
Abstract
Herein, we highlight the significance of molecular modeling approaches prior to in vitro and in vivo studies; particularly, in diseases with no recognized treatments such as neurological abnormalities. Alzheimer disease is a neurodegenerative disorder that causes irreversible cognitive decline. Toxicity and ADMET studies were conducted using the Qikprop platform in Maestro software and Discovery Studio 2.0, respectively, to select the promising skeletons from more than 45 reviewed compounds isolated from mushrooms in the last decade. Using rigid and flexible molecular docking approaches such as induced fit docking (IFD) in the binding sites of β-secretase (BACE1) and acetylcholine esterase (ACHE), promising structures were screened through high precision molecular docking compared with standard drugs donepezil and (2E)-2-imino-3-methyl-5,5-diphenylimidazolidin-4-one (OKK) using Maestro and Cresset Flare platforms. Molecular interactions, binding distances, and RMSD values were measured to reveal key interactions at the binding sites of the two neurodegenerative enzymes. Analysis of IFD results revealed consistent bindings of dictyoquinazol A and gensetin I in the pocket of 4ey7 while inonophenol A, ganomycin, and fornicin fit quite well in 4dju demonstrating binding poses very close to native ligands at ACHE and BACE1. Respective key amino acid contacts manifested the least steric problems according to their Gibbs free binding energies, Glide XP scores, RMSD values, and molecular orientation respect to the key amino acids. Molecular dynamics simulations further confirmed our findings and prospected these compounds to show significant in vitro results in their future pharmacological studies.
INTRODUCTION
Neurological diseases such as Parkinson, Alzheimer, multiple sclerosis, migraine, stroke, brain injuries, spinal cord trauma, and epilepsy are the second-leading causes of death worldwide after heart diseases. 2 They are heterogenous disorders affecting the nervous system leading to a long-lasting disability and/or increased mortality posing huge burden on the global health care system. 2 Till now there is no single medication that can completely eradicate a neurological disease. The currently available drugs can, to some extent, only reduce the severity of the symptoms and enhance the patient quality of life. Therefore, it is noteworthy to search for new chemical entities that can act by different mechanisms and provide better prognosis. Natural products derived from plants, marine organisms, fungi, and bacteria have long been a surplus source of new secondary metabolites. Alzheimer disease (AD) is a neurological disease characterized by progressive deterioration of memory and the loss of a minimum of one other nonamnestic cognitive function that affects the patient socially and occupationally. 3 There are some rare forms of AD that could relatively preserve the memory. The disease affects ca. 24 million persons around the globe, and that number is expected to be doubled every 20 years till 2040. 4 This represented around 70% of all cases of dementia with higher prevalence among the elderly. 3,5 Age is one important risk factor predisposing to AD, suggesting the presence of an age-related physiological process that might be involved in its pathogenesis. The prevalence of AD increased exponentially with age. Hereditary genes could likewise play a role—but with modest contribution—in the transmission of the disease from AD patients to their first-degree relatives. 6,7 Inherited mutations were observed in PSEN1, PSEN2, and APP genes. Carriers of the allelic gene of apolipoprotein E (APOE ε4) were at high risk of AD since it slowed down the clearance of Aβ aggregates. It was observed that increasing level of education could enhance the cognitive reserve which slows down the pathological progression of AD; moreover, administration of anti-inflammatory drugs like NSAIDs showed protective effects against the development of AD. Exposure to head trauma might increase the incidence of neurological deterioration, which raises the susceptibility to AD. Chronic conditions like diabetes, obesity, hypercholesterolemia, and hypertension enhanced the susceptibility to dementia and cognitive dysfunction. 3
Antioxidant diet is largely correlated to disease prevention and treatment. 8,9 Mushrooms display immunomodulatory, antimicrobial, hepatoprotective, anticancer, antioxidant, anti-inflammatory, and neuroprotective effects. 10 As the aging population is rising all over the world, the care about neurodegenerative diseases (NDs) is increasing. The use of diet and nutraceutical approaches to protect from NDs is highly demanded. With a lake of a perfect cure for neurological disorders, the highly nutritious and biologically active mushrooms represent an ideal alternative in the pharmaceutical industry. 11
As the development pathway of new drugs is time and money consuming, there is a directed attention to advanced computational methods that predicts biological activities with low cost and high accuracy. 12 Even though experimental in vitro and in vivo assays are required to elucidate the mechanism in the biological environment, conducting in silico approaches beforehand is largely desirable to reduce costs, evade ethical law problems, and accelerate lead discovery from large chemical libraries; thus, putting more effort to the lead optimization in order to reach the clinical stages.
Reported in vitro studies are limited to neurogenic or antioxidant assays, however, in-depth pharmacological molecular mechanistic studies are lacking. Being expensive and time-consuming, we used structural based molecular docking to provide computational results regarding the binding of a chosen list of chemical molecules in the active pockets of two vital neurodegenerative proteins (i.e., the acetylcholinesterase [ACH] and β-secretase [BACE1]). ACH is a serine hydrolase with a manifested role in the hydrolysis of acetylcholine in the brain cholinergic synapsis while BACE1 is an aspartic protease facilitating the amyloid cascade through the cleavage of Aβ amyloid protein. The inhibition of these two enzymes could ameliorate the symptoms of cognitive decline and brain deterioration in AD. This study aims to screen mushrooms-driven compounds (isolated over the last decade) with promising neuroprotective profiles to study their effects on acetylcholine esterase (ACHE) and BACE1 by structural based molecular modeling and simulations to recognize their mechanistic steps and to identify lead molecules with high selectivity for AD. Additionally, The absorption, distribution, metabolism, excretion (ADME)/tox prediction and drug likeness was conducted using the QikProp module for the selected candidates to further narrow the hits.
Library selection criteria
Recently, plenty of naturally derived compounds were reported to display neuroprotective effects; especially those obtained from mushrooms (Fig. 1), among them the tri- and meroterpenes namely, ganoleucoins Q and R isolated from Ganoderma leucocontextum, a medicinal mushroom cultivated in the Tibet area of China. 13 Sterols isolated from the East Asian edible mushroom Hericium erinaceus 14 along with cyathane diterpenes and drimane sesquiterpenes from the basidiomycete culture Cyathus africanus 15 displayed neuritogenic properties by increasing the number of neurite bearing cells. Erinacines and hericenones present in Hericium mushrooms 16 contributed strongly to their neuroprotective properties during ischemic injury by reducing inflammation 17 and suppressing amyloid deposition. Similarly, the isocoumarin compound “phellxinye A” obtained from the Chaga mushroom, displayed significant neuroprotectant effects. 18 Ergothioneine is a sulfur-containing histidine derivative present in mushrooms and possessed several health promoting effects like antioxidant, anti-inflammatory, antiaging 19 and is used in many parts of the world as a dietary supplement. 20 The extract of Pleurotus giganteus containing standardized amounts of uridine (1.8 g uridine/100 g extract) prolonged the survival and cell viability of neuroblastoma cells after 48 h of pretreatment. 21 Resorcinol derivatives like xylarinigs A-C isolated from the medicinal Chinese mushroom Xylaria nigripes protected PC12 cells from damage induced by the shortage of oxygen and glucose. 22 Pistillarin, a catechol-based siderophore produced by terrestrial and marine fungi, is a strong antioxidant with potent lipid peroxidation inhibitory effect. 23 These antioxidants are neuroprotectants owing to their ability to reduce the accumulation of reactive species that could lead to neuronal cell death and neurodegeneration. 24,25 A group of isoindolinons, namely daldinans, produced by Daldinia concentrica revealed strong antioxidant and neuroprotective activities. 26 Selinane-type sesquiterpenoids like epi-guaidiol A isolated from the edible mushroom Termitomyces albuminosus displayed dose-dependent anticholinesterase activity. 27 The dictyoquinazols (quinazoline alkaloids derived from Dictyophora indusiate) protected the cortical neurons of mice from the excitatory reflex induced by glutamate and NMDA. 28
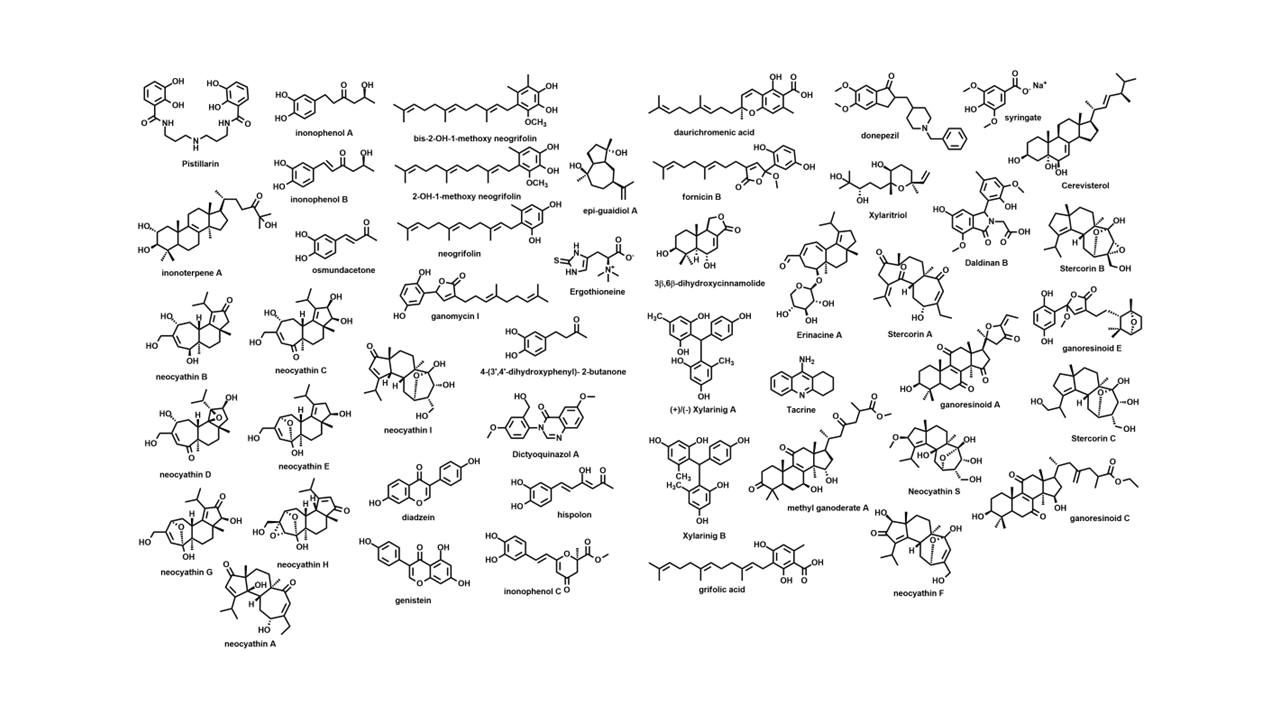
Selected compounds library isolated from mushrooms.
Based on the amyloid cascade hypothesis, inhibition of Aβ peptides aggregation and neuron deposition represented a plausible strategy to combat AD. From the edible mushroom Albatrellus yasudae, 2-hydroxy-1-methoxy neogrifolin, bis-2-hydroxy-1-methoxy neogrifolin, grifolic acid, neogrifolin, 2-hydroxy neogrifolin, and daurichromenic acid were screened for their Aβ aggregation inhibitory effects with IC50 values between 12.3 and 29 µM. 29 Cyathane diterpenes with the unusual 5,6,7-tricyclic structure inhibited the expression of the iNOS and COX-2 to different extents. Activation of microglial cells in the brain could induce iNOS as well as the cyclooxygenase 2 enzymes. Therefore, their mediated inhibition featured a promising strategy to curb neurological injury and inflammation (Fig. 1). 30 Armillaria mellea suppressed the production of NO, IL-6, TNF-α, and other inflammatory mediators in LPS-induced microglial BV2 cells and likewise inhibited phosphorylation of signaling pathways like JNK, MAPK, and NF-κB. 31
Lanostane triterpenes and meroterpenoids isolated from the fruiting bodies of the edible mushroom Ganoderma resinaceum showed IC50 values between 5.41 and 8.91 μM. Ganoresinoid A reduced mitochondrial membrane protein, improved the LPS-induced apoptosis, and controlled the inflammatory mediators and phosphorylation pathways like MAPK and TLR4/NF-κB. 11 Similarly, the new cyathane diterpenes from Cyathus stercoreus and the drimane sesquiterpenes, stercorins D and E, manifested remarkable inhibition of LPS-induced NO production in BV2 cells. The uncommon 9,7-ring in stercorin A was shown to be vital in suppressing NO production and enhancing neurite growth. 32 The basidiomycete Inonotus hispidus showed anti-inflammatory, antioxidant, and neural enhancement activities by diminishing NO generation in the LPS-activated BV2 microglial cells and blocking the expression of the (NF-κB) signaling pathway. Inonophenols B and C induce P-12 cells growth and suppress the expression of TLR-4, NF-kB, COX-2, and iNOS in LPS-induced BV-2 microglial cells. 33 Bioactive cyathane terpenoids, erinacines, and their derivatives, isolated from Hericium erinaceus and H. flagellum, stimulated the brain-derived neurotrophic factor. 34 A total of 47 compounds were selected to conduct the in silico study. They were filtered by similarity check (Figs. 2 and 3), Qikprop, and ADMET properties to exclude 33 compounds whose absorption profile and drug likeness were not favored and limit our next steps to only 14 compounds (Table 1), namely fornicin B, ganomycin I, dictyoqinazol A, genestin, daidzein, hspolon, epiguadiil A, xylainig A, daldinan B, inonophenol A and C, stercoin A, erinacine A, and neocanthin B.
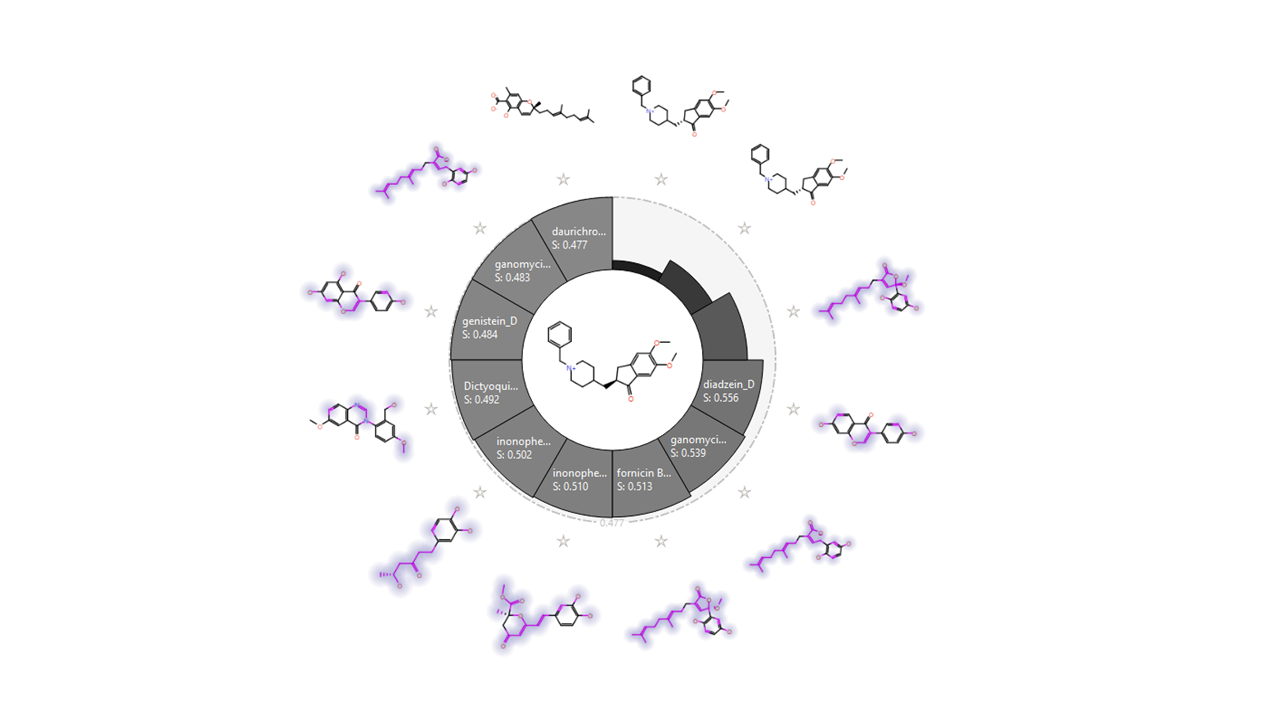
Similarity chart developed through activity miner platform of the selected compounds compared with donepezill native ligand.
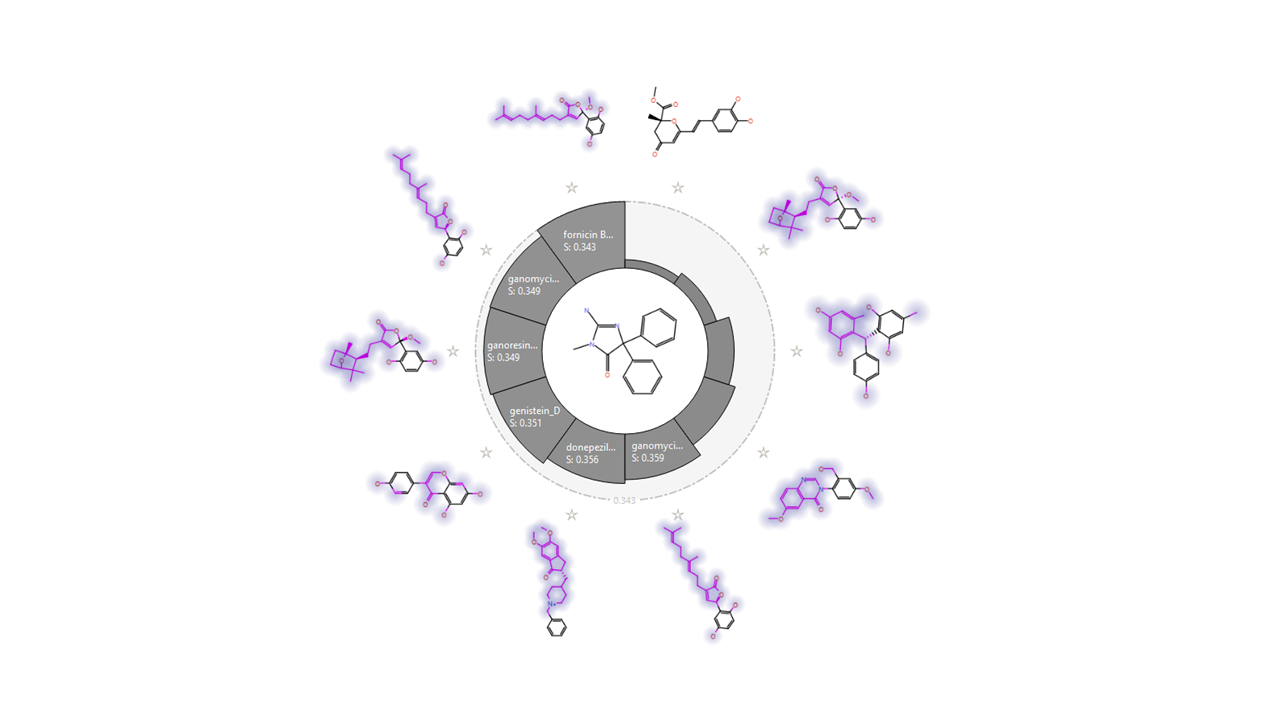
Similarity chart using the BACE1 native ligand as a reference with best similarity compounds compared with OKK native ligand. BACE1, β-secretase; OKK, (2E)-2-imino-3-methyl-5,5-diphenylimidazolidin-4-one.
Predicted Lipinski’s Rule Parameters for the Chosen Compounds’ Library
The excluded compounds were: hericenone C, phellxinye A, N-omega-propyl-
HB, hydrogen bond.
MATERIALS AND METHODS
Protein structure and ligand preparation
The high-quality crystallographic structures of ACHE (Pdb ID:4ey7) and the BACE1 (Pdb ID:4DJU) were chosen for this study. The recombinant human acetylcholinesterase protein with its cocrystallized ligand donepezil (E20) and the structure of the aspartic protease BACE1 bound in complex with 2-imino-3-methyl-5,5-diphenylimidazolidin-4-one (Pdb ID:4DJU) were retrieved from the Protein data Bank (https://www.rcsb.org/). Maestro protein preparation wizard was utilized to optimize the structures by adding hydrogens and correcting residues and loops. Water was kept due to their important role in several interactions. The ionization status was adjusted to pH 7.4 before the optimized preprocessed structures were energy minimized using OPLS-3e force field. 35
Receptor grid generation and ligand preparation
Donepezil (E20) and (2E)-2-imino-3-methyl-5,5-diphenylimidazolidin-4-one (OKK) as the native ligands were picked to define the position and size of the binding site where the docking interactions are performed and calculated using the Glide Receptor Grid generation tool in Maestro. The selected 14 ligands were energy minimized by employing OPLS-3e force field in 2500 iterations at low-energy 3D format using the Ligprep tool.
Molecular docking simulations
Ligands were filtered through the Glide docking tool embedded in Maestro to assess the receptor binding affinity of each ligand to the receptor based on the extra precision (XP) criteria. The glide XP docking score is calculated through an “XP PoseRank” property, governing the way of selecting single ligand poses, and using the following equation considering the Emodel and GlideScore:
The default settings were applied in the grid generation as van der Waals (vdW) factor (1.00), forcefield of OPLS-3e, and a control factor of 0.25 for the workload. A standard grid box with dimensions 14 Å × 14 Å × 14 Å was sufficient for the ligands to fit in. The best poses were selected depending on each ligand’s best docked orientation that provide the lowest Gibbs free energy or donated by glide score. System validation was carried out by redocking the two cocrystallized ligands into their primary protein pockets to measure the RMSD values which revealed to be 2.380 for 4DJU and 0.7040 for the 4ey7. Values of RMSD (measured between the reference ligands before and after docking) below 2 indicated accuracy and precision of the docking protocols used.
Induced fit docking (IFD) procedures
The first step of the protocol was to employ the softened potential Glide in IFD of the selected compounds panel to 4ey7 and 4dju, which accounted for the vdWs radii scaling of 0.7/0.5 between the receptor and ligand, respectively. The achieved poses were arranged and ranked, and the best 20 poses were docked against the protein to model its plasticity using the Prime module of Schrodinger suite 2018. To refine the receptor different conformations and consider its flexible conformations and sample them, residues within the range of 5A of any ligand atom of the top selected 20 ligand poses were searched for their conformational flexibility and energy minimization. Residues lying outside this range were energy minimized on the ligands. Ligands with the lowest energy of around 30 kcal/mol were chosen as template for redocking. All subsequent docking calculations were managed by the Glide SP. The final top poses were produced using a combination of scores developed from both the Glide score functions and prime.
Prediction of the pharmacokinetic properties of the selected skeleton molecules
QikProp wizard tool in Schrodinger was applied (QikProp Schrödinger, 2019) to filter out ligand candidates with high failure probability in clinical studies. Various physicochemical and pharmacokinetic properties were predicted for compounds with top G-glide scores. QPlogS acts as the solubility indicator while QPlogPo/w denotes the ratio between octane to water solubility of molecules; thus, showing the lipophilicity index of ligands. Drug likeness was estimated based on Lipinski’s rule of five depicting the favorable features of drugs such as number of hydrogen bonds (HBs) (≤5), lipophilicity (≤5), molar refractivity (40–130), molecular weight (≤500), and number of H bonds acceptors (≤10). Desmond was employed to conduct molecular dynamics (MD) simulations lasting 100 ns for selected compounds. 36 Protein and ligand complexes were submitted to MD simulations to test their stability. The stability of complexes was assessed through MD simulations, following a series of steps, including preprocessing, optimization, and minimization. The OPLS_2005 force field was utilized for the minimization process. 37 The compounds were solvated in a periodic box with a 10 Å size containing the TIP3P water molecules. 38 Neutralization of the systems was done by adding counter ions and 0.15M NaCl salt as needed to mimic physiological conditions. The NPT ensemble was set to a temperature of 300 K and a pressure of 1 atm. Prior to simulation, systems underwent a relaxation phase. Trajectories were recorded and saved at 40 ps intervals during the simulation, enabling subsequent analysis of the obtained results.
RESULTS
Pharmacokinetics and drug likeness prediction
Before conducting molecular docking experiments, it was necessary to filter out molecules that are of unsuitable ADME, drug likeness, and bioavailability properties.
QikProp descriptors were accurately computed based on molecules’s 3D characters aside from compounds’ nature whether they are previously reported or not (Table 2). These properties were measured to indicate molecules whose values lie within the 95% range of known drugs. In this study, seven compounds were omitted from the list based on the number of stars indicated. The stars index measured the drug likeness and the pharmacokinetics in the 95% range of known drugs depending on 25 properties to be inferred.
QikProp Characteristics for the Chosen Compound’s Library
Compounds excluded were hericenone C, phellxinye A, N-omega-propyl-
Among them was Lipinski’s rule of five to assess if the chosen molecules followed specific parameters: not >5 HB donors, not >10 HB acceptors, molecular weight is up to 500 Da, and a partition coefficient not >5. Jorgensen’s rule of three was employed to measure the bioavailability of the chosen compounds through predicted solubility, liver first-pass effect, and permeability by calculating the aqueous solubility (logSwat >5.7), Caco-2 cell rate permeability (BIPcaco-2 >22), and number of the metabolic products (cannot exceed 7). Furthermore, human oral absorption 2 –4 and skin permeability characteristics (LogKp) were considered before shortlisting lead compounds. The number of discarded compounds was indicated in Tables 3 and 4. The blood–brain barrier (BBB) absorption coefficient indicated by QPlogBB was ranked from −3.0 to 1.2.
ADMET Descriptors for the Chosen Compound’s Library
BBB, blood–brain barrier.
TOPKAT Toxicity Prediction of the Selected Compound’s Library
Molecular docking interactions in acetyl cholinesterase (4ey7) binding pocket
Molecular docking is an indispensable tool to investigate the interactions between ligands and their putative targets. 39 The ACHE (4ey7) active pocket was prepared using Glide program embedded in Maestro software with default setting parameters. All the selected ligands were docked into the 4ey7 protein compared with the native ligand donepezil (E20).
Various inhibitors could adopt three anchoring regions in the 4ey7 binding site, the catalytic anionic site (CAS) (Trp86 and Phe338), catalytic triad (CT) (Ser203, Glu334, His447), and the peripheral anion site PAS (Tyr72, Tyr124 and Trp286). Furthermore, an L-shaped position could be assumed in the entrance spot called the inward–outward orientation. X-ray crystal structure of 4ey7 with the native ligand donepezil was downloaded and prepared prior to conducting molecular docking whose resulting poses were filtered by free-energy binding scores, Glide XP scores, and EC scores. In this enzymatic groove, water molecules played a crucial role affecting the strength of binding energies; particularly, residues Phe295, Tyr121, Tyr341, and Tyr337 that facilitated the native ligand ring flip and the feasibility of several HBs for binding interactions, rarely to be manifested without it, therefore, water molecules were kept during docking. 40,41
IFD in acetyl cholinesterase enzyme (4ey7) binding pocket
Based on their free binding energies, and interactions, top 14 compounds were selected to undergo the IFD, which conferred optimization of the binding interactions between ligands and their active sites through flexible sampling of the protein side chains.
IFD manifested fornicin with an XP Glide score of −10.885 kcal/mol to bind to two anchoring regions in the 4ey7, the CT region represented by HBs to Ser203and Glu202 and the PAS region represented by HBs to Tyr133 and Ser125. Nevertheless, the least steric clashes were revealed by fornicin and the methoxy group in the five-membered lactone ring, which deposited positive electrostatic potential that might suggest the need to introduce some chemical modifications here to impart more stability. Therefore, ganomycin demonstrated a double π–π stacking interaction between its phenolic ring and Trp86 at distance of 3.89 and 4.17 Å analogous to that formed in the standard drug donepezil. HBs were noted between the hydroxyl groups and Tyr124 mediated through water molecules and with Glu202 at 1.41 and 1.62 Å, respectively. The carbonyl group was close to the key amino acid Tyr341 to conduct another HB at 1.89 Å (Figs. 4 and 5) (Table 5).
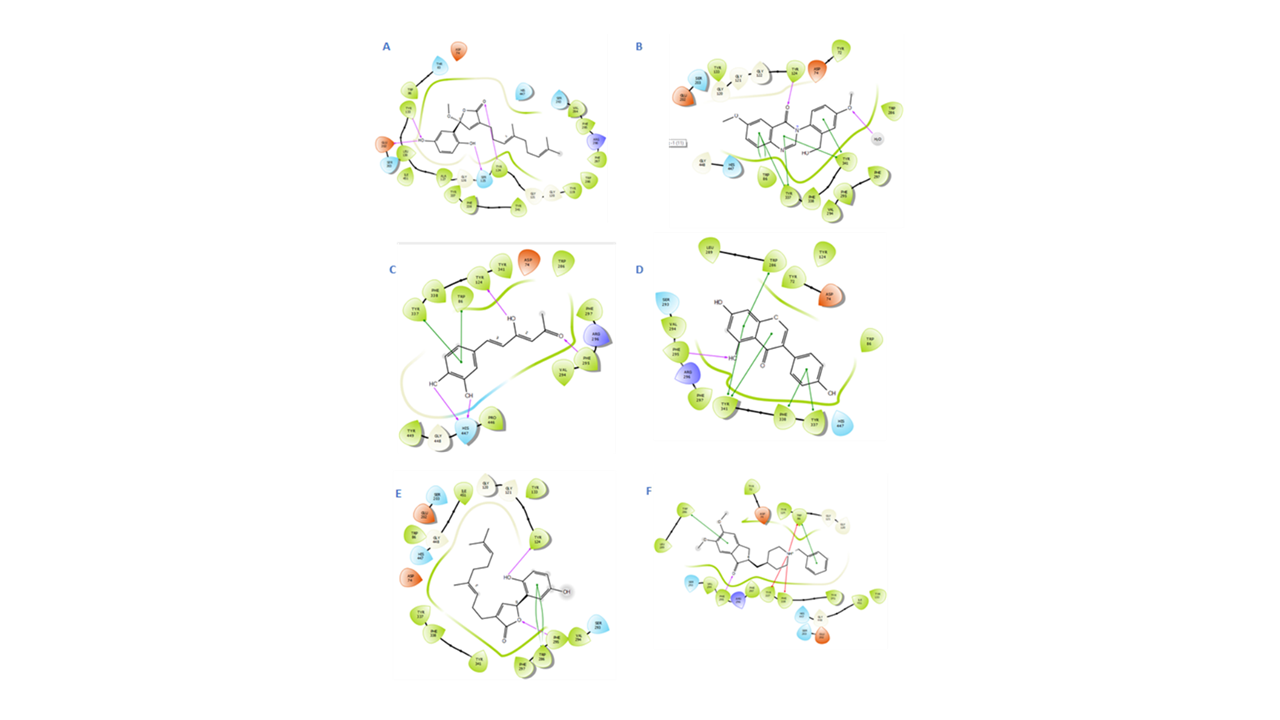
2D IFD interactions in the ACHE binding site:
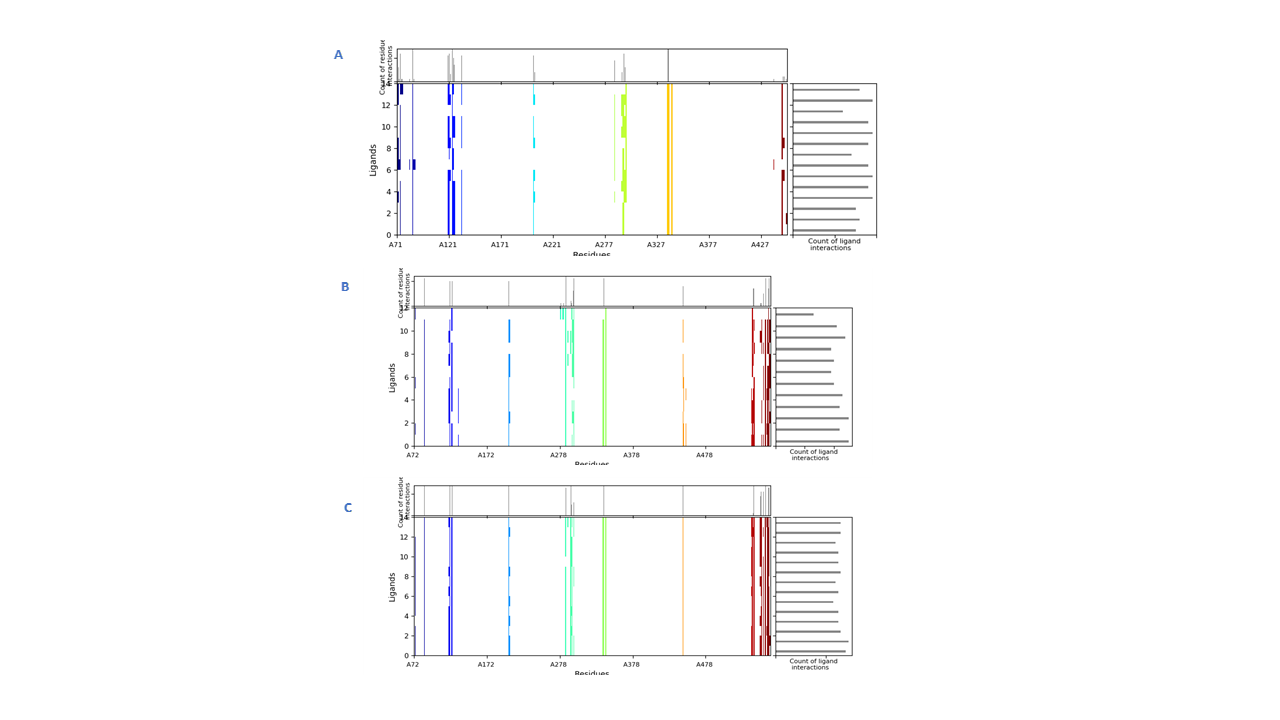
Interactions fingerprints of IFD 20 poses in ACHE binding site:
Scores and Binding Interactions of the Top Selected Compounds Using Induced Fit Docking in the BACE1 Binding Site
IFD, induced fit docking; OKK, (2E)-2-imino-3-methyl-5,5-diphenylimidazolidin-4-one.
Dictyoquinazol A revealed the second best stable GlideXP score of −11.058 kcal/mol, which could be explained through clear interactions with Trp286, Tyr337, and Tyr341 as π–π stacking extending from the quinazolin-4-one ring and HBs between both the ethoxy and methoxy groups to Phe295 and Tyr124, respectively. Genestin showed the least steric clashes and the top Glide XP score with distances as close as 4.00, 3.58, 3.86, and 4.89 Å to most of the core residues Trp286, Tyr341, Phe338, and Tyr337, respectively.
Even though notable noncovalent interactions were lacking with Trp86, the front door and CT component residue His447 was hydrogen bonded to the hydroxyl group C-4′ in ring B at 2.30 Å. These were matching with the native ligand donepezil contacts with Gly120, Tyr124, Glu202, Ser203, Trp286, Phe295, Tyr337, Tyr341, and His447. Hispolon was hydrogen bonded through its terminal carbonyl to Phe295 at 2.00 Å.
MD simulations
The binding mode of donepezil was subjected to a 200 ns simulation for stability analysis of the complex. RMSD of the C-α atoms of protein and ligand fit on protein was computed to find the stability of the complex. It was observed that the C-α atoms of the protein maintained the RMSD values in the range of 1.8–2.1 Å till 100 ns and then increased to 2.4 Å at 125 ns before maintaining this range toward the end of simulation. The RMSD of the ligand was less than the protein till 50 ns before it aligned on the protein atoms at 75 ns, which indicated the stability of the complex (Fig. 5A). The structural dynamics of the protein residues were analyzed by calculating the RMSF values, which showed the flexibility of the protein residues in response to this ligand binding during simulation. The higher RMSF values showed flexibility and the lower RMSF values showed rigidity of the residues. From the RMSF values, it was observed that most of the residues kept rigidity during the simulation except for the loop regions whose values were 4 Å (Fig. 5B). Important interactions between the ligands and the protein revealed during MD simulation analysis were hydrophobic, hydrogen, and ionic bonds. These interactions were essential for maintaining protein and ligand complex’s stability and controlling its functional properties. The residues involved in hydrogen bonding were Trp286, Phe295, and Tyr337 while Asp74 was involved in ionic interactions (Fig. 5C). Trp86 showed the highest tendency to bind all the interacting residues, with interactions being seen in 60% of the total frames (Fig. 5D).
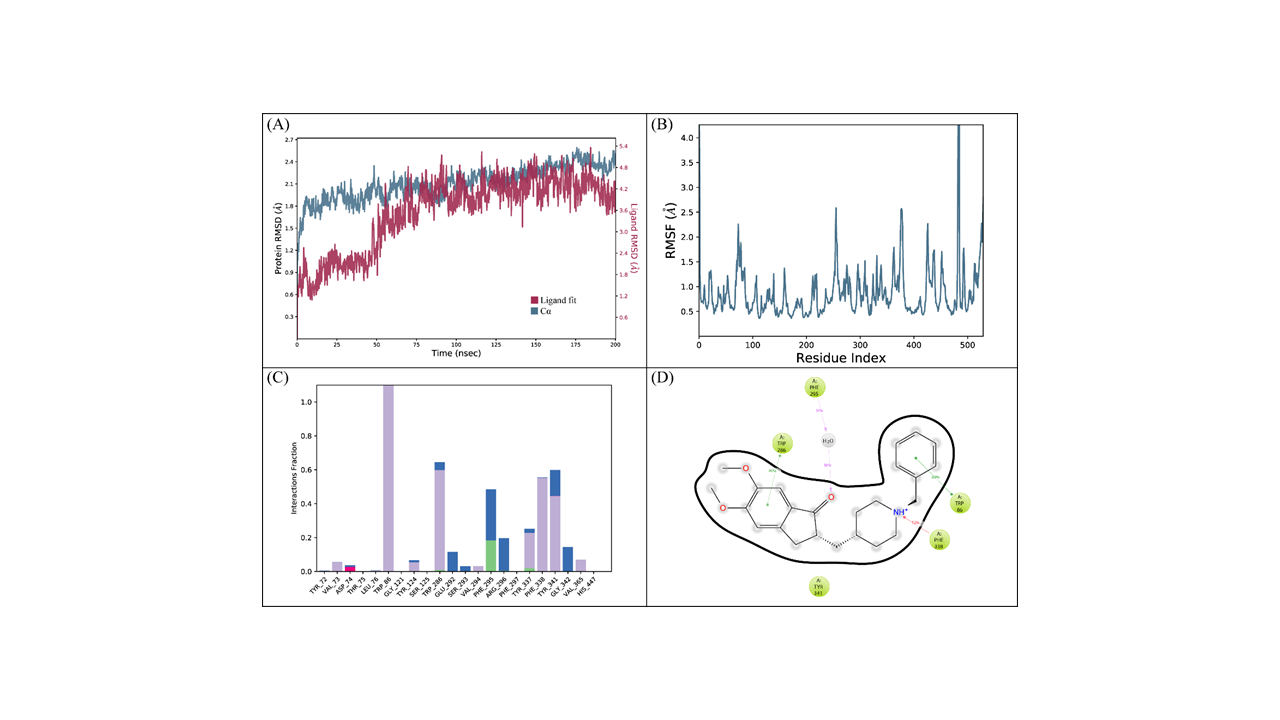
(cont.) The MD trajectory analysis of the ACHE complex with donepezil.
Molecular docking interactions in BACE1 protein binding pocket
The BACE1 played a vital role in the etiological origin of AD, as it was the only enzyme cleaving β-amyloid precursor protein (APP) to yield a 40 to 42 amino acid peptides known as amyloid-β peptides, Aβ, or amyloid plaque. Even though γ-secretase was another protein involved during the processing course of APP, its inhibitors encountered an early failure in their clinical trials. BACE1 is an aspartate protease-like pepsin whose active site comprised mainly two aspartate residues Asp93 and Asp289 with a catalytic water molecule to assist the peptide cleavage. In brief, a nucleophilic attack was initiated toward the carbonyl carbon by the action of one aspartic residue and a water molecule. The carbonyl oxygen was activated by the other aspartate to form a tetrahedral intermediate, subsequently cleaving the amide bond and yielding the products. 42
The conserved binding site in BACE1 was recognized as the catalytic diade formed of Asp93 and Asp289, yet many BACE1 inhibitors located in this site were accompanied by adverse effects. Therefore, allosteric binding sites might provide a better nick for indirect acting BACE1 inhibitors since BACE1 enzyme is endowed by a challenging large enzyme pocket that can accommodate different molecules. The analysis of the docked molecules complexes using Maestro 18.0 software platform manifested three binding models, among which allosteric binding could prove efficacy (Supplementary Table S2).
IFD in BACE1 protein binding pocket
Through IFD approach, the top 14 scored compounds were analyzed to manifest their flexible binding and optimized orientations in the BACE1 binding site. Investigation of different bindings was visualized by interaction finger printing modules, namely 2D and 3D manual inspections. Results showed inonophenol A as the best-scoring compound with Glide XP score of −8.098 kcal/mol. Noncovalent bonding was shown as HBs from the phenolic hydroxyl groups to Asp93 and Asp289 with distances of 1.49 and 1.87 Å, respectively; furthermore, inonophenol A fitted to the SS2 (subsite 2 pocket) as shown by its interactions with Phe169 (4.92 Å), Trp137 (1.72 Å), and Lys168 (1.82 Å) (Figs. 6 and 7).
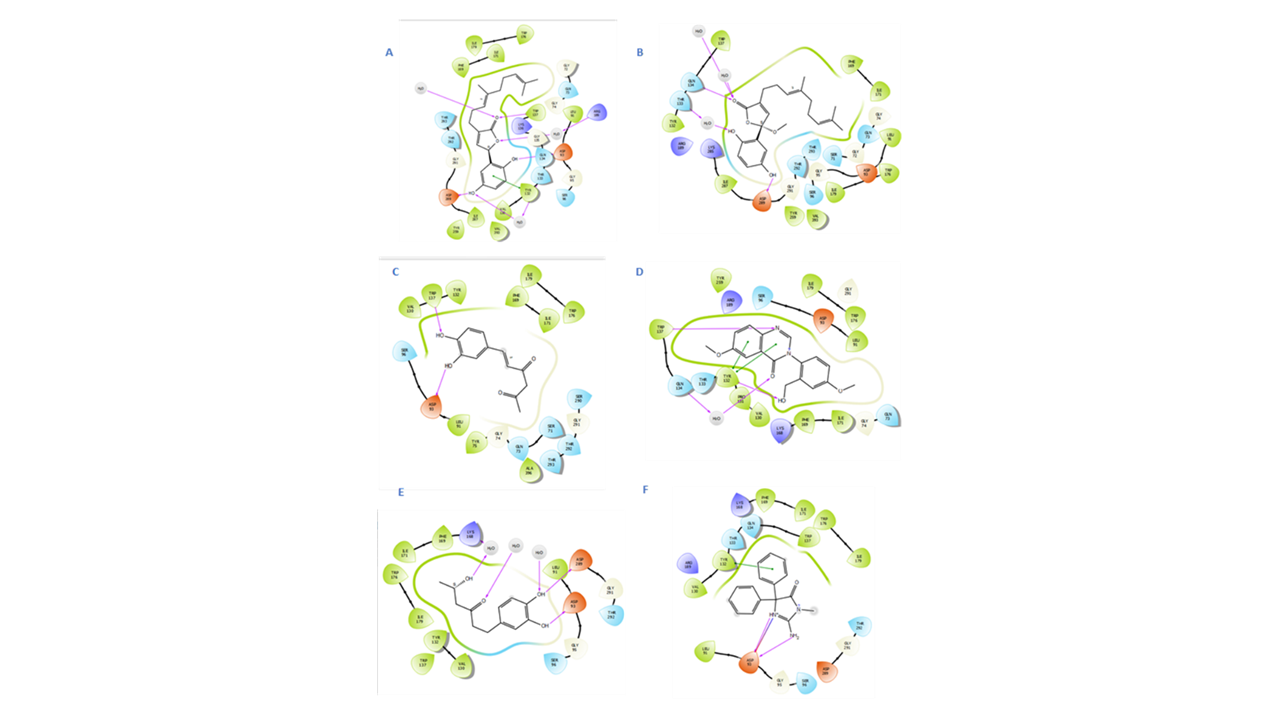
2D IFD interactions in the BACE1 binding site:
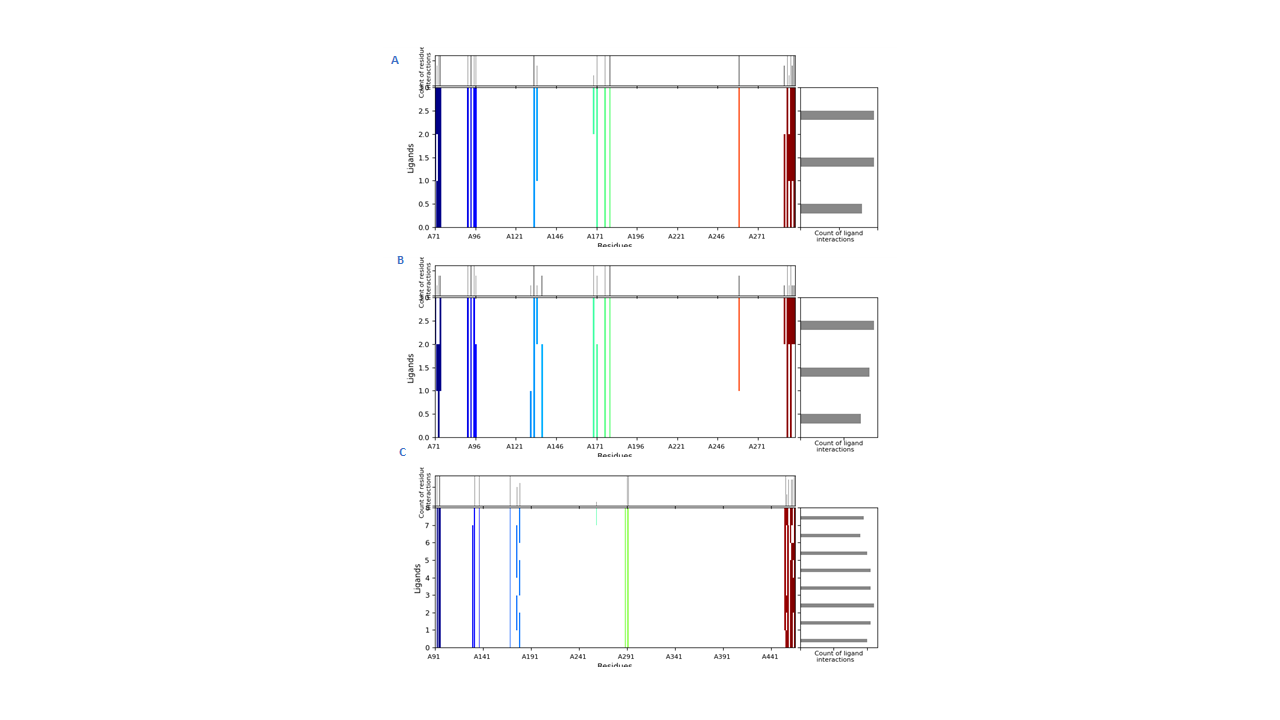
Interactions fingerprints of IFD 20 poses in BACE1 binding site.
Hispolon in the SS1subunit encountered the least steric clashes between its molecules and the active site; thus, rendering better stability and lower free energy. Its binding interactions as shown in the amnio acid fingerprints were as follows: aromatic HB with Trp176 at distance 3.35 Å from the aliphatic carbonyl group, H-bond from Phe169 and Lys168 to the OH group at 2.03 and 2.72 Å, respectively, H-bond from Gly291 at a distance of 2.41 Å to the phenolic OH group, aromatic H-bond from Phe169 to the side chain carbonyl at 4.80 Å, and aromatic H-bond from Trp137 to the side chain carbonyl at 3.24 Å as well as π–π stacking interactions with Tyr137 at 5.11 Å. Hispolon apparently occupied the active pocket in such a way as splitting the nonpolar and polar residues on its two sides, which contributed to its minimal observed steric clashes. Many hydrophobic contacts were also detected to Lys168, Tyr132, Val130, Leu91, and Ile179 (see Table 5 and Fig. 6).
Dictyoquinazol A possessed a quinazolin-4-one ring matching the binding pocket size and facilitating a double π–π stacking from both aromatic rings to Tyr132 at 4.11 Å and HB contacts at 1.96 Å. Aromatic HBs were detected from Asp93 and Trp176 at distances 3.50 and 3.78 Å to the phenyl ring attached to C-3. Contacts with amino acids Leu91, Phe169, Ile171, and Ile179 were also detected through interaction fingerprints.
Ganomycin showed five hydrogen bonding to Trp137 at 1.81 Å, Asp93 at 1.88 Å, Asp289 at 1.82 Å, Arg289 at 2.35 Å, and Thr133 at 2.49 Å; additionally, one π–π stacking with Tyr132 at 5.34 Å was noted with several nonpolar contacts as manifested in the hydrophobic bed by Leu91 Ile171, Ile179. Gly291, Gly95, Trp176, and Phe169.
MD simulations
The binding mode of the inonophenol B with BACE1 was subjected to a 200 ns simulation for stability analysis of the complex. RMSD of the C-α atoms of the protein and ligand fit on protein was computed to find the stability of the complex. It was observed that the RMSD of C-α atoms gradually increased to 2 Å at 50 ns before maintaining this range till 100 ns. After 100 ns, it started to increase and reached 2.4 Å at 125 ns; subsequently, it maintained this range till the end of simulation. While the RMSD of the ligand exceeded the protein till 50 ns, it remained lower thereafter, which indicated the stability of the complex (Fig. 7A). The structural dynamics of the protein residues were analyzed by calculating the RMSF values, which showed the flexibility of protein residues in response to the binding of this ligand during simulation. While high RMSF values showed flexibility, the low RMSF values showed the rigidity of the residues. From the RMSF values, it was observed that most of the residues stayed rigid during the simulation except for the loop regions whose values were 4.5 Å (Fig. 7B). Important interactions between the ligands and the protein discovered during MD simulation analysis were hydrophobic, hydrogen, and ionic bonds. These interactions were essential for maintaining the protein and ligand complex’s stability and controlling its functional properties. The residues involved in hydrogen bonding were Gly72, Gln73, Asp93, Tyr132, Gln134, Gly173, Ser174, Trp176, Gly291, and Thr293. While Gly72 was also involved in ionic interactions (Fig. 7C), Asp93 showed the highest tendency to bind all the interacting residues, with interactions being seen in 98% of the total frames (Fig. 7D).
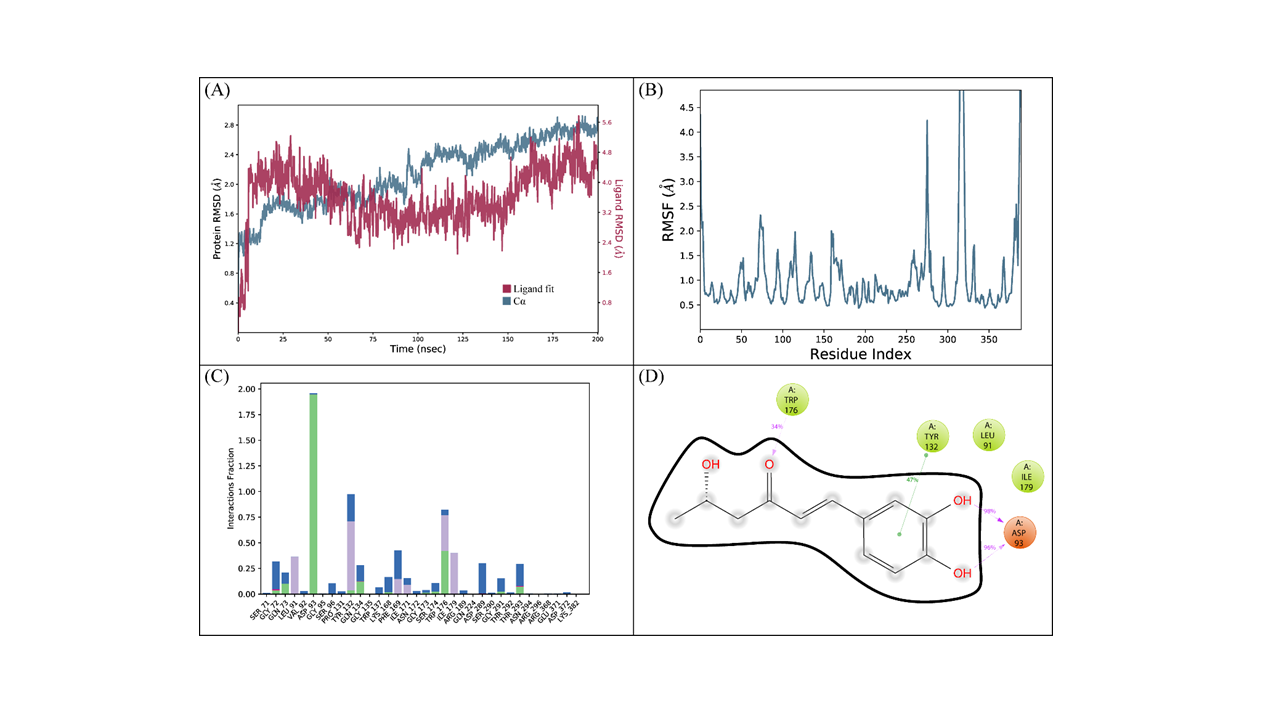
(cont.) The MD trajectory analysis of the BACE1 complex with inonophenol B.
ADMET and TOPKAT analysis
The ADMET analysis showed toxicity, pharmacokinetic and pharmacodynamic properties of the chosen compounds’ skeletons (Tables 3 and 4). Regarding inhibition of P450 cytochrome CY2D6, a few compounds showed binding such as xylarinig A and B, dictyoquinazol A, genestin, fornicin B, ganomycin I, as well as donepezil. BBB absorption was ranked from 0 to 4, signifying the lowest absorption at 4 level and the very high absorption at 0 level. Compounds of interest whose BBB absorption level was outside the 95% eclipse were xylarinig A and B, daldinan B, daurichromenic acid, erincine A, neocanthin A, and fornicin, which suggested the possible medicinal chemist’s work to enhance their nonpolar attributes. Those of moderate absorption lying within the 95% confidence eclipse of BBB absorption were dictyoquinazol A, epiguaidiol A, diadzein, and genstein. With respect to human intestinal absorption, the 99% confidence eclipse encompassed all compounds except eburicol, neogrifolin, and its derivatives, ergothionene, pistillarin, phelixyne, and hericenone (Fig. 8).
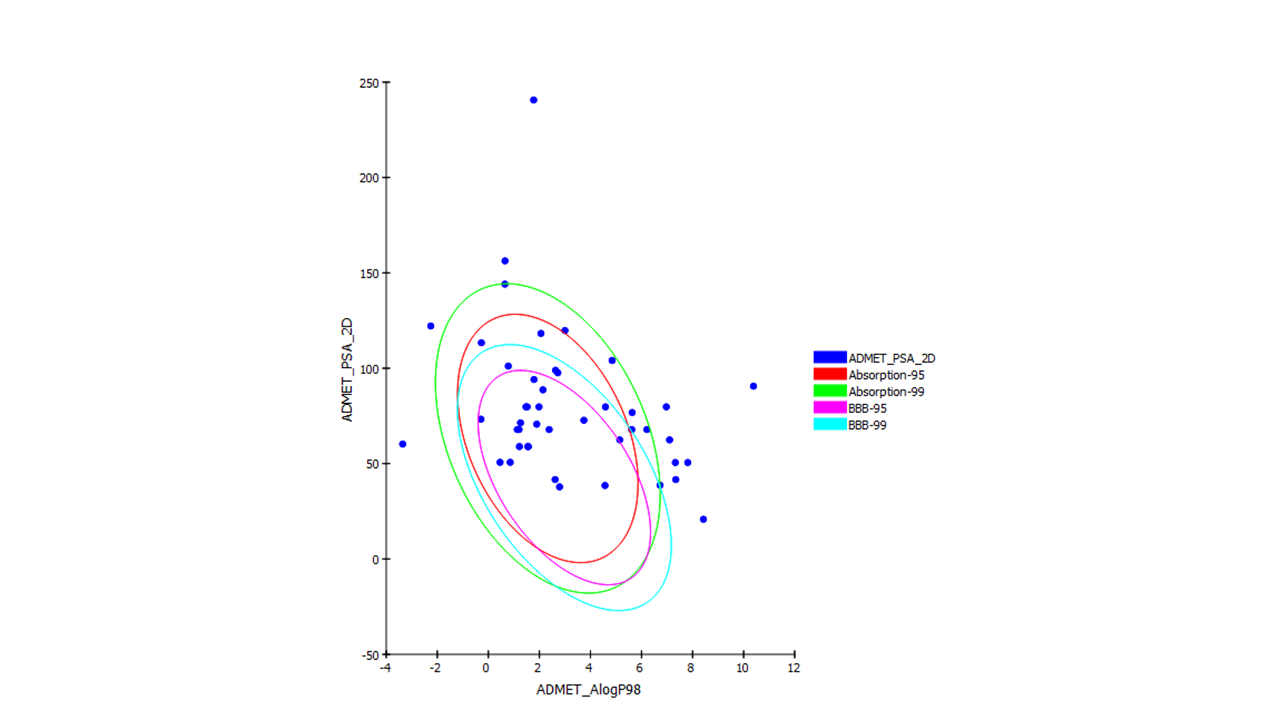
ADMET plot of the screened compounds showing their 95% and 99% confidence limit ellipses with respect to BBB absorption and human intestinal absorption. BBB, blood–brain barrier.
While compounds showing no hepatotoxic adverse effects were epiguaidiol A, daurichromenic acid, ganomycin I, fornicin, and xylaritriol, those with <90% binding to plasma proteins PPB were epi-guaidiol A, dictyoquinazol A, inonophenol A, and hispolon.
Good absorption criteria were manifested by xylarinig A and B, epiguaidiol A, dictyoquinazol A, daldinan B, genestin, ganomycin I, and fornicin using a scale from 0 to 3, which indicated good, moderate, low and very low absorption, respectively (Table 3), whereas solubility levels were seen as moderately soluble (scores 2–3) in xylainig A and B, daurichromenic acid, ganomycinI, forniicn, daldinan B, epiguaidiol a, dictyoquinazol A, and genestin. Hispolon revealed a score 4 indicating its insoluble nature, which could be obviated through suitable formulations to deliver it into the active site. From TOPKAT analysis, most of the potent compounds were of nonmutagenic and noncarcinogenic nature against male and female FDA rats as xylarinig A and B, daldinan B, dictyoquinazol A, daurichromenic acid, genestin, as well as ganomycin I and fornicin (Table 4). The average oral LD50 ranged between 0.78762 and 15.989 g/kg body weight, and the lowest observed adverse effect level was detected between 0.0030 and 0.859 g/kg body weight.
DISCUSSION
Computational studies are important to reveal the binding sites, types of interactions, and even to help determining, in many cases, the unknown targets. 43 The integrated approach of using both in silico and in vitro studies proved success during the previous decade. They were also reported in many combination drug studies to show the effectiveness of various drugs in the same or different binding sites. The revival in natural products research grew in line with the advanced progress in computational approaches. 44 High costs and time consumption associated with the drug discovery timeline, and ethical laws involving the use of animals predisposed the rising interest in in silico studies as the only way to speed up the identification of new targets and leads. Aβ aggregates were in the core pathology of AD, yet their undefined structure and absence of clear binding grooves deferred its study.
Enzymes in the neurodegenerative pathological pathway include β- and γ-secretases whose activity was to generate the Aβ peptides by hydrolyzing the APP. Acetylcholine in the brain served to stimulate both nicotinic and muscarinic receptors relieving memory deficit symptoms in AD, accordingly, ACHE inhibition is a promising strategy to enhance accumulation of acetylcholine. 45 Natural products are at the center of research interest to identify novel molecules, particularly from mushrooms, as they are not subjected to sufficient scrutinization. Besides, unearthing of alkaloids, polyphenols, terpenes, tannins, and saponins with activity in neurodegenerative pathology was hardly experimentally approached to detect their mechanisms of action. 45
Aβ-APPs and their tailoring enzymes directed binding studies were facilitated using computational tools. Experimental binding assays used till now such as thioflavin T-based aggregation kinetics, fluorescence spectroscopy, atomic force of microscopy, transmission electron microscopy, and Western blotting, although available, revealed their drawbacks and costs. 46 Computational strategy is economic, fast, and in most cases, leads to a better drug design experience since the receptor affinity indicators could be used by medicinal chemists to optimize new molecules in a role like what was recognized from the false negative candidates in high throughput experiments, hence, having a higher hit number.
Pharmacokinetics and drug likeness prediction
All compounds showed adequate BBB absorption with more than 90% binding to plasma proteins expressed as the QPlogKhsa coefficient; hence, showing a favorable drug likeness. The human oral absorption score was predicted and expressed as 1, 2, or 3 for low, medium, or high absorption. Most of the studied compounds reported a moderate to high absorption rate except ergosterol peroxide, uridine and bis-2-hydroxy-1-methoxy neogrifolin, which reported poor absorption. The solubility score indicated by QPlogS was in the range from −6.5 to 0.5 and encompassed most of the compounds except for ergosterol peroxide, uridine, bis-2-hydroxy-1-methoxy neogrifolin, and vanillic acid. Toxicity prediction was assessed using the Ame’s Prediction wizard tool in Discovery Studio 2.0.
Molecular docking interactions in acetyl cholinesterase (4ey7) binding pocket
The top nine scoring compounds were daurichromenic acid, ganomycin I, fornicin, genistein, stercoin A, daldinan B, erinacine A, xylarining A, and epi-guaidiol A whose Gibbs free energy ranged between −12.261 and −8.178 kcal/mol. The binding modes were illustrated in Supplementary Table S2 showing that genistein and ganomycin I were bounded in a highly resembling orientation to donepezil. The Glide XP-score and EC scores were used to pick the most favorite pose. With a free binding energy of −9.642 kcal/mol, donepezil showed key SAR of 4ey7 enzyme. The hydrophobic residues in the CAS of the binding pocket featured pronounced importance; for instance, Phe338, Trp86, and Tyr337 as evidenced by donepezil E20 where three π–cation interactions were seen from each of these three residues to the quaternary nitrogen in the piperidine ring. Trp86 extended π–π stacking with the terminal benzyl group of E20 adding more stability to the binding location. Five HBs were also mediated and enforced by water bridging with Tyr341 and Tyr337 within distances in the range of 1.86–2.28 Å. On the contrary, the proximal part of the groove manifested another π–π stacking between Trp286 and benzofuranone. It is worth mentioning that the binding scores were not the only criteria determining the effective fitting of the ligands but HBs, their number, lengths, angles, vdW forces, nature of residues, and positioning inside the pocket can either reward or penalize the energetic cost (Supplementary Table S2).
Genistein with a Glide g-score value of −8.17 kcal/mol formed two π–π stacking interactions with Tyr341 and Trp286 through ring A and B, respectively. Water bridging facilitated interaction with Ser293 at 2.1 Å, perhaps contributing to its favorable ΔG value. In dictyoconazol A binding with 4ey7, a ΔG of −7.763 kcal/mol was calculated by Maestro Glide algorithm where the quinazolinone ring formed π–π stacking with Tyr341 at 3.52 Å.
The aminated phenyl moiety extended π-stacking of length 3.84 Å to Trp286, and further water bridging was seen to the methoxy group rendering more stability to the compounds fitting. Additionally, HBs were formed between the OH group of phenyl methanol to Phe295 and Ser293, yet the latter involved water bridging interaction.
Ganomycin I satisfied the size condition but lacked the appropriate functional groups to interact with the key amino acids. Only the lactone ring formed HBs with Phe295 in the hydrophobic neck of the site, and the hydroxyl group acted as hydrogen donor with Tyr341 through distances of 1.84 and 2.06 Å, respectively. The Glide XP algorithm revealed two π–cationic noncovalent interactions between the daidzein benzopyrone aromatic moiety and Tyr341and Trp286 with a length of 3.95 and 4.16, respectively. The phenyl ring, although revealed no direct receptor interaction, probably contributed to the vdW forces with the hydrophobic bed residues and could assist in lead optimization later.
The four compounds epi-guaidiol A, erinacine A, daldinan B, and genistein interacted with one of the CT amino acids, namely, Ser203, his447, and Ser293, with distances of 1.8, 2.6, 2.2, and 2.1 Å, respectively. All nine molecules showed hydrophobic contacts with the catalytic anionic binding site CAS with at least one of the key residues in the peripheral binding site PAS comprising the hydrophobic bed.
Xylaring A scored −8.284 kcal/mol while the methoxylated phenolic rings aligned well with the donepezil benzyl and piperidinyl rings formed aromatic-aromatic interactions with Trp86 and His447, the third one extended to interact through hydrogen bonding with Asn 87 at 1.9 Å. Another hydrogen bonding contact was detected with Tyr124 (Supplementary Table S1). Epi-guaidiol A with a ΔG value of −9.417 kcal/mol fitted well in the binding spot between the two benzyl and piperidinyl rings of the native molecule forming hydrogen binding to His447 at 2.0 Å, apparently blocking the binding site due to its small molecular size. The EC score of epi-guaidiol was 0.259, which was even better than the native ligand. Daurichromenic acid showed size similarity to donepezil and protracted through the binding pocket over the same axis except that its aromatic moiety lied in the opposite direction to the donzepil’s bicyclic end.
With a Gibbs free energy of −11.892 kcal/mol, daurichromentic acid revealed significant contacts with hydrophobic residues as Phe338, Tyr341, Trp286, Tyr337, and HBs with Ser125 and Tyr133 at 1.9 and 1.8 Å, respectively. π–π interactions were shown between Trp86 and the terminal carboxylated aromatic ring in the binding ligand.
Neocanthin B showed a marked transverse position at the entrance of the 4ey7 groove where it blocked the pocket through ionic interactions with Phe295, Arg296 and Trp86 at distances as close as 2.0, 2.5, and 3.8 Å, respectively. Fornicin extended throughout the whole pocket with noncovalent interactions with Tyr341 and π–π interactions with Trp286 in the peripheral anionic site PAS giving rise to its favorable free binding energy of −10.099 kcal/mol. Both stercoin A and daldinan B featured a back rear orientation blocking the exposure to the CT residues Ser203 and His447. Erinacine A stood at the front entrance of the groove and displayed hydrogen bonding to Try124 and Tyr72 at 1.8 and 2.7 Å, respectively, from the hydroxyl group in its sugar moiety while the cyathane seven membered ring encountered nonpolar interactions with Trp286.
The electrostatic complementarity EC score was measured for all the molecules and revealed effectiveness as an activity indicator since it matched in many cases with free binding energy results. Although not the only criteria predicting activity, this score was calculated through a mixture of complex dielectric constants and formal charges as an accurate index regarding the electrostatic potential similarity between the ligand and protein contact surface inside the binding pocket.
IFD in acetyl cholinesterase enzyme (4ey7) binding pocket
Through flexible sampling of the acetyl cholinesterase protein side chains, fornicin B demonstrated binding to the CT region and the PAS via HBs to Ser203, Glu202, Tyr133, and Ser125, respectively, with an XP Glide score of −10.885 kcal/mol.
Some steric clashes were noted due to the electrostatic forces of the fornicin methoxy group.
Ganomycin I showed double π–π stacking interaction analogous to the standard donepezil with water-mediated HBs within distances below 2.0 Å. Dictyoquinazol A manifested a GlideXP score −11.058 kcal/mol mainly through its quinazolin-4-one ring interactions and HBs formed with Phe295 and Tyr124. Genestin displayed matched interactions with the native ligand donepezil with low steric clashes to Trp286, Tyr341, Phe338, and Tyr337 as well as hydrogen bonding to the CT component residue His447. It is worth mentioning that the structural configuration of the aliphatic chain double bonds as E and Z facilitated the orientation of the hydroxyl group to form HB with Tyr124 with 1.95 Å.
Per the characteristics of this binding site, phenolic rings were crucial for effective binding where His447 extended two HBs to the hydroxyl groups at distances of 1.85 and 1.65 Å. π–π stacking was formed with Tyr337 and Trp86 at 3.74 and 5.26 Å, respectively. Most of these consistent binding modes were not shown in the rigid docking module, which necessitated using IFD. No data is available till now about the in vitro assays of these molecules; therefore, further investigations are recommended to confirm the docking results through cell based biological assays.
Molecular docking interactions in BACE1 protein binding pocket
It is worth mentioning that BACE1 was structurally classified into ten subsites (S1–S10), according to the protein ligand interaction maps. Each subsite denoted a group of residues involved in different noncovalent interactions like hydrogen bonding, π–π interaction or π–cation interactions with aliphatic chains, aromatic rings of inhibitors or charged moieties. This explained the different interaction modes illustrated for 4dju and their binding energies (Supplementary Table S1). The first mode was seen in the native ligand OKK through its HB interactions with Asp289 and Asp93 at distances 2.0 and 1.7 Å, respectively; moreover, a π–π stacking was noted between Tyr132 and the two aromatic rings. This binding mode coincided with the SS2 (subsite 2 pocket) in the BACE1 enzyme. In the same way, daldinan B, fornicin and xylarinig A belonged to this binding model. While daldinan B showed a forked anchor to both Asp93 and Asp289 at distances 2.31 and 1.80 Å, it rested over the hydrophobic Trp137 revealing aromatic π–π interactions with its phenyl rings. Fornicin molecule was oriented differently in the same subsite, thus developed only binding to Asp289, which explained its low binding affinity. Xylarinig A manifested two different poses where it was outward directed to Arg189 and Pro131 with salt bridge formation in the former and hydrogen bonding in the latter, and it revealed hydrogen bonding to Asp289, Arg189, and Tyr259. Another set of compounds fitted in subsites SS4 and SS5 whose binding features originated from the Gly291 structure that lacked side chain and only interacted through its backbone with ligands. Daldinan B, epi-guaidiol A, inonophenol A, and tacrine noncovalent bonds coincided with the key amino acids in these subsites manifested as Gly291/Trp 137 or Trp132. Moreover, hydrophobic interactions as well as HBs were important and suggested the nature of binding ligands to possess large hydrophobic moieties.
Thirdly, binding to subsite 8 (SS8) was noted in inonophenol C and ganomycin I. In this subsite, compounds with largely hydrophobic nature fitted into the nonpolar cavity with residues like Trp137 and Pro131, and their one or two polar groups interact with polar residues such as Arg296. Inonophenol C showed hydrogen bonding with Trp137 and Pro131 both at 1.75 Å, and ganomycin I displayed the same HBs to the same amino acids but at farther distances of 2.44 and 1.83 Å, respectively. The Cresset Flare discovery showed xylarinig A as of the lowest binding energy level −7.613 kcal/mol. Upon visualization of the 2D and 3D structures, it was evident that xylarinig A interacted with the core aspartate amino acids in the BACE1 binding site Asp93 and Asp289 through four HBs of lengths 2.1, 2.8, 2.1, and 1.8 Å, respectively. The bridging water molecules formed HBs with hydroxyl groups and carbonyl of the ligand, which shed light on these features in the SAR analysis of xylarinig A together with its three phenolic rings that facilitated fitting in the same groove and contacting with the same residues as the OKK. Binding to both asparate residues Asp93 and Asp289 as well as hydrophobic interactions with Tyr132 were common characteristics in xylarinig A and the native ligand OKK. Despite the fact that other factors contributed to the affinity of the ligand to the receptor (other than the binding score; among which are the number of HBs, their lengths, orientations, binding residues, hydrophobic contacts, and polar residues), it is speculated that the free energy negative high scores reflected more stability, yet the biological efficacy remains to be proved through in vitro and in vivo studies.
IFD in BACE1 protein binding pocket
Per the amino acid fingerprints, hispolon showed better stability and HBs to key amino acids as Trp176, Phe169, Lys168, Gly291, and Trp137. Furthermore, it approached many hydrophobic contacts and splitted the active site into clear nonpolar and polar pockets.
Multiple poses conferred from the IFD of fornicin showed hydrogen bonding with Asp93 at 1.87 Å, Asp289 at 1.50 Å, Gly95 at 1.75 Å, and π–π stacking with Phe169. These contacts were verified visually and through interaction fingerprinting. The methoxy group attached to the five membered lactone ring showed no added stability than ganomycin I; on the contrary, its electro positivity caused unfavorable contacts between nonpolar linker and both water molecules and its adjacent residues in many poses, Tyr132, Ser96, Ile179, Tyr259, Thr292, and Gly291. The fingerprint correlation revealed noticeable similarity with the native ligand OKK with evident interactions with Asp93, Gly95, Tyr132, Trp137, Asp289, and Gly291.
Footnotes
AUTHORS’ CONTRIBUTIONS
A.Y.M.: Conceptualization, methodology, and analysis. A.Y.M. and S.F.: Writing original draft. A.R.A.: Methodology and revising. M.R.: Methodology and revising.
AUTHOR DISCLOSURE STATEMENT
The authors declare that they have no conflict of financial interests or personal relationships that could have appeared to influence the work reported in this article.
FUNDING INFORMATION
The authors gratefully acknowledge the
SUPPLEMENTARY MATERIAL
Supplementary Data S1
Supplementary Table S1
Supplementary Table S2