
Abstract
Background:
Infants born with occipital encephalocele carry a high morbidity and mortality risk. Palliative care plays a central role in their management, which is difficult due to variability in outcomes and prognosis.
Case Description:
An infant with an inoperable giant occipital who had remained admitted to the hospital since birth experienced a progressive decline in mental status and quality of life. Her family made the decision to stop enteral nutrition at 18 months of age. In contrast to the expectation of progressive fading of consciousness, the patient became increasingly alert and agitated with notable improvement in neurological status. Simultaneously, her encephalocele significantly decreased in size.
Conclusions:
The patient’s protracted end-of-life course with signs of sustained hydration, along with improved mentation and encephalocele size reduction, suggests that fluid within the defect was systemically resorbed. This case demonstrates how individual disease pathophysiology can impact the end-of-life course.
Keywords
Introduction
Encephalocele is a type of neural tube defect in which a skin or membrane-covered sac containing brain tissue and/or meninges protrudes through a bony defect in the skull. The estimated incidence of congenital encephaloceles is 1 in 10,000 live births. 1 Encephaloceles can be classified by location of the defect and vary significantly in size and symptoms. 2 About 60% of infants with encephalocele will have a chromosomal defect or other birth malformation, and 60%–80% have associated structural abnormalities. 3 The majority are diagnosed prenatally with ultrasound or fetal magnetic resonance imaging (MRI) and are confirmed by postnatal exam and imaging. 4
Occipital encephaloceles, a posterior form of the defect, account for about 74% of all encephaloceles. 2 They are further classified as giant if the sac is larger than the newborn’s head. 5 Posterior encephaloceles carry a higher risk of neurological problems such as seizures, spasticity, developmental delay, and vision impairment, as well as a poorer prognosis.1,4
Treatment for occipital encephaloceles is surgical.1,4 Risks and benefits of surgical intervention must be extensively contemplated, as postoperative complications such as hydrocephalus requiring a ventriculoperitoneal shunt are frequent, and patients with microcephaly or vital brain tissue within the sac have a poor prognosis regardless. 4 Additionally, surgery may not improve neurological deficits caused by an underlying congenital disorder. 4
Mortality rates for occipital encephaloceles have been reported between 29% and 33%.6,7 Encephaloceles containing more brain tissue beget worse outcomes; however, encephalocele size as a prognostic indicator is variable in the literature.4–7 In those who are not deemed surgical candidates, it remains challenging to predict life expectancy, as this can range from days to many years.4,5
The wide variability in patient functioning, complications, and survival necessitates early and ongoing discussions with the family about their priorities. Medical decision making should be a collaborative effort driven by the family and supported by the multidisciplinary team, and ideally should start with extensive counseling during the prenatal period. 8 At centers in which it is available, the pediatric palliative care team can be helpful in navigating care goals, optimizing communication, and providing continuity. 8 Unexpected survival time or level of functioning, and complications, such as hydrocephalus or infection, may alter goals and hopes, making treatment decisions dynamic and complex.6,8
The care of infants with inoperable encephaloceles is palliative in nature. While there is some literature to guide the general approach to comfort care, including stopping artificial nutrition, there is a lack of published work describing the challenges that may arise for patients with inoperable encephaloceles and how to best navigate them. Here, we discuss the palliative management and end-of-life course of an infant with giant occipital encephalocele. As this report involves the review of a single patient’s medical care without active intervention or interaction with the subject, it does not meet the Department of Health and Human Services definition of human subject research and therefore was exempt from full review by Cincinnati Children’s Institutional Review Board. The details and images of this case have been shared with the family’s permission.
Case Description
The female patient was born at 37 weeks’ gestational age to a 31-year-old mother, whose pregnancy was complicated by poorly controlled type 2 diabetes mellitus and a prenatally diagnosed large fetal occipital encephalocele with Chiari III malformation. Her mother was followed by an academic fetal care center, where she underwent multidisciplinary evaluation and was counseled on her infant’s projected severe neurological impairment. Prenatal consultation with the pediatric palliative care team was recommended; however, no appointment was ever scheduled. The patient was delivered via scheduled cesarean section, with planned direct admission to the neonatal intensive care unit (NICU) for further management. Her initial exam was notable for a large occipital encephalocele with overlying dysplastic skin, hypotonia, spontaneous extremity movement but no eye opening, and no other congenital abnormalities. Initial NICU management included intubation and mechanical ventilation, intravenous (IV) hydration, hypoglycemia correction, and neurosurgery and genetics consultations. Brain MRI on day of life two demonstrated a giant occipito-cervical encephalocele with extensive tissue within the encephalocele likely representing the occipital lobes, cerebellar hemispheres, and possibly the dorsal brainstem (Fig. 1). The consulting neurosurgical team determined that because the encephalocele contained vital brain tissue, surgical resection carried a high risk of morbidity and mortality and therefore would not be offered. Additionally, palliative surgical intervention was decided to likely be physiologically futile, as any drained cerebrospinal fluid (CSF) would gradually reaccumulate given the encephalocele’s connection with the ventricular system. Genetic testing with microarray analysis was normal, indicating environmental factors as the most likely etiology of her encephalocele. The palliative care team became involved during the first week of life to provide psychosocial support for the family and explore goals of care.
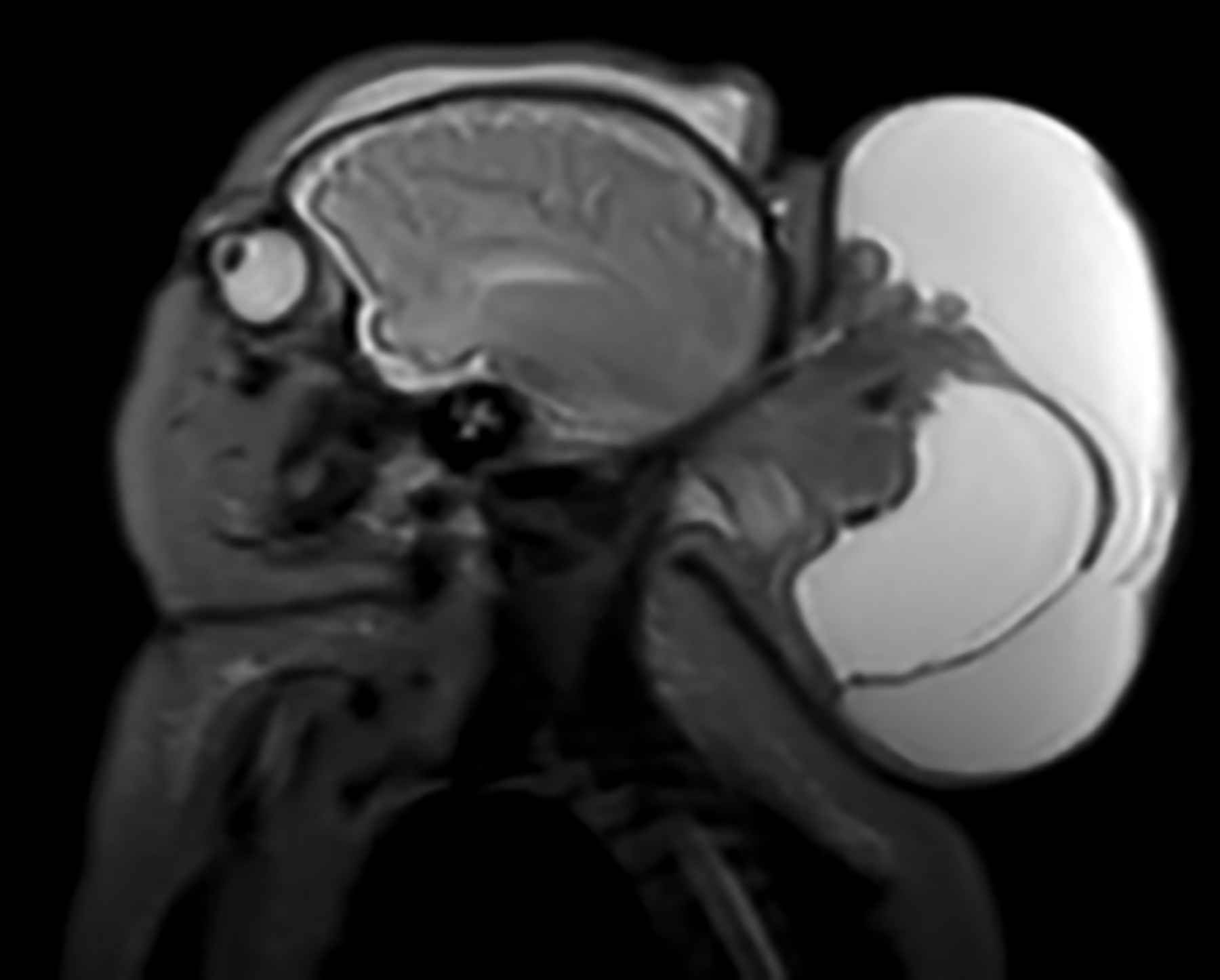
Brain magnetic resonance imaging obtained on day of life two demonstrating a giant occipito-cervical encephalocele measuring approximately 7.1 cm × 7.9 cm × 11.0 cm, with dorsal extension of the brainstem to the encephalocele base. The encephalocele also appears to contain midline cerebellum, occipital lobes, and cystic components representing trapped ventricles.
In exploring parents’ initial goals, her father voiced not wanting her to suffer if she could not have a real childhood; however, he deferred to her mother, who desired cure-directed and aggressive treatment. Her mother expressed wanting to feel like they had tried something and were not “giving up.” As the family sought a second opinion for intervention, nasogastric tube (NGT) feeds were initiated given her inability to feed by mouth. The patient was approved for surgical debulking at another institution; however, transfer was ultimately cancelled when she developed signs of increased intracranial pressure with concern for imminent herniation. She clinically stabilized but remained admitted due to her complex care needs and parental preference.
At seven months of age, imaging to reassess surgical options showed a significant increase in the size as well as brainstem presence within the encephalocele (Fig. 2). The encephalocele was again deemed inoperable. While her respiratory status remained stable and she was tolerating enteral feeds, her encephalocele continued to enlarge. In concordance with her family’s wishes, the care team prioritized pain control and comfort.
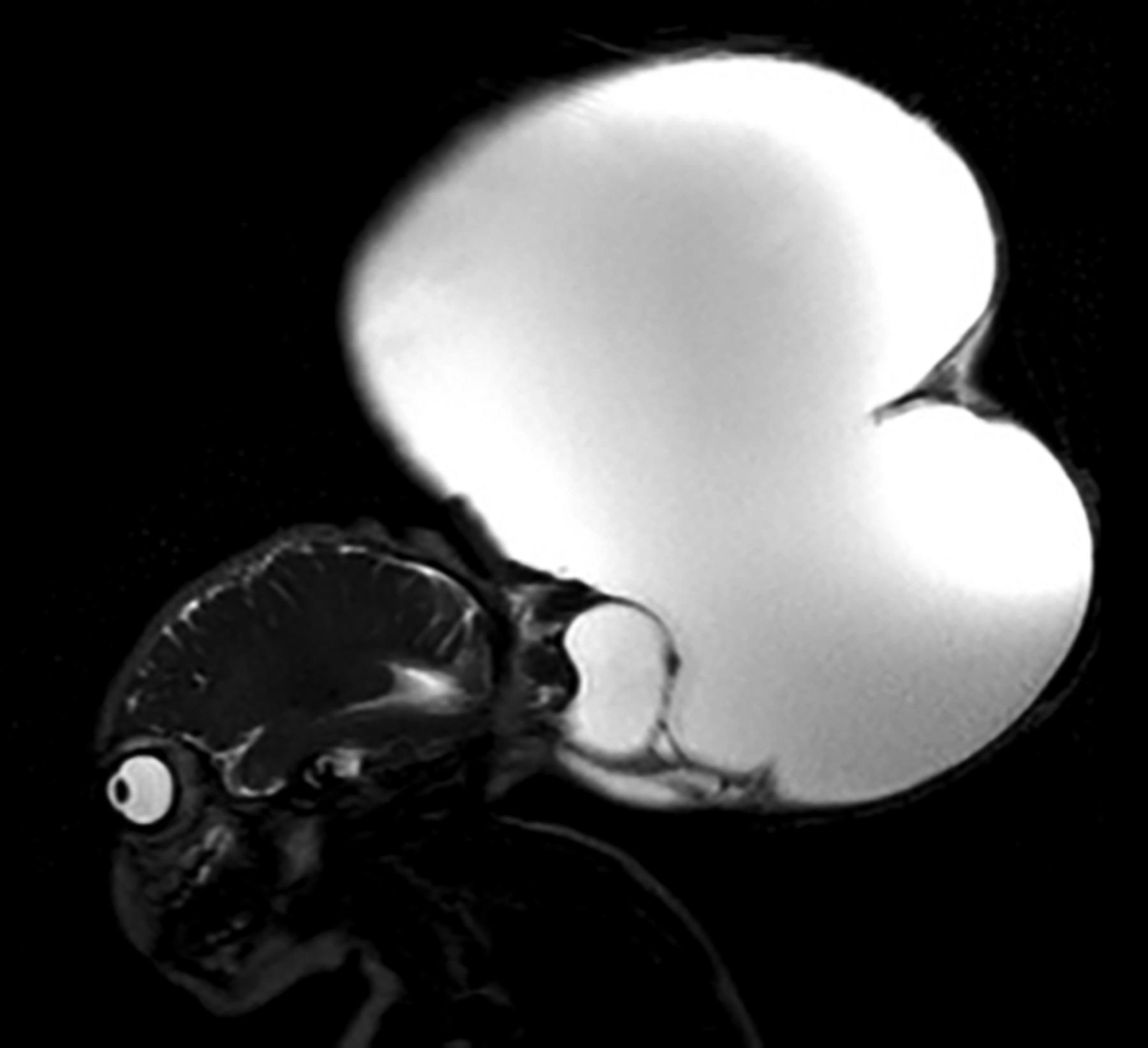
Repeat brain magnetic resonance imaging (MRI) obtained around 7 months of life to reassess surgical options. MRI shows a significant interval increase in the cystic component of the encephalocele (21.0 cm × 14.6 cm × 27.0 cm), which contains trapped ventricles and venous structures in addition to components of the occipital lobes, cerebellum, and dorsal brainstem.
By 18 months of age, the patient had become minimally responsive with only brief periods of wakefulness secondary to continued encephalocele growth. She was not interactive and did not cry, but often seemed uncomfortable with irregular respirations and a high secretion burden. Her parents recognized the extent of decline in mental status and overall quality of life and agreed that she was actively suffering. After extensive discussion among the care team and family and with acknowledgment of the patient’s inability to feed orally and lack of hunger cues, the patient’s NGT feeds were discontinued per the parents’ wishes. Parents were counseled about expected changes following the removal of artificial nutrition and hydration, and that anticipated death would likely be within two weeks.
At this time, she remained admitted to a general pediatric inpatient unit. The palliative care team was managing her pain and agitation with scheduled enteral methadone, gabapentin, diazepam, and nortriptyline with intermittent morphine and lorazepam for breakthrough symptoms. Her NGT was maintained for medication administration. As time progressed following feed discontinuation, she seemed to have an overall improvement in neurological status. She was spending a significant portion of the day alert and with her eyes open, vocalizing frequently, and moving her arms and legs. Her improvement in mental status coincided with increased agitation despite breakthrough medications, requiring escalation in her methadone, nortriptyline, and diazepam more than 20 days following feed withdrawal. She was requiring more frequent nursing staff presence at the bedside to manage her agitation, and bedside staff conveyed distress about their lack of ability to comfort her.
As her mental status improved, she was simultaneously noted to have significant changes on physical exam. Her encephalocele had substantially decreased in size by roughly half since the withdrawal of enteral nutrition. Despite very limited intake from medications and flushes, stool and urine output continued. Nearly four weeks from cessation of feeds, she was producing up to 0.6 mL/Kg/hour of output.
At over four weeks following care redirection, the patient was awake, alert, and crying despite aggressive increases in comfort medications. She required central line placement due to her inability to tolerate enteral medications and failed peripheral IV attempts. Following return from the operating room, she had an abrupt change in clinical status from being awake and crying to unresponsive and somnolent. The patient died shortly after at 19 months of age, 33 days following withdrawal of artificial nutrition and hydration (WANH).
Discussion
Withholding or withdrawing medically provided fluids and nutrition is ethically and legally permissible, including in the pediatric population, when it is determined that the patient will gain no net benefit from the intervention.9,10 This includes such instances as when these interventions would prolong the dying process and increase the risk of harm or suffering or when a child has permanent unawareness and inability to interact with the environment. 9 While acceptable, WANH in children is a relatively infrequent practice, even at large high-acuity centers. 11 Shared decision making with the parents or caregivers is essential, as cessation of these interventions is not morally required.9,10 The health care team should discuss the child’s expected course, including common symptoms and potential complications, and how these will be managed to ensure comfort. 12
Symptom prevalence in children during the final days of life can be high and frequently includes lack of energy, irritability, pain, extremity swelling, dyspnea, and gastrointestinal disturbances. 13 In comparison, the process of dying following WANH has been described in both adult and pediatric populations as high quality and peaceful.14–17 In this circumstance, the immediate cause of death is dehydration, which is hypothesized to induce the accumulation of ketones and other metabolites that create a sedative effect.17,18 Observations of patients following the decision to forgo nutrition and hydration describe increased comfort and peacefulness, development of dry mucous membranes, and confusion or lethargy with eventual fading of consciousness.14,19–21 Importantly, parents also report satisfaction with the decision for WANH and perceive their child’s death as good quality and peaceful. 12 Time to death following WANH is highly variable; Hellmann et al. found a range of 2–37 days in infants, and the majority of patients die within three weeks.14,15,18
In our case, the patient’s 33-day duration of life after fluid withdrawal is still within the expected range; however, her increasing alertness and neurological improvement were not consistent with what is described in the literature. As previously discussed, progressive dehydration is suspected to create a state of tranquility, as opposed to the increasing agitation and irritability seen in this patient. Additionally, her change in mental status and more frequent periods of conscious awareness that surpassed her condition prior to cessation of nutrition were unexpected. Her neurological improvement and sustained hydration paralleled and were suspected to be a direct consequence of her shrinking encephalocele.
While there are several documented cases of spontaneous resolution or resorption of encephalocele variants, they involve an adult patient with a post-traumatic defect or children in which the defect resolved within six months or less of birth.22–24 None of these cases specifically discusses an occipital encephalocele. Furthermore, there is a lack of investigation into the potential physiological mechanism for such a process. After conferring with multiple subspecialists at our institution, the proposed etiology for our patient’s clinical course is a hypernatremic state induced by dehydration. An elevation in plasma osmolality creates an osmotic gradient, which pulls water out of the brain parenchyma and into the intravascular space.25,26 This transition of fluid from the central nervous system into the bloodstream would align with the decreasing size of her defect; it would also cause a reduction in intracranial pressure that could explain her improved mentation and theoretically prolong survival by providing systemic hydration. An alternate theory is that dehydration led to decreased CSF production, resulting in encephalocele size reduction; however, this would not account for her maintained urine output and longer survival. Despite uncertainty about the exact pathophysiology of this patient’s course, the resorption of fluid from her encephalocele is likely what sustained her and reversed her decline in neurological status. Her abrupt change in clinical status just prior to death was suspected to be related to anesthesia during line placement attempts.
Given that the extensively deliberated and difficult decision to stop enteral feeding was made with the goal of ending her suffering, this patient’s increasing awareness and discomfort requiring medication escalation and procedural intervention was anxiety provoking for both the medical team and family. Many team members had anticipated that the patient would instead become progressively more somnolent, and they shared feelings of distress from the incongruence between their experience and preconceived hopes and expectations. WANH is an accepted though infrequent practice at our institution, and this stark change in mentation is not an effect that has been seen in previous patients or reported elsewhere to our knowledge. Potential limitations to this study include that it is only a single case with findings that may not be generalizable, and that the case is described from the perspective of only a few providers and does not include other staff or family perceptions. Additional reports are needed to better understand the end-of-life course in children with neurological defects.
Conclusions
Careful consideration and ongoing reassessment of each patient’s disease pathophysiology and clinical status are essential for providers to tailor individualized care that maximizes quality and minimizes distress at the end of life. For patients with neurological disease, end-of-life anticipatory guidance should include the possibility of improvement in existing neurological deficits and mental status. Additionally, a potentially protracted end-of-life course should be anticipated in patients with any reservoir of fluid (intracranial or otherwise), and caregivers should be counseled accordingly.
This case also highlights the importance of early palliative care involvement, and prenatal consultation if available, for children with anticipated severe neurological impairment. In such cases in which a family’s initial goals are most in line with aggressive intervention but a comfort-focused approach would be reasonable, palliative care support and guidance are still warranted. Although cases like this are rare, they contribute to a better understanding of the dying process, especially in the practice of WANH, and more broadly of how to optimize the delivery of palliative care across a child’s lifespan.
Footnotes
Author Disclosure Statement
The authors have no conflicts of interest to disclose.
Funding Information
No funding was received for this article.