
Abstract
Purpose:
To determine whether the adiponectin receptor (AdipoR) agonist AdipoRon inhibits glutamate-induced neuronal cell death and to investigate the neuroprotective mechanism of AdipoRon in rat primary retinal ganglion cells (RGCs).
Methods:
The expression pattern of AdipoR1 and AdipoR2 in rat retina and primary RGCs was examined by immunostaining. The neuroprotective effect of AdipoRon on glutamate-induced cell death was evaluated in rat primary RGCs. Cellular levels of reactive oxygen species (ROS) were also measured. Peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α), estrogen-related receptor-α (Esrra), mitochondrial transcription factor A (TFAM), peroxisome proliferator-activated receptor α (PPARα), and catalase mRNA levels were examined.
Results:
The expression of AdipoR1 and AdipoR2 was confirmed in rat retina and primary RGCs. AdipoRon significantly increased the survival rate of glutamate-induced cell death and decreased ROS production. Additionally, the mRNA levels of PGC-1α, Esrra, and TFAM were upregulated by AdipoRon.
Conclusions:
These results suggest that AdipoRon has a neuroprotective effect by inhibiting ROS production via upregulation of PGC-1α, Esrra, and TFAM against glutamate-induced RGC death.
Introduction
The only reliable evidence-based treatment for glaucoma, including normal tension glaucoma, is reducing intraocular pressure (IOP).1,2 However, some patients are resistant to a reduction of IOP, and the disease progresses. 3 Neuroprotective therapy, which involves inhibiting the death of retinal ganglion cells (RGCs), are gaining increased attention for their potential to be developed as a treatment for glaucoma.4,5
Adiponectin is a protein secreted by adipocytes and is maintained at high levels in serum. 6 It plays an important role in glucose and lipid metabolism,7–10 inflammation,11–13 and oxidative stress.14,15 Adiponectin has neuroprotective effects and inhibits neuronal death induced by excitotoxicity in vitro, as reported previously in primary hippocampal cells and the HT22 hippocampal cell line.16,17 Adiponectin exerts these effects by activating 5′ adenosine monophosphate-activated protein kinase (AMPK) and proliferator-activated receptor α (PPARα) via the adiponectin receptor (AdipoR) 1 and AdipoR2.
AdipoRon was a small molecule agonist of the AdipoR. 18 Many studies have reported the various effects of AdipoRon, including its protective effects in diabetic nephropathy and hepatic injury.15,19–22 We hypothesized that AdipoRon would have a neuroprotective effect on glaucoma by inhibiting the death of RGCs, as reported for adiponectin.
In this study, we assessed the neuroprotective effect of AdipoRon in rat primary RGCs and investigated its possible mechanism of action.
Methods
Animals
Wistar rats were purchased from Tokyo Laboratory Animals Science (Tokyo, Japan). All animal experiments were performed in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and the National Institutes of Health Guiding Principles for the Care and Use of Animals (DHEW Publication, NIH 80–23) and were approved by the Institutional Animal Research Committee of the University of Tokyo.
Immunohistochemistry
Postnatal day 5 (P5) and 7-week-old rats were euthanized, and their eyes were enucleated. The enucleated eyes were fixed for 1 h in 4% paraformaldehyde in phosphate-buffered saline (PBS) (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) after puncturing the cornea and immersed overnight in PBS containing 30% sucrose. Further, the eyes were embedded in OCT compound (Tissue-Tek, Tokyo, Japan), frozen, and the posterior retina was cut into 10-μm-thick sections. The slides of posterior retina and primary RGCs on chamber slides II (AGC, Inc., Tokyo, Japan) were permeabilized with a 0.05% Triton X-100 solution in PBS and blocked with Blocking One Histo (Nacalai Tesque, Kyoto, Japan). The slides were incubated with anti-AdipR1 or AdipoR2 antibody (1:100, sc-46748, sc-46755; Santa Cruz Biotechnology, Dallas, TX) and anti-Brn-3a antibody (1:100, MAB1585; Merck, darmstadt, Germany) overnight at 4°C, washed with PBS, and incubated with Alexa594-conjugated anti-goat IgG secondary antibody (1:200, A11058; Thermo Fisher Scientific, Waltham, MA) and Alexa488-conjugated anti-mouse IgG secondary antibody (1:200, A21202; Thermo Fisher Scientific) for 1 h with 4′,6-diamidino-2-phenylindole (1 μg/mL; Dojindo Molecular Tech, Kumamoto, Japan) at room temperature. The slides were washed with PBS and embedded with VECTASHIELD Mounting Medium (Vector Laboratories, Burlingame, CA). Fluorescent images were observed under a fluorescence microscope (Keyence, Osaka, Japan).
Purification and culture of RGCs
RGCs were isolated by a 2-step immune-panning procedure as described previously. 23 In brief, P5- to 6-old rats were euthanized, and the dissected retinas were dissociated into a cell suspension using a papain dissociation system (Worthington Biochemical, Lakewood, NJ). The cell suspensions were incubated in a 50 mL tube coated with anti-macrophage antibody (MAB1407P; Millipore, Bedford, MA) for 30 min at room temperature. Cells not bound to the antimacrophage antibody were transferred to incubate in a 50 mL tube coated with anti-Thy1.1 antibody (MAB1406; Millipore) for 60 min at room temperature. Cells binding to the anti-Thy1.1 antibody were suspended in serum-free Neurobasal medium (Thermo Fisher Scientific) supplemented with 2% B27 supplement (Thermo Fisher Scientific), 1 mM L-glutamine (Sigma-Aldrich, St. Louis, MO), 40 ng/mL brain-derived neurotrophic factor (450-02; PEPROTECH, Rocky Hill, NJ), 40 ng/mL rat ciliary neurotrophic factor (NBP2-35289; NOVUS Biologicals, Littleton, CO), 10 μM forskolin (FUJIFILM Wako Pure Chemical Corporation), 100 U/mL penicillin, and 100 μg/mL streptomycin (FUJIFILM Wako Pure Chemical Corporation). RGCs from a total of 8 eyes were used per each experiment. RGCs were seeded on plates coated with 50 μg/mL poly-L-lysine (Sigma-Aldrich) and 1 μg/mL laminin (Thermo Fisher Scientific) at 2 × 105 cells/mL. The purity of cells was confirmed by RGC marker, Brn-3a staining. The RGCs were used for the experiments after a 3-day incubation.
RGC survival assay
The RGC survival assay was conducted as described previously. 23 RGCs were pretreated with 1, 2, 5, and 10 μM AdipoRon (Adooq Bioscience, Irvine, CA) diluted in DMSO for 1 h and then exposed to 25 μM glutamate (Glu; FUJIFILM Wako Pure Chemical Corporation) for 72 h. The RGCs were incubated in 2 μM Calcein-AM solution for 20 min, and the number of living RGCs was counted in each well with a fluorescence microscope. A surviving RGC was defined as a cell with a Calcein-AM-stained cell body and a process extending at least 3 cell diameters from the cell body. Dead cells were stained with Ethidium Homodimer 1 (Thermo Fisher Scientific).
Measurement of reactive oxygen species
Reactive oxygen species (ROS) production was measured using the oxidative stress indicator, 5-(and-6)-chlorometyl-2′7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA; Thermo Fisher Scientific). RGCs were pretreated with 2 μM AdipoRon for 1 h 3 days after isolation and then exposed to 25 μM Glu. After exposure to Glu for 24 h, the cells were incubated with 5 μM CM-H2DCFDA for 1 h and then observed with a fluorescence microscope; fluorescence was analyzed at 485 nm excitation/535 nm emission wavelengths.
Quantitative real-time polymerase chain reaction analysis
Total RNA was extracted 5 h after the AdipoRon treatment using ISOGEN (Nippon Gene, Tokyo, Japan). Complementary DNA was prepared with the ReverTra Ace® qPCR RT Master Mix with gDNA remover (Toyobo, Osaka, Japan). Quantitative polymerase chain reaction (qPCR) was carried out with the Thermal Cycler Dice Realtime System (Takara Bio, Shiga, Japan) using SYBR® Premix Ex Taq™ II (Tli RNaseH Plus) (Takara Bio). The primer sequences used in qPCR were rat glyceraldehyde-3-phosphate dehydrogenase (Gapdh): forward, 5′-GCTGCCTTCTCTTGTGACAAAG-3′ and reverse, 5′-TGACTGTGCCGTTGAACTTG-3′; rat PPARG coactivator 1 alpha (Ppargc1α): forward, 5′-ATGTGTCGCCTTCTTGCTCT-3′ and reverse, 5′-ATCTACTGCCTGGGGACCTT-3′; rat estrogen related receptor, alpha (Esrrα): forward, 5′-GCTTTTGCTGCTGCTTACTC-3′ and reverse, 5′-AGCCTGCTTGGAGTTATTGC-3′; rat transcription factor A, mitochondrial (Tfam): forward, 5′-ATCAAGACTGTGCGTGCATC-3′ and reverse, 5′-AAAGCCCGGAAGGTTCTTAG-3′; rat peroxisome proliferator activated receptor alpha (Pparα): forward, 5′-TGGCAATGCACTGAACATCG-3′ and reverse, 5′-TGCAGCTTCGATCACACTTG-3′; rat catalase: forward, 5′-TTTGACCCAAGCAACATGCC-3′ and reverse, 5′-GTGTCTGGGTAAGCAAAAAGGC-3′. The values for each gene were normalized to the level of GAPDH.
Statistical analysis
Data are expressed as mean ± standard error. The statistical analysis was performed with a 2-tailed Student's t-test or Dunnett's test. P values of P < 0.05 were considered significant.
Results
AdipoR1 and AdipoR2 protein expression in rat retina and primary RGCs
First, we examined the expression of AdipoR1 and AdipoR2 in the rat retina and primary RGCs. We co-immunostained P5 and 7-week-old rat retinal sections for AdipoR1 or AdipoR2 and the RGC marker Brn-3a. Expression of AdipoR1 and AdipoR2 was confirmed in the retina both at P5 and 7-week-old rat; At P5 and 7-week-old rat, AdipoR1 was expressed in the entire retina (Fig. 1A, left), and AdipoR2 was expressed at relatively high concentration in the ganglion cell layer (Fig. 1A, right). AdipoR1 and AdipoR2 were detected in cytoplasm of isolated rat primary RGCs (Fig. 1B).
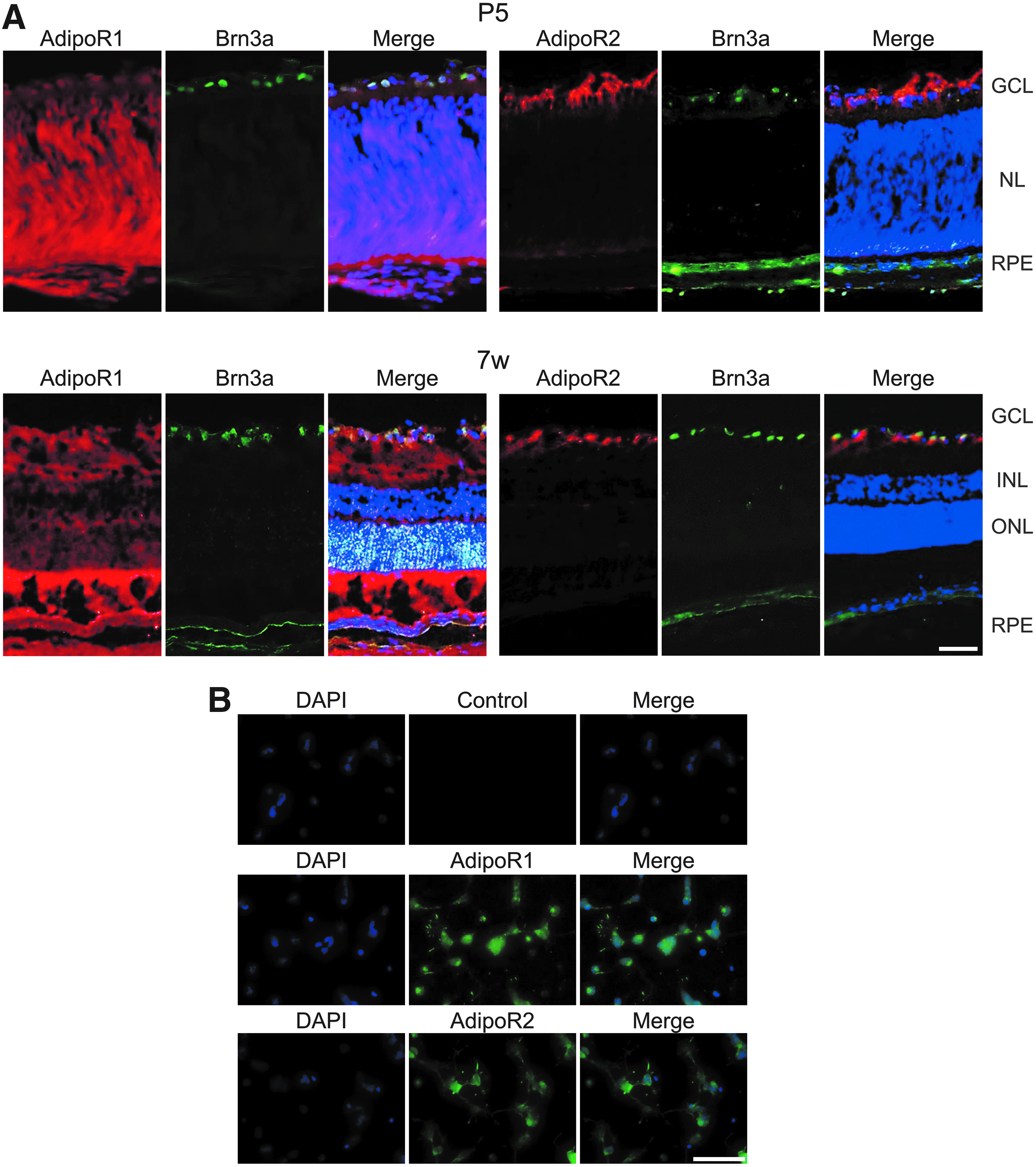
AdipoR1 and AdipoR2 expressions in vitro and in vivo.
Neuroprotective effect of AdipoRon on Glu-induced neurotoxicity in rat primary RGCs
We evaluated the neuroprotective effect of AdipoRon on Glu-induced neurotoxicity in rat primary RGCs. The neurotoxic effect of 25 μM Glu was confirmed, and pretreatment with 2 μM AdipoRon significantly reduced Glu-induced neurotoxicity as demonstrated by microscopy (Fig. 2A, B). Neuroprotective effects of AdipoRon were lost at 5 and 10 μM concentrations.
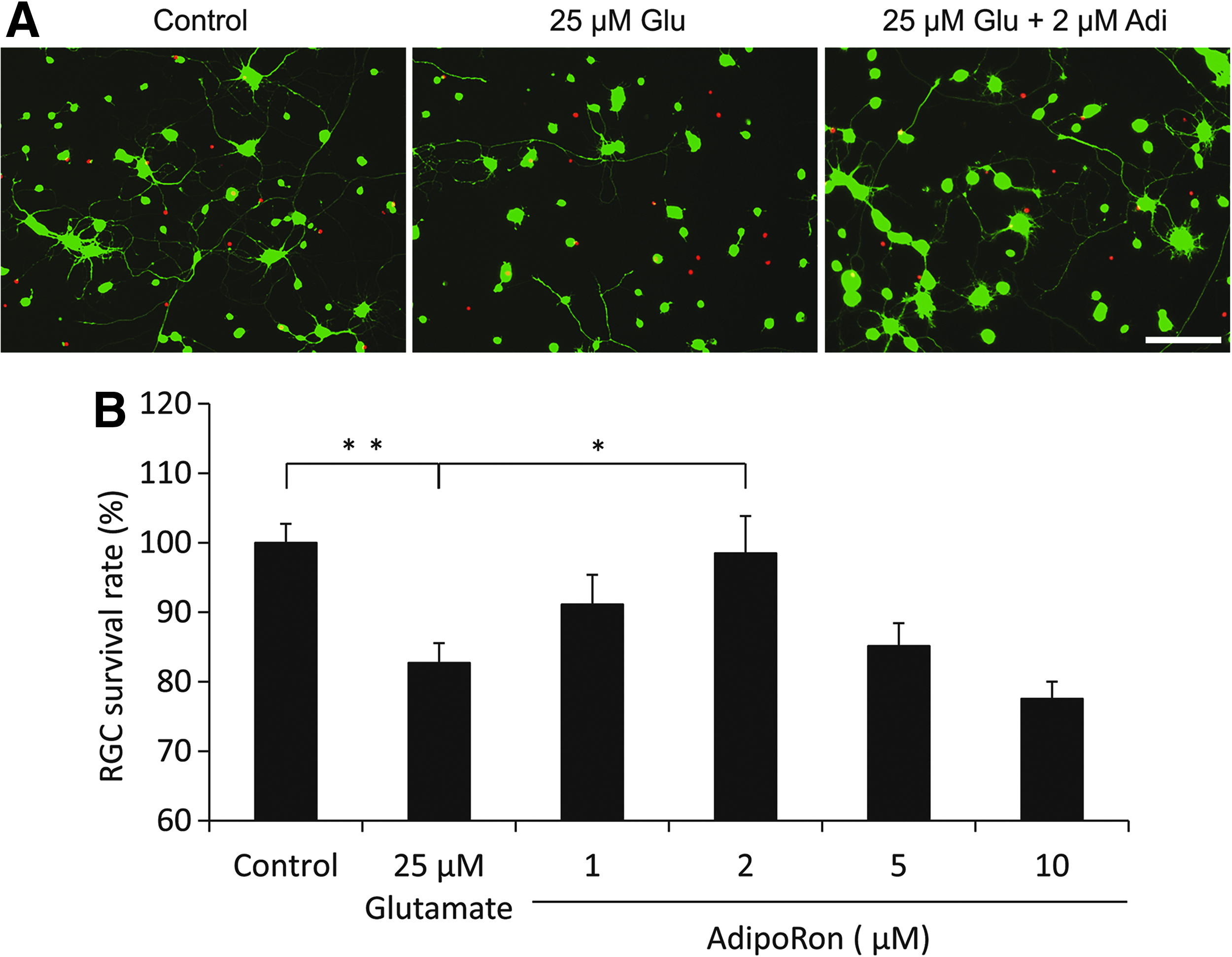
Neuroprotective effect of the AdipoR agonist AdipoRon against Glu-induced RGC death.
Effect of AdipoRon on Glu-induced ROS production
Glu-induced cell death results in overproduction of ROS, which causes oxidative damage and cell death. 24 We used the ROS indicator CM-H2DCFDA to assess ROS production. Glu treatment significantly increased ROS production in rat primary RGCs, and pretreatment with 2 μM AdipoRon significantly reduced Glu-induced ROS production as demonstrated by microscopy (Fig. 3A, B).
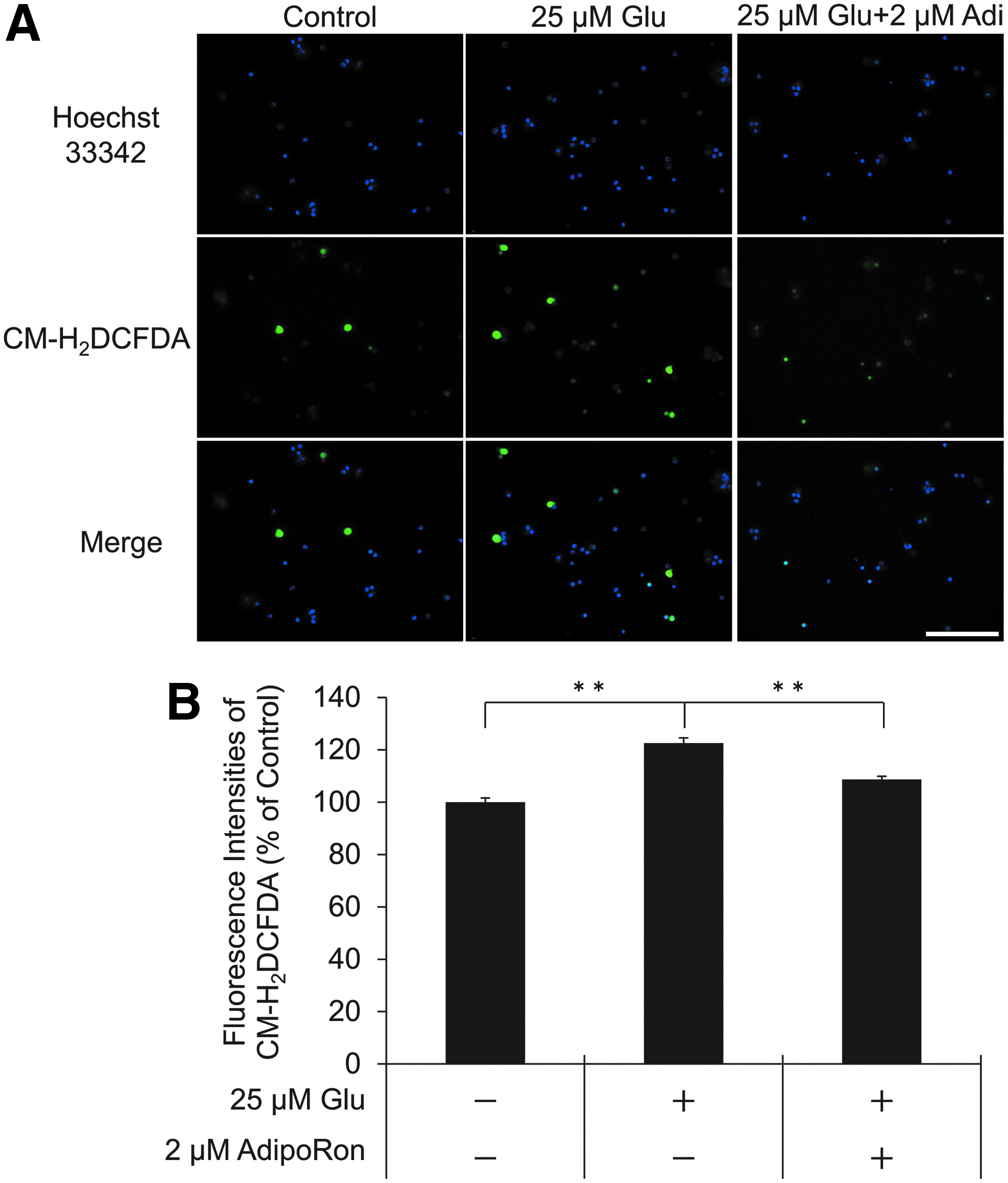
Effect of AdipoRon against Glu-induced ROS production.
Effect of AdipoRon on mRNA level of genes related to mitochondria biogenesis and antioxidation in RGCs
It has been previously reported that adiponectin activates and increases the expression of peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) via AdipoR1 and its target genes Esrra and Tfam.25,26 Adiponectin also increases the expression of PPARα via AdipoR2, and PPARα activates antioxidant genes, catalase, and superoxide dismutase1 (Sod1) at mRNA level. 27 Accordingly, we conducted a qPCR analysis to determine whether AdipoRon activates the expression of these genes through the AdipoR1 and AdipoR2 in rat primary RGCs. As a result, AdipoRon significantly increased the mRNA levels of PGC-1α, Esrra, and TFAM, whereas PPARα and catalase mRNAs remained unchanged in response to AdipoRon (Fig. 4).
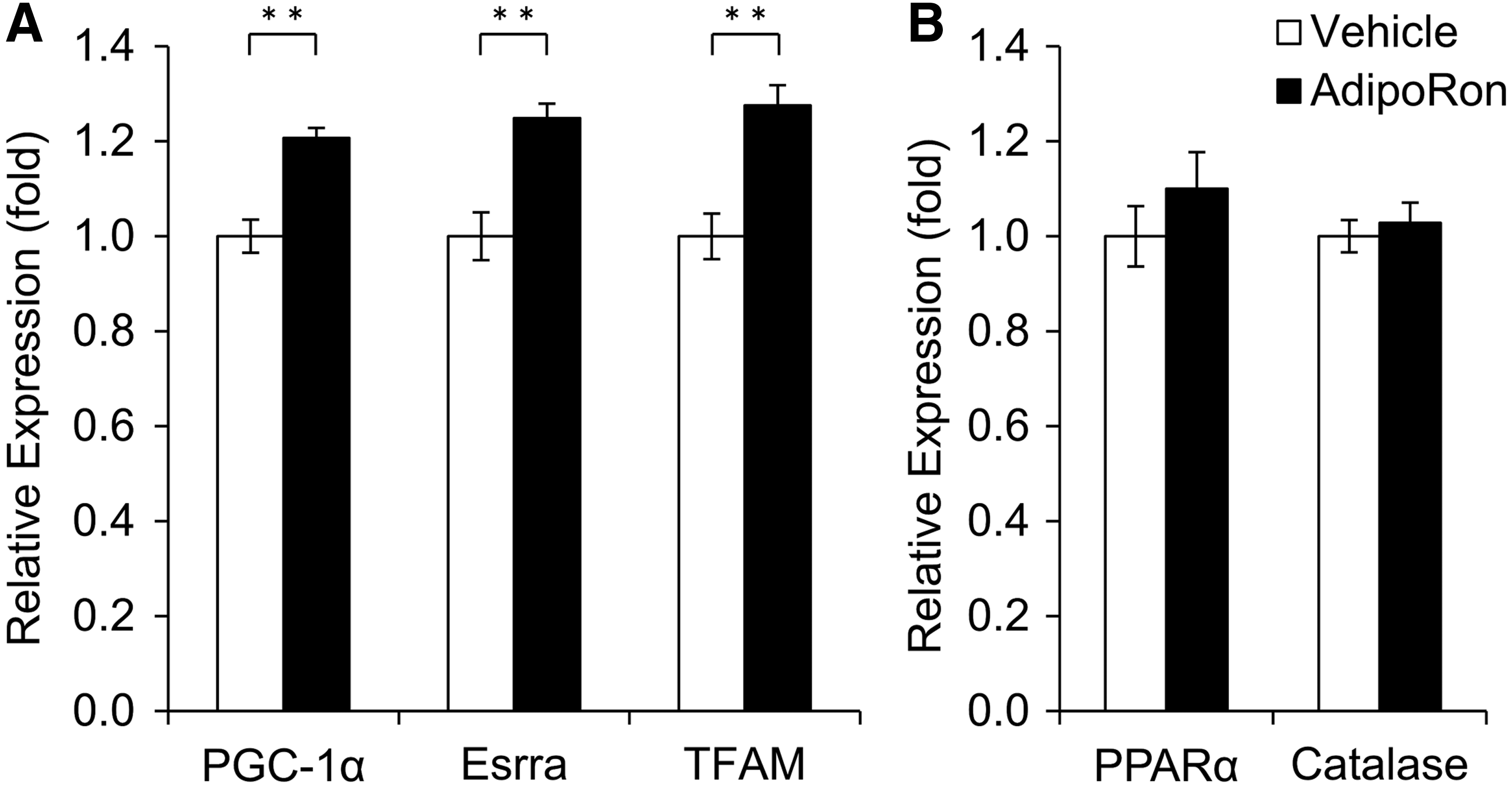
Effect of AdipoRon on mRNA levels of genes related to mitochondrial biogenesis and antioxidants in RGCs.
Discussion
In this study, we demonstrated the protein expression patterns of AdipoR1 and AdipoR2 in P5 rat retina and isolated RGCs. The AdipoR agonist AdipoRon significantly prevented the Glu-induced RGC death and ROS production. Furthermore, the neuroprotective effect of AdipoRon was linked to the increase in the mRNA expression of genes related to mitochondria biogenesis, such as Ppargc1α, Esrra, and Tfam.
It has been previously revealed that AdipoRon binds to AdipoR1 and increases the expression of genes involved in mitochondrial biogenesis such as Ppargc1α, Esrra, Tfam, and those involved with oxidative phosphorylation such as cytochrome c oxidase subunit II (mt-Co2) in skeletal muscle. 18 In this study, the mRNA expression levels of PGC-1α, Esrra, and TFAM increased in RGCs in response to the AdipoRon-treatment. PGC-1α is a master regulator of mitochondrial biogenesis and a potent inhibitor of oxidative stress.28–30 An increase in the PGC-1α level is known to protect cultured neural cells from oxidative stress-mediated death and this protection is correlated with increased expression levels of mRNA for several ROS-detoxifying enzymes, including SOD, glutathione peroxidase (GPx), and uncoupling protein (UCP). 30 Furthermore, Esrra and TFAM have been reported to be downstream effectors of PGC-1α.25,26 Esrra activated by PGC-1α induces genes that play a role in mitochondrial biogenesis31,32 and oxidative stress defense. 33 Additionally, overexpression of TFAM or treatment with recombinant TFAM protects against oxidative stress diseases in vitro and in vivo.34–36 Therefore, we speculated that the overall decrease in ROS level in AdipoRon-treated RGCs in this study occurred via increased expressions of PGC-1α, Esrra, TFAM, and their downstream ROS-detoxifying proteins, SOD, GPx, and UCP.
Several studies have showed that adiponectin can exert a modulatory effect on oxidative stress other than the regulation of PGC-1α, Esrra, and TFAM. NADPH oxidase, which is one of the main enzymes for generating superoxide ROS, is inhibited by adiponectin37–39 and also involved in glaucoma model.40,41 In addition, adiponectin induces nuclear translocation of nuclear factor-erythroid-2-related factor-2 (Nrf2), which increases transcription of heme oxygenase-1 (HO-1) and attenuates cellular oxidative stress.42–44 Therefore, we performed a qPCR analysis of the NADPH oxidase 1-4 and HO-1; however, AdipoRon did not affect the mRNA levels of the NADPH oxidase family and HO-1 (data not shown), suggesting that NADPH oxidase family and Nfr2/HO-1 would not be related to the regulation of oxidative stress by AdipoRon in RGC.
AdipoRon was originally developed as an AdipoR agonist and has been confirmed to have the same effect as adiponectin. 18 However, there are reports that AdipoRon acts by mechanisms distinct to adiponectin and exhibits a different effect. In human pancreatic cancer cell lines MIAPaCa-2, both 100 μM AdipoRon and 20 μg/mL Adiponectin elicits survival signals in an AdipoR-dependent manner, but only AdipoRon induces rapid mitochondrial dysfunction to lead cell death independently of AdipoRs. 45 In this study, the neuroprotective effect was canceled in AdipoRon at 5 μM or more. These suggest that AdipoRon produces a protective effect, but high concentrations of AdipoRon simultaneously induce cell death strongly, suggesting that the death signal may dominate the survival signal. Therefore, our immunohistochemistry data suggest the distinct expression of AdipoR2 in RGCs, but the mRNA levels of AdipoR2 target genes in rat primary cultured RGCs were not affected by the AdipoRon treatment. Further studies are needed to determine the precise mechanism by which AdipoRon exerts its neuroprotective effect.
Several possibilities and limitations of this study should be discussed. We showed that AdipoRon protects against Glu-induced RGC death. However, RGC death can be caused by various stresses including excess glutamate, oxidative stress, deprivation of neurotrophic factors, and deficits in axonal transport, and these causes of RGC death occur at different stages of various forms of glaucoma. 46 Therefore, it is necessary to determine whether AdipoRon can suppress RGC death caused by stresses other than excess glutamate in glaucoma animal models. Based on our results of relatively narrow therapeutic window for AdipoRon to suppress RGC death, efficacy and safety should be carefully evaluated in more clinically relevant glaucoma models, such as ischemia and ocular hypertension in vivo. On the other hand, considering that AdipoRon exerts a regulatory effect on oxidative stress in RGC, it may be used for other oxidative stress-related eye diseases. Because the administrations of antioxidant have the potential to prevent RGC death in the early stages of diabetic retinopathy, 47 AdipoRon treatment may also be a potential drug therapy aiming to treat early stages of diabetic retinopathy.
In conclusion, this study provides evidence that AdipoRon has a neuroprotective effect, and that this protective effect involves suppressing ROS production by upregulating PGC-1α, Esrra, and TFAM, which are involved in mitochondrial biogenesis and oxidative stress defense. Our data demonstrate that AdipoRon may be a candidate for treating glaucoma and diabetic retinopathy and that the AdipoR signaling pathway contributes to the protective effects of RGCs against death. These data provide a new approach and idea to understanding the mechanisms of RGC death and will contribute to the development of neuroprotective therapies.
Footnotes
Acknowledgments
We thank J. Nishida and F. Takeda for their help with cell culture and histopathology, and the staff of the Department of Ophthalmology, the University of Tokyo, for their assistance. The English in this document has been checked by at least 2 professional editors, both native speakers of English. For a certificate, please see: ![]()
Author Disclosure Statement
T.U. is an employee of Senju Pharmaceutical Co., Ltd. The other authors have no conflicts of interest in this study.
Funding Information
No funding was received for this article.