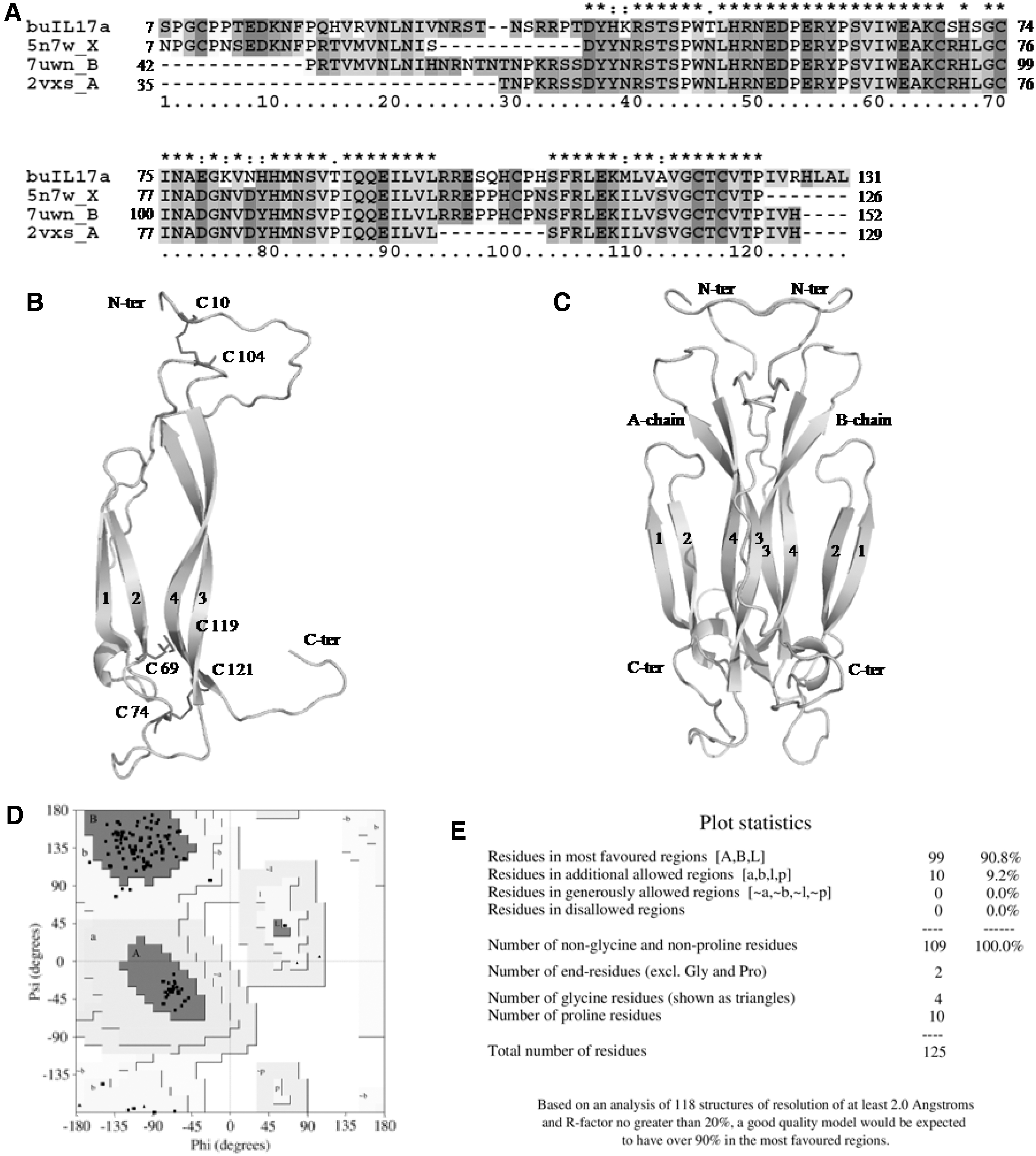
The translated buIL-17A nucleotide sequence was compared with the corresponding IL-17A sequence in human (hsIL-17A) to understand their closeness (Fig. 3). In buIL-17A protein, initial 22 amino acids formed signal peptide, compared to 23 in human and 17 in swamp buffalo. The length of trimmed IL-17A protein in Murrah buffalo and human was 132 amino acid residues. The percent identity between buffalo and human protein sequences was found to be 75%, whereas Murrah and swamp buffalo have 100% identical sequences.

Amino acid sequence alignment of buIL-17A and hsIL-17A. Comparing the sequences of buffalo and human, 75% identity was observed and there were only two amino acid residue insertions found in hsIL-17A sequence positioned between 36th and 37th position of buffalo sequence. Total of six cysteine residues were observed, which are important to form three disulfide bonds to maintain the fold of the structure (Signal peptide regions were removed before multiple sequence alignment). hsIL-17A, human IL-17A.
BuIL-17A protein sequence was compared with orthologous protein sequences to understand the amino acid conservation for structural and functional aspects. The orthologous sequences of 28 species were included for MSA using Clustal × 2.1 (Larkin et al., 2007) (Fig. 4). The predicted secondary structural regions of buIL-17A sequence were mapped on MSA to analyze the region-specific conservation. The predicted secondary structure of buIL-17A protein sequence presented four β-strands, one small helix and coil regions. A high sequence diversity was observed due to amino acid substitutions at the N-terminal regions. However, few substitutions were noticed in other places too, which are important for structural and functional properties.

MSA of IL-17A orthologous protein sequences. Mapping of the predicted secondary structural regions of IL-17A was performed on MSA. There were four β-strands, one small helix and coil regions were predicted and marked based on previous publications. The β-strands found at N and C-terminal regions, as well as two small β-strands found between β2 and β3 strands, were not considered while numbering as found in other published literature. MSA, multiple sequence alignment.
The residues found at all predicted secondary structural regions were mostly conserved, including six cysteine residues. The residue Ser at position 7 was observed to be conserved in almost all the species of the orders Artiodactyla, including buffalo, as well as species from other orders such as mouse, rat, swine, and rhinoceros, whereas in other species, Ser 7 was replaced by Asn. The residue, Asp, at position 15 was observed to be conserved in almost all the species except mouse, rat, and guinea pig. The residue Asn 25 was observed to be conserved in all the species except rabbit. The residue Arg 36 (in human: Lys 38) was observed to be conserved in all the species except human and camel, where it was replaced by Lys. The residues Pro 38 and Thr 39 (in human: Ser 40 and Ser 41) were replaced with Ser/Ala and Ser/Leu, respectively. However, Asp 40, Tyr 41, and Arg 44 were found to be conserved across the species.
The residue Ser at position 70 and 72 in Artiodactyla species, including buffalo (in human: Arg 72 and Leu 74), were positioned between the second and third Cysteines, which were replaced by Arg in primates, rodents, and chiroptera species and Leu in carnivores at position 70 and Lue in canidae species, Trp in felidae, and Gln in rat and mouse at position 72. The residue His 83 (in human: Tyr 85) in buffalo sequence was substituted by similar aromatic amino acids, such as Tyr in canidae family members and Phe in zebra and horse; however, it was also replaced by Val in Rhinoceros. The residue His at position 84 was observed to be conserved in all the species except in pangolins and emperor penguin, where they are substituted by Asp and Ser, respectively.
The residues, Gln 92, Glu 93, Ser 107, Phe 108, and Leu 110 (in human: Gln 94, Glu 95, Ser 109, Phe 110, and Leu 112), were observed to be conserved in all the species. Another residue, Ala 130, was observed to be conserved in several species; however, they are replaced by Val in cheetah, cat, tiger, Gly in zebra, horse, guinea pig, and Ser in Swine and rat. These residues were found to have structural importance while examining the model structure, which are discussed later. The BuIL-17A protein sequence was subjected to epitope prediction through the Immune Epitope Database (IEDB) (Jespersen et al., 2017). IEDB prediction shows that there are four epitope regions, which are named Epi1 − 4. The range and name of the residues that fall under each epitope are given in Table 1.
Predicted Epitopes for Buffalo Interleukin-17A
Evolutionary relationship
The IL-17A sequence of buffalo, cattle, sheep, goat, and camel was found to form a clade in the order Artiodactyla, whereas the marine animals like whale, prawn, and dolphin formed a separate clade within the same order. However, swine and common warthog form a separate clade, which was found to be away from both the clades within Artiodactyla order. The buffalo sequence was closely related to cattle in the phylogenetic tree; however, it tends to have more substitutions compared to cattle. Although hsIL-17A sequence was forming a clade along with few more species under the Primates order, the human sequence was diverged much compared to other species within the clade. Among the species included for evolutionary analysis, the emperor penguin and mouse sequences were found to have highly diverged sequences followed by rat, common warthog (Phacochoerus africanus), wild boar, bat, South African small mongoose, elephant, plateau pika, human, alpine marmot, and buffalo (Fig. 5).
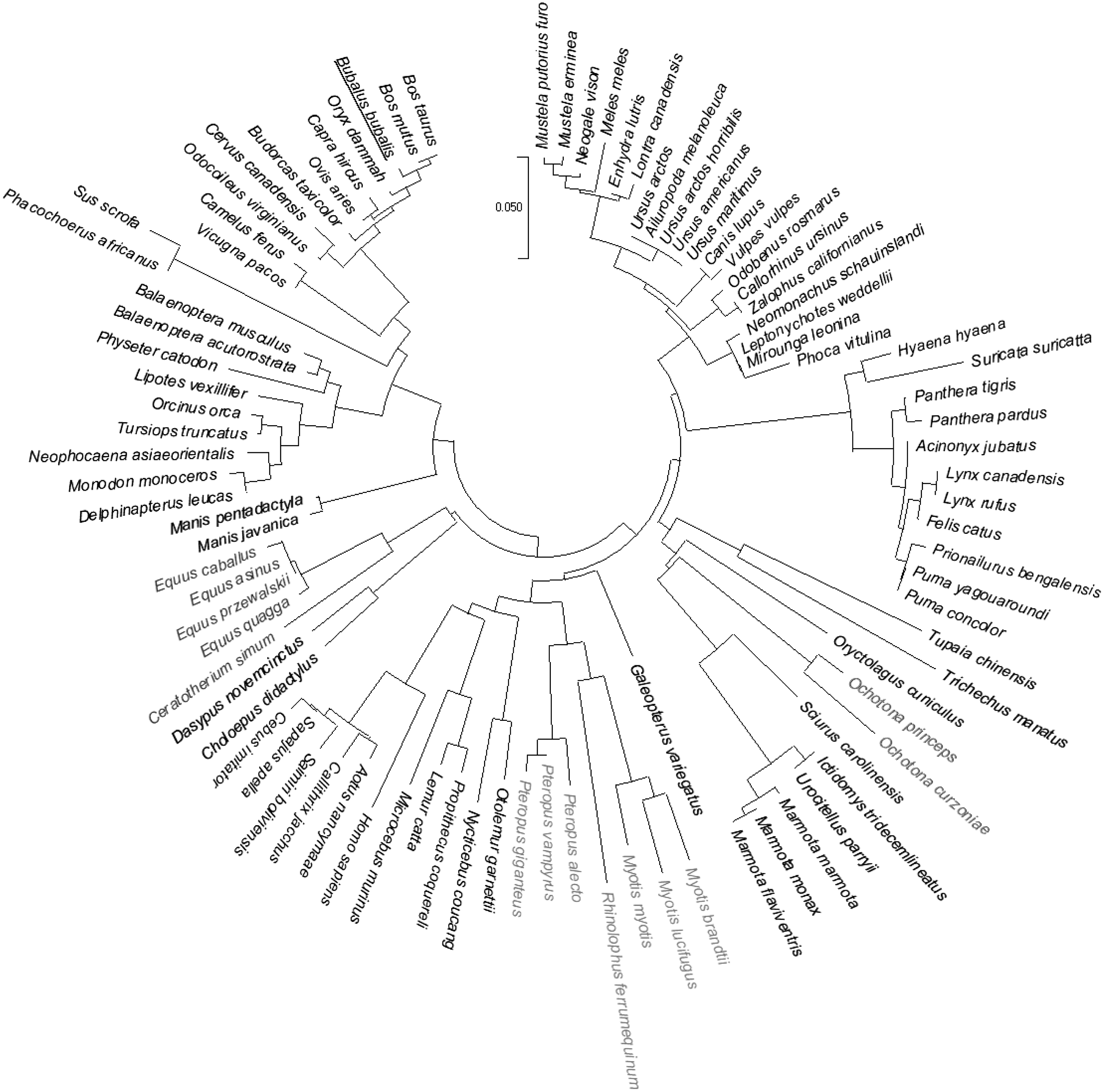
Phylogenetic relationship of IL-17A orthologous protein sequences of 92 species. buIL-17A protein sequence is colored red and underlined.
In-silico structural analysis
The multitemplate model algorithm of the modeler package has been used to build the three-dimensional structure of buIL-17A protein. The input multitemplate sequence alignment was produced by the Clustal × 2.1 (Fig. 6). The superimposition of the final refined model on to the template structure shows the root mean square deviation of 0.45 Ao.
3D model of buIL-17A and its validation.
The model structure of the buIL-17A protein has four β-strands, one helix and coiled/loop region. Apart from two β-strands, in which one was found at N-terminal region and another was at C-terminal region, two more small β-strands were observed between second and third β-strands. However, only β-strands present at the core regions were considered for numbering as per hsIL-17A crystal structure, (pdb id:2vxs; Gerhardt et al., 2009; Fig. 6). The cysteine residues were conserved at the relative site in buIL-17A compared to hsIL-17A and form three disulfide bridges in the molecule.
The dimer form of the model was generated, and the examination of dimer shows that it was symmetrical in nature as noticed in hsIL-17A (Liu et al., 2013). The binding free energy value of the buIL-17A dimer was calculated to be −16.8 kcal/mol (Krissinel and Henrick, 2007). The dimer was examined for interfacing residues of contact between the chain A and B (Table 2), which were considered to be important for maintaining the dimer interface of IL-17A protein.
Comparison of Human Interleukin-17A and Buffalo Interleukin-17A Protein Sequence
BuIL-17A, buffalo IL-17A; HsIL-17A, human IL-17A; PDB, Protein Data Bank.
The interface hydrogen bond forming residues were identified using hbplus algorithm (McDonald and Thornton, 1994). The hydrogen bonded residues between chain A and chain B of human (crystal structure) and buIL-17A dimer (model) were examined and compared, in which the bold residues (Table 2) were found to be conserved between buIL-17A and hsIL-17A, whereas other residues were found in either of them due to either lack of residue information at crystal structure or substitution of amino acid. Some of the residues observed to be conserved through sequence analysis in this study were found to be involved in dimer formation. The residue Ser at position 7 of chain A in buIL-17A was found to have h-bond interaction with Asp 15 of B chain during dimer formation. However, substitution of Ser with another polar residue Asn in hsIL-17A does not show h-bond interaction, which is probably due to either minor structural deviation or they might not be captured in that form due to their dynamic motion.
The residue, Asn 25, of chain A found to form h-bond interaction with Ser 107, Phe 108 (in human: Ser 109 and Phe 110) of chain B and the reverse was also true in dimer, and interestingly, these residues were observed to be conserved across the species through sequence analysis. However, in case of human, Asn 27 instead of Asn 25 was found to have interaction with Ser 109 and Phe 110. Similarly, Gln 92 of β3-strands of chain A was forming h-bond interaction with Glu 93 of β3-strands of chain B and vice versa during dimer formation; however, in human Asn 32 instead of Gln 94 of chain A was found to have interaction with Glu 95. The amino acid Glu 93 (in human: Glu 95) was found to be conserved across the species.
The 7uwn crystal structure consists of hsIL-17A and its reported receptors, human IL-17RA and human IL-17RC. The buIL-17A dimer was superimposed on to the hsIL-17A dimer. After superimposition of the dimer model of bovine IL-17A on 7uwn crystal structure, hsIL-17A dimer was removed. However, other portion of the crystal structure such as human IL-17RA and human IL-17RC was retained to see interaction of buIL-17A with human IL-17RA and human IL-17RC (Fig. 7). The binding free energy value of the whole complex (buIL-17A dimer with hsIL-17RA) was calculated to be −28.8 kcal/mol (Krissinel and Henrick, 2007).
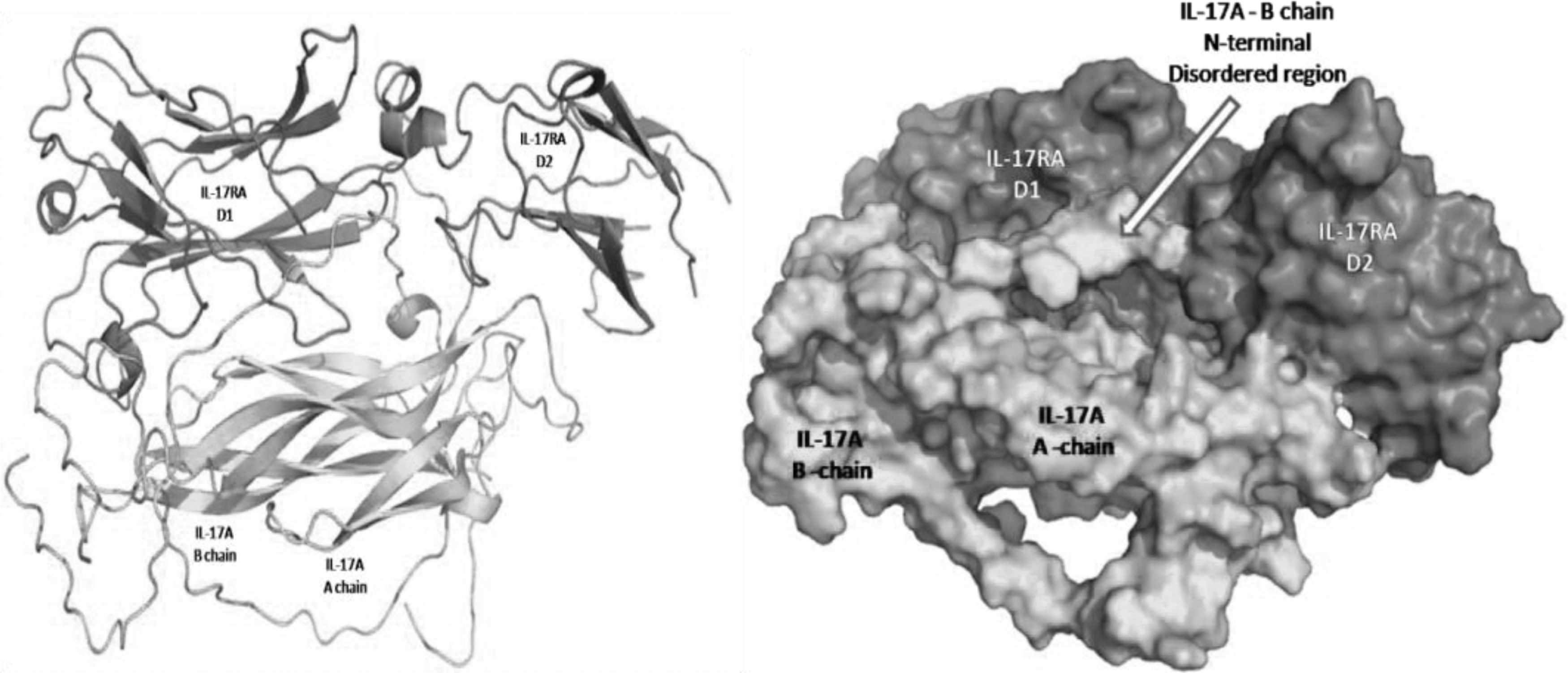
buIL-17A crystal structure and its interaction with reported receptors (IL-17RA and IL-17RC) in human.
Physiochemical properties of buIL-17A
Physiochemical properties calculated using Prot-Param prediction server were shown in Table 3. The negative GRAVY value of this protein indicates its nonpolar nature consisting of more hydrophobic residues indicating its secretory nature. pI value is at 9.4. The theoretical pI indicates that buIL-17A is basic in nature which is useful for wet laboratory extraction (through chromatographic methods). The half-life of buIL-17 was 30 h (mammalian reticulocytes, in vitro), >20 h (yeast, in vivo), and >10 h (E. coli, in vivo) assumed as the time taken for half of the amount of protein in a cell to disappear after its synthesis in the cell. This will be helpful in the estimation of the residual time of this protein after its expression in any system.
Physiochemical Properties of Buffalo Interleukin-17A
EC, extinction coefficient; GRAVY, grand average hydropathy.
Discussion
Discovery of IL-17A role in autoimmune diseases, inflammation, and host defense directed us to use this molecule in humans, as well as in animals, for therapeutic purpose (Patel et al., 2013). It has already been approved for treatment of psoriasis by FDA in 2015 (Beringer et al., 2016; Toussirot, 2018). In humans, role of IL-17A has been explored in many diseases like asthma, chronic obstructive pulmonary disease, host defense, and also in cancer (Chen and Kolls, 2017).
In case of animals, characterization of IL-17A gene has been performed in many species like chicken, duck, sheep, and cattle. However, in buffalo, which is one of the major milch animals in India, no molecular study has been performed. Hence, in the present study, we have sequenced the IL-17A gene from Indian Murrah buffalo (GenBank Accession No.: OM524556). Cloned gene sequence in expression vector was further expressed and purified in denaturing conditions. Furthermore, in-silico analysis confirmed its suitability to use it as immunobiological reagent for diagnosis and therapeutic purposes.
Sequence analysis revealed the characteristic feature of amino acids. The length of putative orthologous protein sequences is 132 amino acids after removing the signal peptide, which is as same as in cattle sequence. The predicted secondary structure is similar to hsIL-17A crystal structure available at protein data bank, 7uwn, 5n7w.
MSA for sequences of various species gives a clue about the conserved and varying residues across the species. The residue Ser 7 was observed to be conserved in rat and mouse despite its conservation found in the species present in the order Artiodactyla; however, Ser 7 was replaced by Asn in hsIL-17A. The residue, Asp 15, was observed to be conserved in almost all the species except mouse, rat, and guinea pig. The amino acid Asn 25 was found to be important in making hydrogen bond with the residues present at β4-strand. According to the crystal structure report, the amino acids 19–28 were reported to form β-strand in the consistent disordered N-terminal region and form anti-parallel β-sheet with another chain.
The residue Asn 25 was observed to be conserved in all the species except rabbit, where it was replaced by Ser. Two of the Asn 25 interacting residues in the opposite chain, Ser 107, Leu 110 were observed to be conserved almost in all the species except rat and mouse where it was replaced by Ile and Val, respectively. However, another Asn 25 interacting residue, Phe 108, was observed to be conserved in all the species. Another hydrogen bond forming residues from opposite β3-strands such as Gln 97 and Glu 98 were observed to be conserved in all the species. Arg 26, another hydrogen bond interacting residue present at the disordered N-terminal region, was observed to be conserved in all the species except human and camel, where it was replaced by Lys. The Arg 26 interacting residue at C-terminal, Ala 130, was observed to be conserved in several species; however, they are replaced by Val in cheetah, cat, tiger, Gly in zebra, horse, guinea pig, and Ser in Swine and rat.
Based on the analysis, it was observed that residues like Asn 25, Glu 93, Phe 108 found at β0-strands, β3-strands, and β4-strands, respectively, were involved in dimer formation.
The amino acids present at the secondary structural regions are conserved; hence, they are mostly involved in maintaining the secondary structural regions and motifs. The amino acids present at the N-terminal regions are highly varying in nature and are important for maintaining the biological function like dimerization, interaction with its receptor(s). The amino acids in A chain that were found to interact with amino acids from B chain are Ser 7, Asp 15, Val 22, Val 24, Asn 25, Arg 36, Gln 92, Glu 93, Ser 107, Phe 108, Leu 110, and Ala 130. Among them, amino acids that are shown in bold form are found to be conserved with hsIL-17A (Table 2). Superimposition of buIL-17A dimer onto the hsIL-17A dimer and further interaction with hsIL-17A receptors, that is, hsIL-17RA and hsIL-17RC, suggested that the interaction is conserved.
The binding free energy value of the whole complex (buIL-17A dimer with hsIL-17RA) was calculated to be −28.8 kcal/mol (Krissinel and Henrick, 2007) which was higher than the binding free energy of the buIL-17A dimer. The interacting hydrogen bond residues between the buIL-17A dimer and hsIL-17RA were examined and compared with the crystal structure interaction between hsIL-17A dimer and hsIL-17RA. Several hydrogen bond interactions were found to be conserved between both complexes. Residues such as His 84, Asn 86 from A-chain of bovine IL-17A and Arg 44, Arg 53, Val 63, Trp 65 from B-chain of buIL-17A, and Trp 31, Asn 89, Asp 262 from hsIL-17RA were observed to preserve the hydrogen bond interaction as noticed in the crystal structure, 7uwn. Human IL-17RC structure, which was retrieved from the crystal structure, 7uwn, was bound to A-chain of hsIL-17A in 7uwn. Similarly, it was incorporated to bind with A-chain of buIL-17A. So the buIL-17A dimer is found to have human IL-17RC on one side and IL-17RA on the other side.
This is the first report on molecular characterization of buIL-17A protein. Characterization of buIL-17A protein with different bioinformatics tool suggested that the protein can be further studied for expression and antibodies can be raised for diagnostic purposes in buffalo immunology. In conclusion buIL-17A protein can be used as a potential immunobiological tool for diagnosis of inflammatory diseases.
Footnotes
Acknowledgment
The authors are thankful to the Head, Department of Veterinary Microbiology, LUVAS, Hisar for providing the necessary infrastructure and facilities to carry out the Research work.
Authors' Contributions
S.K.K. designed the research study and was responsible for funding acquisition; S.B. performed the investigation and the writing of the experimental part; K.K. performed the investigation and the writing of in silico section, A.K., V.Y., and A.K.G. helped with formal analysis of the data and reviewed the article. All the authors have critically reviewed and approved the article.
Disclaimer
The funders played no role in the study design, data collection or analysis, the decision to publish, or article preparation.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The work was supported by Indian Council of Agricultural Research, New Delhi, India under-Emeritus Scientist scheme (5531-C(b)-Micro-08(ES)-ICAR).