
Abstract
Abstract
Thrombospondin-1 (TSP-1) is a matricellular glycoprotein that belongs to a family of evolutionary highly conserved calcium-binding proteins consisting of 5 members (TSP-1–TSP-5). In the eye, TSP-1 is expressed by several ocular cell types and is also detectable in the aqueous humor and the vitreous body. So far, TSP-1 is one of the major activators of TGFβ, suggesting a strong influence on various important cellular functions and interactions such as differentiation, migration, and wound healing. TSP-1 is also a key endogenous inhibitor of hem- and lymphangiogenesis. Several lines of evidence indicate a crucial role of TSP-1 in maintaining the ocular immune and angiogenic privilege, for example, by regulating T lymphocytes and the tolerance-promoting properties of ocular antigen-presenting cells. This review discusses the role of TSP-1 in dry eye disease and corneal graft rejection through its effects on hem- and lymphangiogenesis, as well as on the underlying immune responses. Recent work will be reviewed showing by which molecular mechanism TSP-1 modulates inflammatory processes during ocular diseases. This opens potential new treatment avenues in inflammatory and (lymph)angiogenic ocular diseases.
Introduction
T
Affecting a multitude of cellular functions such as intracellular signaling, proliferation, and migration, TSPs are involved in wound healing, angiogenesis, and inflammation.
TSP-1 is expressed by various cells types such as endothelial cells, fibroblasts, adipocytes, smooth muscle cells, monocytes, and macrophages. 4 In respect to the eye, TSP-1 is expressed in several tissues such as the trabecular meshwork, the cornea, the conjunctiva, as well as the retinal pigment epithelium.5–8 Furthermore, TSP-1 is detectable in the aqueous humor and vitreous body. 9 By binding protein components of the ECM-like integrin or fibronectin, TSP-1 is stored in the ECM.3,10 Due to this wide distribution, TSP-1 is assumed to have an essential influence on the ocular environment.
In the following sections, recent work will be reviewed showing by which molecular mechanism TSP-1 modulates corneal inflammation and (lymph)angiogenesis specifically in (1) dry eye disease (DED) and (2) corneal graft rejection. Due to the high prevalence of these ocular diseases and the crucial role of TSP-1 in their underlying immune response potential, new treatment avenues in inflammatory and (lymph)angiogenic ocular diseases can be indicated.
Dry Eye Disease
DED is one of the most common ocular diseases affecting millions of people. 11 Meanwhile, it is considered as a multifactorial disease of the ocular surface and the lacrimal functional unit (LFU). The LFU is defined as an integrated system comprising the ocular surface (tear film, corneal and conjunctival epithelia, and meibomian glands), the lacrimal glands, and nerves. 12 DED is accompanied with epitheliopathy, hyperosmolarity, tear film instability, and an inflammation of the ocular surface resulting in symptoms of discomfort and visual disturbance. 13 The inflammatory processes are considered to be essential in the pathogenesis of dry eye and can be understood as both cause and consequence. 14
Due to various etiological factors, DED is classified in 2 main groups: aqueous tear-deficient dry eye (ADDE) and evaporative dry eye (EDE). 13 ADDE is caused by a failed lacrimal gland function, leading to reduced lacrimal tear secretion and volume. It is divided in 2 subclasses depending on their underlying immune response: Sjögren's syndrome dry eye and non-Sjögren's syndrome dry eye. The former is characterized by an autoimmune-driven response that affects lacrimal, salivary, and potentially other exocrine glands. 15 The loss of glandular epithelial cells and the accumulation of inflammatory CD4+ T cells in the lacrimal gland are attributed to different viral infections such as the Epstein–Barr virus, hepatitis C virus, and human T-cell leukemia virus type 1. Although the clinical picture of non-Sjögren's syndrome DED proceeds without an autoimmune reaction, it is classified to the ADDE due to its etiological lacrimal gland dysfunction. Most commonly, it appears as an age-related form as a result of decreased tear volume and flow, increased osmolarity, 16 decreased tear film stability, 17 and alterations in the lipid layer of the tear film. 18 In contrast, EDE is hallmarked by a normal tear secretion with a concurrent excessive evaporation rate. Due to a meibomian gland dysfunction, the lipid layer shows quantitative or qualitative alterations in the lipid components.
For a long time, it was assumed that the ocular surface inflammation is primarily mediated by pathogenic T cells. This was supported by findings such as increased conjunctival infiltration of T cells in clinical as well as experimental DED and the development of DED in nude mice after the adoptive transfer of primed CD4+ T cells from dry eye mice.19,20 Especially TH1 T cells, identified by their activation in the draining lymph nodes, their increased secretion of interferon-γ (IFN-γ), and an elevated expression of TH1-type chemokine receptors (CCR5, CXCR3) and cytokine receptors (IL-12Rβ2), 21 were assumed to mediate the immune response.
However, recent data revealed that the immune response to dry eye is also associated with an inefficient function of regulatory T cells (Tregs) to suppress activation of effector T cells, which leads to an unrestricted generation and activation of self-reactive T cells by antigen-presenting cells (APCs) in the regional lymph nodes.20,22,23 Resident APCs become activated and migrate toward the draining lymph nodes where they stimulate cognate naive T cells. In this context, Tregs are well known to suppress inappropriate inflammation and regulate autoimmunity, 24 in which TH17 T cells also play a key modulatory role. 25 Reconstitution of T-cell-deficient nude mice with CD4+CD25+Foxp3+ Tregs results in nonsignificant disease development after injection of DED-specific primed pathogenic CD4+ T cells. 20 In addition, in vitro expanded CD4+CD25+Foxp3+ Tregs show similar suppressive behavior. 26 Thus, Tregs primed for the specific antigen responsible for initiating the disease provide a new platform of treatment as already shown in several animal models of inflammatory disease.27,28
Interestingly, while the potential to suppress effector T cells is reduced, the Treg homeostasis is not affected. 23 Thereby, only the TH17 T cell subpopulation seems to be resistant to Treg suppression. 23 This is corroborated by the detection of mainly interleukin 17 (IL-17)-secreting proliferative CD4+ T cells in an experimental model of DED. 23 Furthermore, typical TH17 promoters such as inteleukin-6, IL-23, IL-23R, and TGFβ were significantly increased in mice exposed to desiccating stress (constant airflow with a room humidity of 30% 29 ). 30 Due to the attenuated induction and severity of the disease after blocking IL-17 in vivo, the functional relevance of TH17 T cells in DED is further confirmed. In vivo blockade of IL-17 not only leads to a reduced generation of TH17 T cells but more importantly to a recovery of Treg function. 23 Thus, IL-17 supports the inefficient Treg suppression which, in turn, promotes further expansion of TH17 T cells that migrate to the ocular surface.
However, the cytokine IL-17 (TH17 T cells) as well as the cytokine IFN-γ (TH1 T cells) contribute to corneal barrier disruption, and an increased expression level of both cytokines could be observed in human and experimental DED.30–33 As these 2 cytokines could directly activate macrophages, the infiltration of the corneal stroma, the limbus, as well as the lacrimal and salivary glands could be detected in human specimens and experimental DED.34–36 Recent study evaluates that these macrophages are polarized in a proinflammatory M1 phenotype. 37
Since TSP-1 promotes the cleavage of TGFβ to its active form,38,39 which has an anti-inflammatory effect and plays a crucial role in the induction of regulatory T cells, 40 TSP-1-deficient mice resemble the autoimmune Sjögren's syndrome, indicated by inflammatory infiltrates in the lacrimal gland, as well as the damaged corneal epithelial barrier that correlates with the lacrimal gland dysfunction and reduced goblet cell density. 41 The presence of anti-Sjögren's syndrome antigen A (SSA) and anti-Sjögren's syndrome antigen B (SSB) indicates a potential autoimmune mechanism underlying the ocular surface inflammation. In addition, TSP-1-deficient mice showed a conjunctival inflammation similar to those observed in Sjögren patients.42–44 Not only an increased expression of the typical TH1 T cell- and TH17 T-cell-associated inflammatory cytokines (IFN-γ, TNF-α, IL-6, and IL-17A) but also an increased expression of related transcription factors (Tbet, RORyt) was detected in TSP-1 null conjunctiva. 42 The detected CD4+ T cells in the lacrimal gland as well as those in the periphery were shown to mainly secrete IL-17. 41 The increased amount of effector cells in the draining lymph nodes can be explained by the missing inhibitory effect of TSP-1 on the maturation of DCs, 45 including corneal DCs 46 as well as the upregulated expression of the c-c chemokine receptor type 7 (CCR7).42,46,47
As it has been reported that the induction of peripheral immune tolerance by APCs exposed to TGFβ depends on TSP-1, 48 it could be expected that TSP-1-deficient APCs in the lacrimal gland are incapable of inducing tolerance. In addition, it has been demonstrated that macrophages secrete anti-inflammatory TGFβ during removal of cellular debris. Thus, it could be assumed that TSP-1-deficient macrophages that fail to activate latent TGFβ can be supportive for an inflammatory activation.
As TGFβ is important for the development of Tregs, 40 it is not surprising that TSP-1-deficient mice reveal a decline in splenic Tregs. 41 In addition, an imbalance between TH17 T cells and Tregs in young TSP-1-deficient mice could be detected, which is consistent with the presence of chronic inflammatory autoimmune disease. 49 In addition, TSP-1 itself is shown to inhibit the activation and maturation of T cells into TH1 T cells 50 and to facilitate the generation of thymus-derived CD4+CD25+ Tregs. 51 Thus, it could be assumed that TSP-1 indirectly drives the immune response to a TH17 T cell phenotype.
Interestingly, Gandhi et al. reported that TSP-1-deficient mice failed to show any worsening of the ocular surface parameters above their baseline after desiccating stress. 52
It is worth mentioning that in this study, before exposure to desiccating stress, nearly a 3-fold higher CD4+ T cell infiltration was detected in the conjunctiva of TSP-1-deficient mice compared to wild-type (WT) mice, consistent with the spontaneous ocular surface disease associated with TSP-1 deficiency.41,42,52 In addition, comparison between desiccating stress-exposed WT and TSP-1-deficient mice indicates increased CD4+ T cell infiltration in the latter. 52 This study reports no change in the corneal barrier disruption in TSP-1-deficient mice after desiccating stress. However, differences in methods of evaluating corneal barrier between studies may account for the seemingly contradictory results. The desiccating stress study used the larger fluorescent dye OGD 488 (70 kDa) in enucleated eyes compared to use of the traditional smaller fluorescein (376 Da) dye in live mice, as used in previous studies in TSP-1-deficient mice.
Apart from this, it is known that the corneal angiogenic privilege, 53 which maintains the transparency of the cornea and preserves high visual acuity, is disturbed in DED. In a mouse model of DED, Goyal et al. could show an isolated ingrowth of lymphatic vessels, but not blood vessels, into the physiologically avascular cornea. 54 Nevertheless, little is known about the mechanism leading to corneal lymphangiogenesis occurring during DED. Investigations in a suture-induced inflammatory corneal neovascularization model revealed that lymphangiogenesis correlates with the number of CD11b+ macrophages recruited by vascular endothelial growth factor A (VEGF-A), 55 which in turn express VEGF-A, -C, and -D.55,56 The interaction of these growth factors with both VEGF receptor 2 (VEGFR2) and VEGF receptor 3 (VEGFR3) leads to the induction of hem- and lymphangiogenesis in the injured cornea. 57 Consistent with the TH17 dominant autoimmune response in DED, it was shown that IL-17 also has relevance in desiccating stress-induced corneal lymphangiogenesis. 58 In vivo blockade of IL-17 in an autoimmune DED model not only leads to a significant reduction of corneal lymphangiogenesis but also to a decreased expression of VEGF-D and VEGF-C. 58 Furthermore, in vitro lymphatic endothelial cell tube formation was significantly decreased when either IL-17 or VEGFR3 was blocked, implicating that IL-17 directly induces corneal lymphangiogenesis by the VEGF-C/D-VEGFR3 signaling pathway. 58 Thus, IL-17 not only antagonizes Treg homeostasis but also promotes inflammatory lymphangiogenesis, thereby facilitating progression and amplification of the (auto)immune response.
In the healthy cornea, several mechanisms such as the specialized anatomy of the cornea, the angiostatic effect of corneal cells, and the expression of antiangiogenic molecules like TSP-1 contribute to its angiogenic privilege. In general, TSP-1 seems to have immunomodulatory and anti(lymph)angiogenic effects, which help in maintaining both the corneal immune as well as the angiogenic privilege.59,60 Already in 1990 it was shown that TSP-1 regulates migration and proliferation of blood endothelial cells. 61 Nowadays, several studies imply a directly mediated anti(lymph)angiogenic effect of TSP-1 by engaging CD36 and CD47 receptors on endothelial cells. Thus, TSP-1 is demonstrated to disrupt the direct binding of CD47 to VEGFR2, which in consequence avoids autophosphorylation and, thereby, inhibits downstream signaling. 62 Furthermore, by engaging CD36, TSP-1 inhibits endothelial tube formation in vitro 63 and mediates macrophage phagocytosis of apoptotic and endothelial cells. 64
Only recently, experimental studies in TSP-1-deficient mice demonstrated the crucial role of TSP-1 in the regulation of inflammatory corneal lymphangiogenesis. Thus, 6-month-old TSP-1-deficient mice develop spontaneous corneal lymphangiogenesis (Fig. 1A, B). Furthermore, corneal suture placement induces a significantly increased ingrowth of lymphatic vessels in young TSP-deficient mice compared to WT mice (Fig. 1C), which could be reversed by topical application of recombinant human TSP-1. 65

Absence of Thrombospondin-1 (TSP-1) promotes spontaneous corneal lymphangiogenesis. Corneal flat mounts of 6-month-old TSP-1-deficient mice show isolated lymphatic vessels (red)
Coincident with the spontaneous corneal lymphangiogenesis in 6-month-old TSP-1-deficient mice, enhanced expression levels of VEGF-C in the cornea and an increased number of corneal CD11b+ macrophages were detectable (Fig. 2A, B). Moreover, it was shown that resident CD11b+ macrophages in the cornea express CD36, a potential TSP-1 receptor. While WT macrophages exposed to TSP-1 revealed a suppressed expression of VEGF-C and VEGF-D in vitro, the blockade of CD36 on these macrophages partially reversed the inhibition of the VEGF-C and VEGF-D expression. Thus, TSP-1 acts as an indirect inhibitor of lymphangiogenesis by binding CD36 on macrophages which, in turn, regulates the expression of VEGF-C at the mRNA level 65 (Fig. 2C).
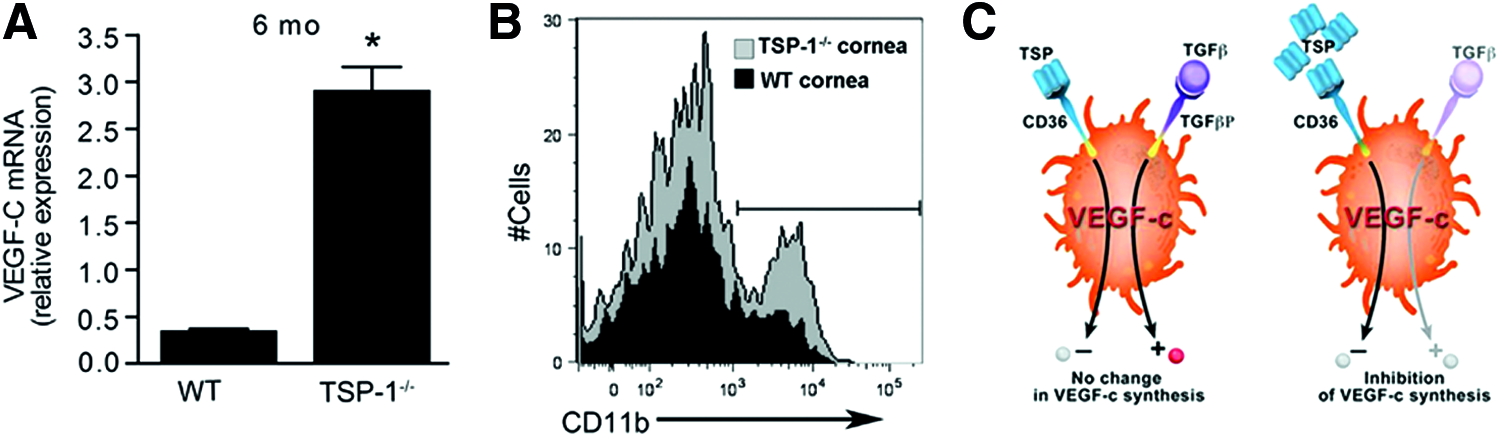
TSP-1 regulates VEGF-C expression through CD36 on macrophages.
Despite these experimental findings, recent data reveal an association between the development of postrefractive dry eye in humans and polymorphisms in the TSP-1 gene (THBS1). 66 Single-nucleotide polymorphisms (SNPs) in the regulatory 5′-untranslated region (UTR) of THBS1 were identified and shown to correlate with a reduced expression of TSP-1 in the ocular surface and an increased expression of the inflammatory cytokine IL-1β. 66 Thus, polymorphisms in the TSP-1 gene seem to provide a genetic basis for the development of postsurgical DED.
Corneal Graft Rejection
Orthotopic corneal transplantation (also called keratoplasty) is the oldest and most frequently performed tissue transplantation worldwide, with >40,000 transplantations in a year alone in the United States. 67 Even without HLA-matching, a 2-year graft survival rate of 90% is achieved when corneal grafts are placed in avascular and noninflammatory recipient beds (normal-risk keratoplasty). 68 These excellent graft survival rates are caused by the angiogenic and immune-privileged properties of the eye. The avascularity of the cornea represents a kind of anatomical barrier suppressing both arms of the immune reflex arc.69,70
Blood vessels providing a route of entry for immune effector cells to the graft (efferent arm) as well as lymphatic vessels enabling the effective access of antigenic material and APCs to the regional lymph nodes (afferent arm) are not connected to the donor tissue, 71 resulting in an altered immune reaction and enhanced transplant acceptance.67,72,73 Not surprisingly, it is demonstrated that in case of prevascularized recipient beds (high-risk keratoplasty), the graft survival rate is below 50%. 70 Due to the enhanced interaction of the graft and the vascularized recipient bed, alloimmunization and subsequent graft rejection can be induced more easily.
For many decades, pre-existing blood vessels were considered as the most important risk factor for immune-mediated graft rejections. This assumption was fostered by several findings such as (1) a significant decrease of graft survival by the presence of pre-existing blood vessels in high-risk eyes74,75 and (2) a significant decrease of graft survival when corneal neovascularization occurred after normal-risk keratoplasty.76,77 Furthermore, it has been argued that due to the absence of mature bone marrow-derived APCs78,79 and the unique property of only MHC class II negative (MHCII−) APCs in the center of the cornea, 80 allosensitization is induced by the indirect pathway where recipient-derived APCs present processed allopeptides.81,82 However, recently, several studies have also revealed the great importance of the lymphatic system for the outcome of corneal transplantation (Fig. 3).
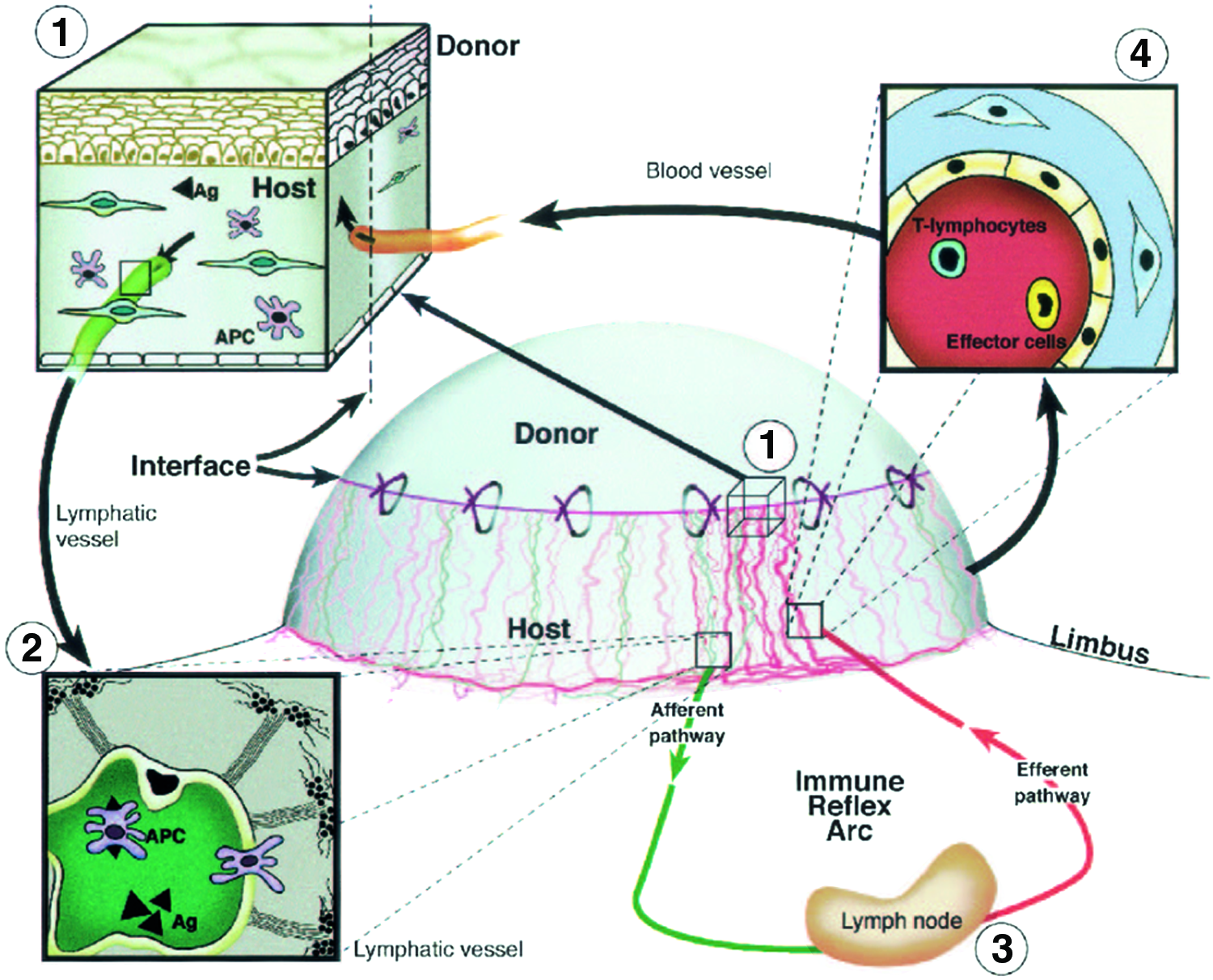
Role of blood and lymphatic vessels in the high-risk corneal host bed as exit and entry routes of the immune reflex arc leading to immunologic graft rejection.
In this context, it was demonstrated that cervical lymphadenectomy (the surgical excision of draining regional lymph nodes) in mice results in 100% graft survival after normal-risk keratoplasty and 90% graft survival after high-risk keratoplasty.83,84
Another important finding was the migration of donor APCs to the draining lymph nodes after transplantation. Liu et al. demonstrated for the first time that resident donor-derived MHCII− APCs are bone marrow-derived dendritic cells (CD45+CD11c+) that become MHCII+ after keratoplasty. 85 Apart from this, however, it was demonstrated that whether allosensitization occurs directly or indirectly might depend on the graft bed microenvironment. 86
With the discovery of novel specific lymphatic markers (eg, Lyve-1), evidence of lymphangiogenesis in both human corneas vascularized secondary to several diseases (eg, keratitis, graft rejection, limbal stem cell insufficiency, and chemical burns) and mouse corneas after experimental keratoplasty was provided.87,88
Using the mouse model of normal-risk keratoplasty, the simultaneous outgrowth of both blood and lymphatic vessels into the avascular recipient bed after keratoplasty was demonstrated. 88 Both vessel types reached the graft on day 7 and no differences in postkeratoplasty neovascularization between syngeneic and allogenic transplantation were detectable. 88 Comparable increased densities in blood and lymphatic vessels after high-risk keratoplasty could be observed. 89
As VEGF-A has been demonstrated to indirectly stimulate inflammatory lymphangiogenesis by macrophage recruitment, 55 inhibition of VEGF-A was considered as a potential strategy to promote long-term allograft survival. Following this consideration, several studies have been performed to analyze the effect of VEGF-A inhibition regarding corneal transplant survival. In both normal-risk and high-risk keratoplasty, neutralization of VEGF-A not only inhibited surgery-induced hem- and lymphangiogenesis but also hampered mononuclear cell recruitment,55,89,90 thereby promoting graft survival.
With the development of the specific pharmacological lymphangiogenesis inhibitors, VEGFR3 antibody mF4-31C1 91 and the integrin α5β1 inhibitor JSM6427, 92 the specific inhibition of lymphatic vessels was enabled and the role of lymphatic vessels in graft rejection further delineated. By selectively inhibiting lymphangiogenesis without affecting hemangiogenesis in the suture-induced corneal neovascularization model, differently prevascularized recipient beds were generated and revealed a similar graft survival rate for alymphatic high-risk recipient beds compared to normal-risk recipient beds, whereas recipients containing lymphatic vessels rapidly rejected the grafts. 93 Accordingly, lymphatic vessels are primarily responsible for the high-risk status of prevascularized recipient beds. 93
Nevertheless, not only the avascularity but also the immune privilege of the eye promotes graft survival. 53 Since TSP-1 has anti(lymph)angiogenic as well as immunomodulatory properties,59,60 it facilitates both the corneal avascularity as well as the corneal immunity. In relation to corneal transplant outcome, the binding of CD36 and its involvement in the induction of the anterior chamber-associated immune deviation (ACAID) are the characteristics emphasized. Due to the binding of CD36, TSP-1 downregulates the expression of VEGF-C, 65 thereby promoting the alymphatic status of the eye and the corneal transplant survival. In addition, it is heavily involved and essential for the induction of the ACAID, which is responsible for the peripheral tolerance against eye-derived antigens.67,94
Following corneal transplantation, ocular APCs capture donor-derived antigens and migrate through the bloodstream to the marginal zone of the spleen where they induce the unique setup of T cells found in ACAID.95–97 By secreting the macrophage inflammatory protein 2 (MIP2 or CXCL2), natural killer (NK) cells are recruited to the site98,99 and both together, NK cells and APCs, generate a microenvironment that is rich in TSP-1 and TGFβ.100,101
As TSP-1 promotes the cleavage of TGFβ in its active form,38,39 the enhanced expression of TSP-1 may also contribute to ensure the enhanced TGFβ production by APCs. 100 Ocular APCs exposed to a TGFβ-rich microenvironment exhibit an uncommon phenotype that leads to a systemic immune response, in which antigen-specific Tregs are involved.102,103 While specialized CD4+ T cells suppress the initial activation and differentiation of naive T cells into TH1 effector cells, CD8+ T cells inhibit the expression of TH1-mediated immunity, such as alloantigen-specific delayed-type hypersensitivity, 104 and promote long-term survival. 94 In previous studies, it was demonstrated that neutralization of TGFβ restores the ability of TGFβ-treated APCs to activate the immune response in a TH1 phenotype and fails to suppress TH1-mediated inflammatory delayed hypersensitivity.105,106
Several lines of evidence pointed out that TSP-1 is involved in the adaptive immune response due to its ability to interact with receptors on T cells (CD47) and APCs (CD36 and CD47),48,107,108 therefore forming a bridge that holds T cells and APCs together. This would stabilize the typical TGFβ-rich microenvironment at the surface of APCs and simultaneously diminish the generation of CD40 and IL-12 stimulation that is necessary for the induction of TH1 immune response100,101,109 (Fig. 4). Thus, ocular APCs use TSP-1 to orchestrate the required TGFβ-rich microenvironment concurrently facilitating their tolerance-promoting properties. 48
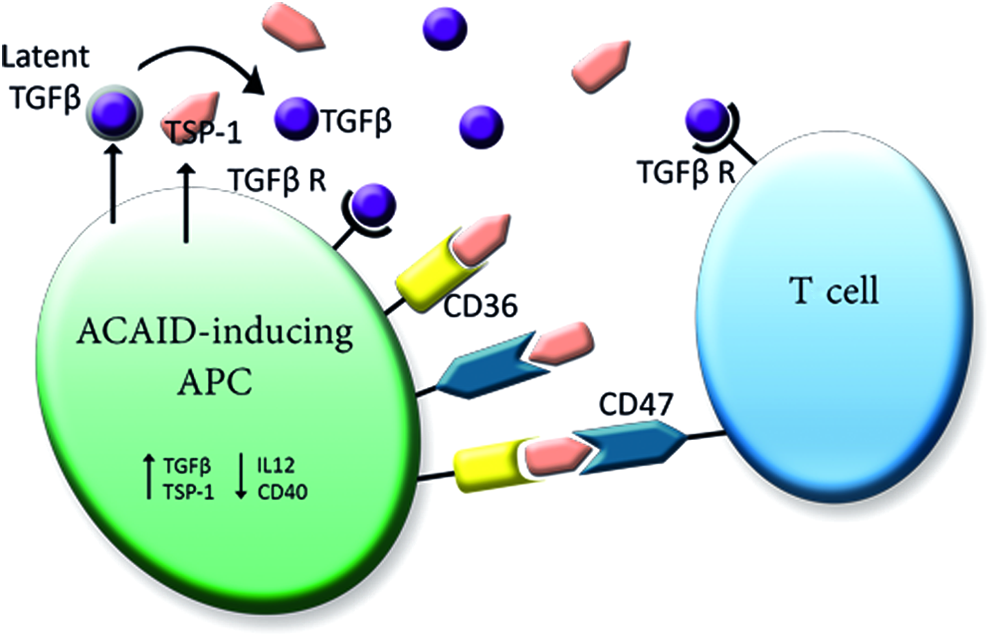
Model of the assumed interplay between TSP-1, APCs, and T cells. Macrophages/APCs exposed to a TGFβ-rich environment upregulate expression of TSP-1 and TGFβ. In turn, TSP-1 promotes the cleavage of latent TGFβ in its active form to ensure the required TGFβ microenvironment. As TSP-1 has the ability to interact with receptors on T cells (CD47) and APCs (CD36 and CD47), TSP-1 can facilitate the interactions of T cells and APCs by forming a bridge that holds both together. This would stabilize the TGFβ-rich environment at the surface of APCs and simultaneously reduce CD40 and IL-12 expression, which is necessary for the TH1 immune response. Adapted from Masli et al. 100 Color images available online at www.liebertpub.com/jop
Consistently, it was demonstrated that most of the TSP-1 null allografts in a corneal transplantation model were rejected 46 (Fig. 5). Thereby, graft-derived TSP-1 was shown to regulate allosensitization by donor APCs, but not host APCs, leading to the assumption that the relevant source of TSP-1 is donor APCs. 46 In concordance to this, an association between TSP-1 gene polymorphisms and the outcome of corneal high-risk transplantation in humans was demonstrated. 110 The identified SNPs in the 5′-UTR of THBS1, which differs from the SNPs found in DED (see above), increased the risk for corneal transplant rejection. 110
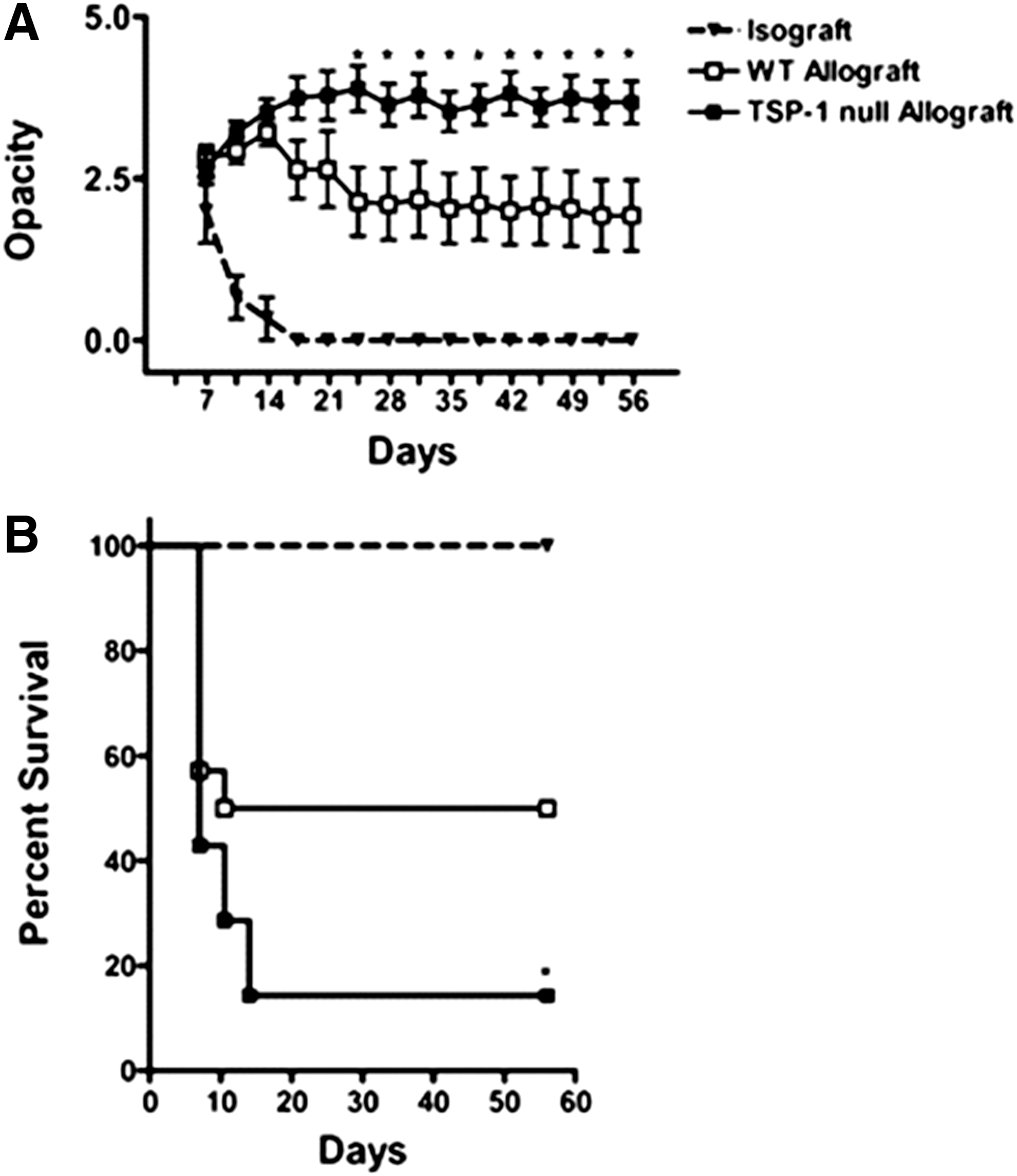
Graft-derived TSP-1 protects transplants from immune rejection. Corneal isografts (BALB/c; triangles), WT allografts (C57BL/6; open squares), or TSP-1 null allografts (C57BL/6; filled squares) were transplanted into BALB/c mice. Graft survival was followed biomicroscopically for 8 weeks (n=14 per group).
Conclusion
TSP-1 plays an important role in ocular inflammatory processes such as DED and corneal graft rejection.
TSP-1 is involved in several molecular and cellular mechanisms that contribute to the angiogenic and immune-privileged status of the eye indicating a crucial role of TSP-1 in regulating ocular immune responses.
Due to its indirect anti(lymph)angiogenic effect, TSP-1 regulates the outgrowth of vessels in the avascular cornea, thereby affecting both arms of the immune reflex arc. This is primarily mediated by its interactions with CD36 and CD47.
Since TSP-1 is one of the major activators of TGFβ, it is actively involved in generating the TGFβ-rich microenvironment that, in turn, is essential for ACAID. ACAID subsequently allows emergence of Tregs that suppress systemically the induction of T-cell-mediated immune responses.
Thus, TSP-1 may be an effective novel therapeutic tool in diminishing severity of DED and promoting corneal transplant survival.
Overall, TSP-1 plays an important role in maintaining the avascular status of the cornea and the immune privilege of the eye.
Footnotes
Acknowledgments
This work was supported by DFG FOR 2240 (Lymph) Angiogenesis And Cellular Immunity In Inflammatory Diseases Of The Eye; DFG Cu 47/4-1; DFG Cu 47/10-1; EU COST BM1302 Joining Forces in Corneal Regeneration; and the Bayer Graduate School in Pharmacology Cologne.
Author Disclosure Statement
No competing financial interests exist.