
Abstract
Methicillin-resistant Staphylococcus aureus (MRSA) is a multidrug-resistant bacterium that causes serious infections worldwide. This pathogen is resistant to all beta lactam antibiotics due the presence of PBP2a, a transpeptidase enzyme that presents very low beta-lactam affinity. Here we report the generation and characterization of mouse monoclonal antibodies to PBP2a of MRSA strains. Two clones were obtained and characterized by immunoassays (ELISA, avidity index determination, and immunoblotting), isotyping, association/dissociation rate constants by surface plasmon resonance (SPR), and flow cytometry. Clone 38, which showed the best avidity and affinity, bound to PBP2a located on the bacterial surface by flow cytometry. Further studies are warranted in order to evaluate if these antibodies may help inhibit bacterial growth and be used to treat infections by MRSA.
Introduction
N
In this study, we report the generation and characterization of specific murine monoclonal antibodies (MAbs) to PBP2a from MRSA strains. These results may be the first step toward the development of passive immunotherapy to treat infections caused by these bacteria.
Material and Methods
Bacterial strains
MRSA COL and MRSA COLΔmecA(16) strains were received from Unité des Agents Antibactériens–Institut Pasteur. BEC (Brazilian epidemic clone)(17) was received from the Instituto de Microbiologia Paulo de Goés, Universidade Federal do Rio de Janeiro. A clinical methicillin-sensitive Staphylococcus aureus (MSSA) strain was received from the Hospital de Pronto Socorro-Porto Alegre.
Recombinant PBP2a fragment
The PBP2a fragment corresponding to amino acids 372–447 was obtained as previously described(18) with minor modifications in protein expression and purification.
Monoclonal antibody production and purification
Four female BALB/c mice, age 4–8 weeks, received an initial dose of 100 μg of pCI-Neo plasmid: fragment of MRSA mecA gene,(14) followed by a 10 μg dose of purified recombinant protein 14 days later, corresponding to an internal region of the MRSAPBP2a,(21) emulsified in complete Freund's adjuvant (Sigma, St. Louis, MO). This was followed by a second dose 14 days later, this time emulsified in incomplete Freund's adjuvant. Fourteen days later, the animal with the best immune response (evaluated by enzyme-linked immunoassay [ELISA]) received an intravenous dose of 10 μg of purified protein diluted in phosphate-buffered saline (PBS). Three days after the IV injection, the animal was subjected to euthanasia by asphyxia in CO2 (protocol approved by Oswaldo Cruz Foundation Animal Ethics Committee, no. L0009-07), and the spleen was aseptically removed for cell fusion. The serum of the animal with the best immune response was used as a positive control for other immunological tests.
Lymphocytes prepared from each spleen were fused to an SP2/0-Ag14 (CRL-1581, ATCC, Sigma-Aldrich) myeloma cell line and subsequently plated in hypoxanthine-aminopterin-thymidine selection medium at 37°C and CO2 5%. Polyethylene glycol-induced cell fusion, subsequent plating, and feeding were all performed in accordance with the production of MAbs protocol described by Yokoyama and colleagues.(19) Resulting hybridomas were screened 14 days following fusion by ELISA for antibody recognition of the PBP2a fragment, as described below and subsequently cloned by limiting dilution. Hybridoma cells were grown in DMEN (Dulbecco's Modified Eagle Medium, Life Technologies, Carlsbad, CA) with 10% of fetal bovine serum (FBS) in bottles of 100 mL at 37°C and 5% of CO2. The positive clones were cloned a second time. Hybridoma supernatants were harvested by centrifugation and passed through a 0.2 mm pore-size filter before HPLC purification with mAB Select sure protein A resin (GE Healthcare, Uppsala, Sweden). Purified antibodies were dialyzed against diluted PBS (50 times), concentrated with Amicon ultrafiltration units (Merck Millipore, Cork, Ireland) quantified at 220 nm using a Nanodrop (Thermo Scientific, Waltham, MA) apparatus and stored at −20°C.
Isotyping
The antibody isotype produced by the selected hybridoma cell line was determined using the Mouse MonoAB ID isotyping kit (HRP, Zymed, San Francisco, CA), according to the manufacturer's instructions.
ELISA
Enzyme-linked immunosorbent assay (ELISA) was used to determine the presence of specific anti-PBP2a antibodies in the serum of immunized mice and hybridoma supernantants. Plates were coated with 10 μg/mL recombinant protein, diluted in 0.1 M carbonate/bicarbonate coating buffer (pH 9.6), and incubated overnight at 4°C. After blocking (PBS containing 5% non-fat milk, for 2 h at 37°C) the plates were incubated for a further 2 h at 37°C with serum or antibody supernatants. Subsequently, the plates were washed (PBS 0.05% Tween-20, Sigma-Aldrich) and incubated at 37°C for 2 h with the secondary antibody (anti-mouse polyvalent peroxidase-conjugated; Sigma-Aldrich) diluted 1:10,000. To develop the reaction, plates were washed as previously done and incubated for 5 min at room temperature with a chromogenic substrate solution (TMB peroxidase, Bio-Rad, Hercules, CA). To stop the reaction, 50 μL/well 2 M H2SO4 was added and the optical density (OD) was read on a microplate reader Benchmark (Bio-Rad) at 450 nm.
Immunoblotting
A MRSA (BEC) strain, a methicillin-sensitive Staphylococcus aureus (MSSA) strain, and the BL-21 DE3 (Life Technologies, Carlsbad, CA) Escherichia coli strain were grown to exponential phase. One mL of each sample was centrifuged and lysed by agitation in glass beads in a mini-Bead Beater device (Biospec Products, Bartlesville, OK) three times for 30 s. One aliquot of each sample was subjected to electrophoresis in 12% denaturing polyacrylamide gel (SDS-PAGE), and the proteins were later transferred to a nylon membrane (Hybond N, Bio-Rad). The membrane was blocked under mild agitation for 23 h in PBS buffer containing 10% skimmed milk and 1% BSA (bovine serum albumin). The membrane was washed three times in PBS and Tween-20 (0.05%) and three times in PBS. Subsequently, the latter was incubated for 2 h, with supernatant of anti-PBP2a monoclonal antibody diluted in PBS at a 1:1 ratio. After the incubation, the membrane was washed as previously described, and incubated for 90 min with the alkaline phosphatase conjugate (murine anti-IgG antibody, Sigma A3688) at a 1:15,000 ratio. After this period, the PBS was repeated, and development with Western Blue substrate for alkaline phosphatase (Promega, Madison, WI) was performed.
Determination of association and dissociation constants for MAbs by surface plasmon resonance
To determine the affinity of the anti-PBP2a monoclonal antibody, surface plasmon resonance (SPR) measurements were performed using research-grade CM5 sensor chips on a Biacore X® instrument (BIAcore AB). Coupling reagents and HBS-EP buffer (10 mM Hepes, 150 mM NaCl, 3 mM EDTA, 0.005% P20 [Tween-20], pH 7.4) were from GE Healthcare. Immobilization of the PBP antigen was performed by amino coupling at 25°C using HBS-EP as running buffer and a flow rate of 5 μL/min. High density surfaces were prepared for assay development and low density surfaces were prepared for kinetic analyses. All kinetic measurements were made at 25°C with a flow rate of 10 μL/min. The samples were diluted in running buffer (HBS-EP) by serial dilution, and long association and dissociation time (9 min and 20 min, respectively) were employed in order to detect the slow dissociation of the samples. The regeneration was achieved by a 30 s injection of a 10 mM glycine solution (pH 1.5). In order to correct the data for bulk refractive index changes, the reference surface responses were subtracted from the responses of the active surface with the immobilized PBP antigen. All kinetic data were also submitted to double referencing,(20) a process in which the response from a blank injection is subtracted from all binding curves, correcting the data by removing artifacts that may systematically occur in every injection. All kinetic data were analyzed by global fitting of at least 10 different sensorgrams, including duplicates (Fig. 2), using the BIAevaluation software (v4.1), and employing the simple 1:1 Langmuir model.
Flow cytometry—PBP2a recognition at bacterium surface
Flow cytometry analyses were performed to confirm binding of monoclonal antibodies to PBP2a on the bacterial surface. Single colonies of MRSA COL and MRSA COLΔmecA were grown overnight at 37°C in Luria broth containing oxacillin (COL) or tetracycline (COLΔmecA) at 10 μg/mL. On the next day, a pre-inoculum of each bacteria was grown to mid-log phase (optical density at 600 nm [OD600] of 0.6) and 1 mL of each culture was centrifuged for 5 min at 13,000 g. Sediment was washed and suspended in 1 mL PBS, being further sonicated at 4 kHz for 120 s to promote cluster and aggregate dissolution. Two hundred μL of each bacterial suspension were centrifuged and the sediment suspended in 50 μL of PBS and 0.5% bovine serum albumin (BSA). Samples were incubated for 30 min at 4°C with 25 ng of the purified monoclonal antibody. Samples were washed, suspended in 50 μL of PBS containing 2.5 μL of fluorescein isothiocianate (FITC) conjugated-goat anti-mouse IgG (anti-IgG-FITC; Sigma Chemical Co.), and incubated at room temperature for 30 min in a dark chamber. Bacteria were washed twice with PBS and fixed for 15 min at 4°C in PBS containing 2% paraformaldehyde. Bacterial samples without primary (purified monoclonal antibody) and secondary antibodies (anti-IgG-FITC) and samples incubated only with secondary antibody were used as negative controls.
Flow cytometric acquisitions were performed using a BD FACSCalibur (Becton-Dickinson, Franklin Lakes, NJ) equipped with a 488 nm argon laser and 530/30 nm band pass filter for the detection of FITC emission. For each sample, a gate of the bacteria population was set according to size (FSC) and granularity (SSC), thus removing all debris. Both linear and log mode acquisitions for FSC and SSC were performed during the protocol setup. A total of 20,000 events in the bacteria gate were acquired. The acquisition was done using CellQuest software (Becton-Dickinson), and data analyses were performed using FlowJo software (TreeStar, Ashland, OR). The results were reported as the percentage of stained bacteria (FITC+) from MRSA COL and MRSA COLΔmecA (negative control) samples.
Results
Generation of murine anti-PBP2a monoclonal antibodies
After the fusion process, the supernatant from 96 wells (hybridomas) was analyzed by ELISA. From this total, the five best samples were selected, the cells were expanded (cloning), and once again, the resulting supernatants were analyzed by ELISA. Positive samples were validated by immunoblotting against the purified recombinant protein (PBP2a). The two selected clones (clones 10 and 38) were isotyped as IgG2b and IgG1, respectively.
In vitro PBP2a recognition—immunoblotting
In order to investigate the specificity and binding capacity, immunoblotting was performed with proteins from bacteria presenting or not presenting PBP2a (CEB and COL MRSA, a MSSA methicillin-sensitive S. aureus strain, and an E. coli strain in which the recombinant protein was generated). The results show that antibody 38 recognized a protein with molecular weight of approximately 76 kDa in the MRSA strain, which corresponds to the molecular weight of PBP2a. No reactivity of the monoclonal 38 antibody was seen with proteins of methicillin-sensitive S. aureus (PBP2a-negative) nor with the E. coli strain (data not shown). The result of the immunoblotting test of MRSA and MSSA lysates against the supernatant containing anti-PBP2a monoclonal antibodies is shown in Figure 1. Immunoblotting of MRSA lysate against supernatants containing anti-PBP2a MAbs from clones 10 and 38 is included as supplementary data (Supplementary Fig. 1).
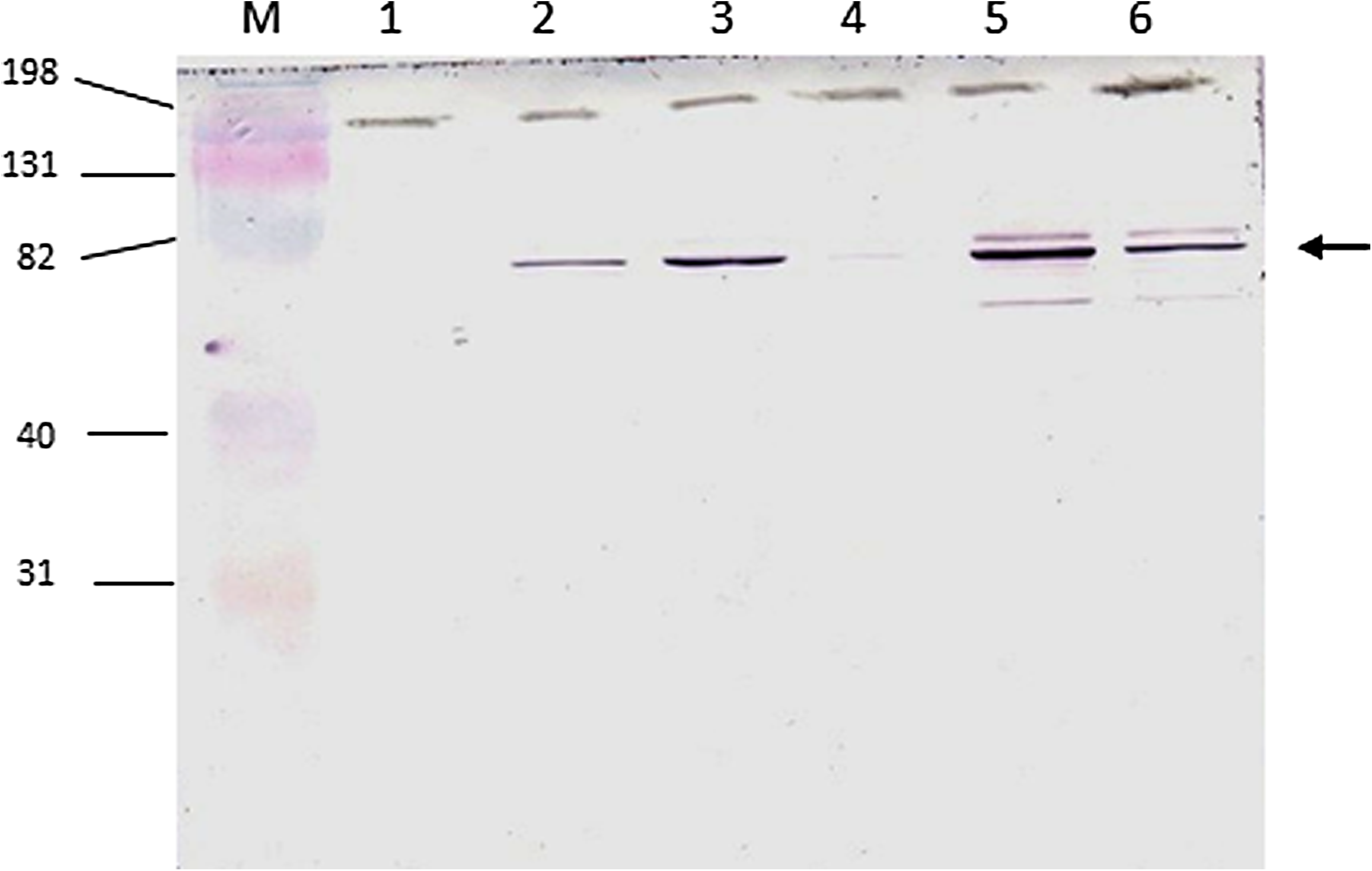
Immunoblotting of bacterial lysates and supernatant containing monoclonal antibody anti-PBP2a (clone 38). M, protein molecular weight marker (Kaleidoscope 161-0324, Bio-Rad); lane 1, MSSA (methicillin-sensitive S. aureus) sample grown in exponential phase; lanes 2, 3, MRSA samples grown in exponential phase; lane 4, MSSA sample grown in stationary phase (overnight); lanes 5, 6, MRSA samples grown in stationary phase. The arrow indicates molecular weight of PBP2a (76 kDa). Molecular weight markers are indicated on the left side.
Analysis of antibody binding and dissociation constants by surface plasmon resonance
The results obtained by surface plasmon resonance (SPR) confirmed the preliminary results of antibody avidity (ELISA), where the clone 38 antibody displayed higher avidity than that from clone 10. According to the data shown in Table 1, clone 38 shows an affinity that is 450 times higher than clone 10. This affinity is mainly due to its higher association rate, which is about 100 times higher than clone 10, although there is also a minor contribution due to a slightly slower dissociation rate. Due to the very high affinity of clone 38, which lies close to the equipment detection threshold, considerable care in assay design and execution were necessary in order to obtain an excellent adjustment to the experimental data using Langmuir's model, indicating reliability in the derived parameters.
It is worth noting that a sensor chip presenting a very low antigen density was used to perform all the kinetic measurements. This was necessary to reduce avidity effects since bivalent antibodies were used as the species in solution and the data were subsequently fitted to a 1:1 Langmuir model. Therefore, this information should be considered when analyzing the binding rates and constants.
Figure 2 shows the interaction between recombinant PBP2a (antigen) and clone 38 (Fig. 2A) and clone 10 (Fig. 2B) monoclonal antibodies. Dashed curves represent SPR data in concentrations noted in the right key. All samples were analyzed in duplicate, and the 1:1 Langmuir's theoretical model is shown in black under each curve. Response units (RU) are represented in the vertical axis, and time is represented on the horizontal axis in seconds. The closest lines to the horizontal axis represent the baseline for each sample (negative control).
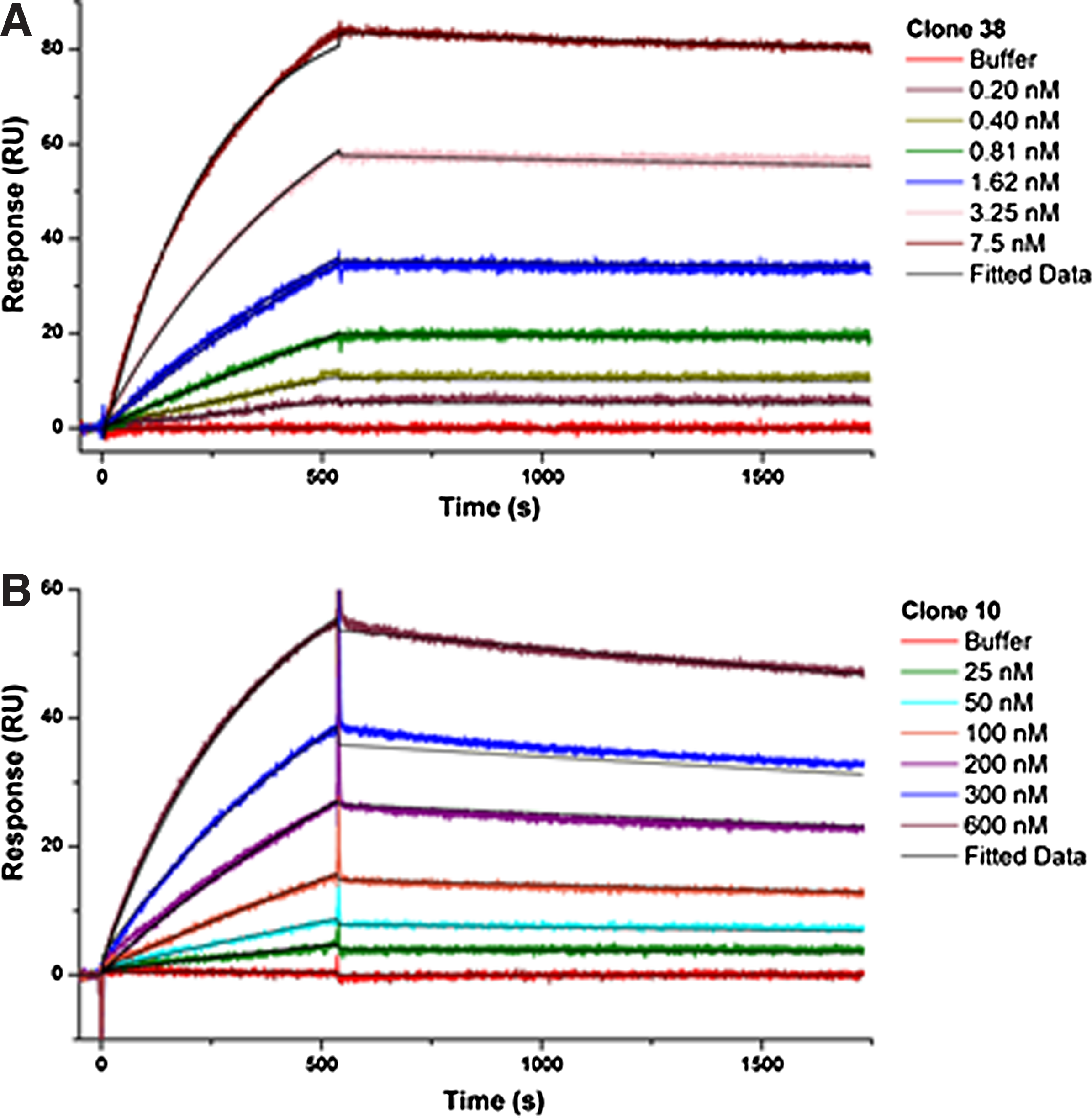
Interaction of antigen PBP with clone 38 (
PBP2a is recognized on bacterium surface—flow cytometry
The objective of the flow cytometry test is to validate the capacity for target recognition on the bacterium in its native form by the monoclonal antibody. In previous tests (immunoblotting), we observed target recognition with proteins subjected to a denaturation process, which occurs during protein separation by electrophoresis in denaturing polyacrylamide gel. Again, we analyzed a negative control strain (PBP2a-negative—MRSA COLΔmecA) and a MRSA strain (COL) grown in exponential and stationary phases. The results show that antibody (clone 38) recognized the target on the bacterial surface. In order to block the protein A of S. aureus, a previous treatment with murine serum was performed and no differences were observed (data not shown), indicating that the presence of protein A on the surface of S. aureus was not able to inhibit the antibody binding to PBP2a. These results can be seen in Figure 3.
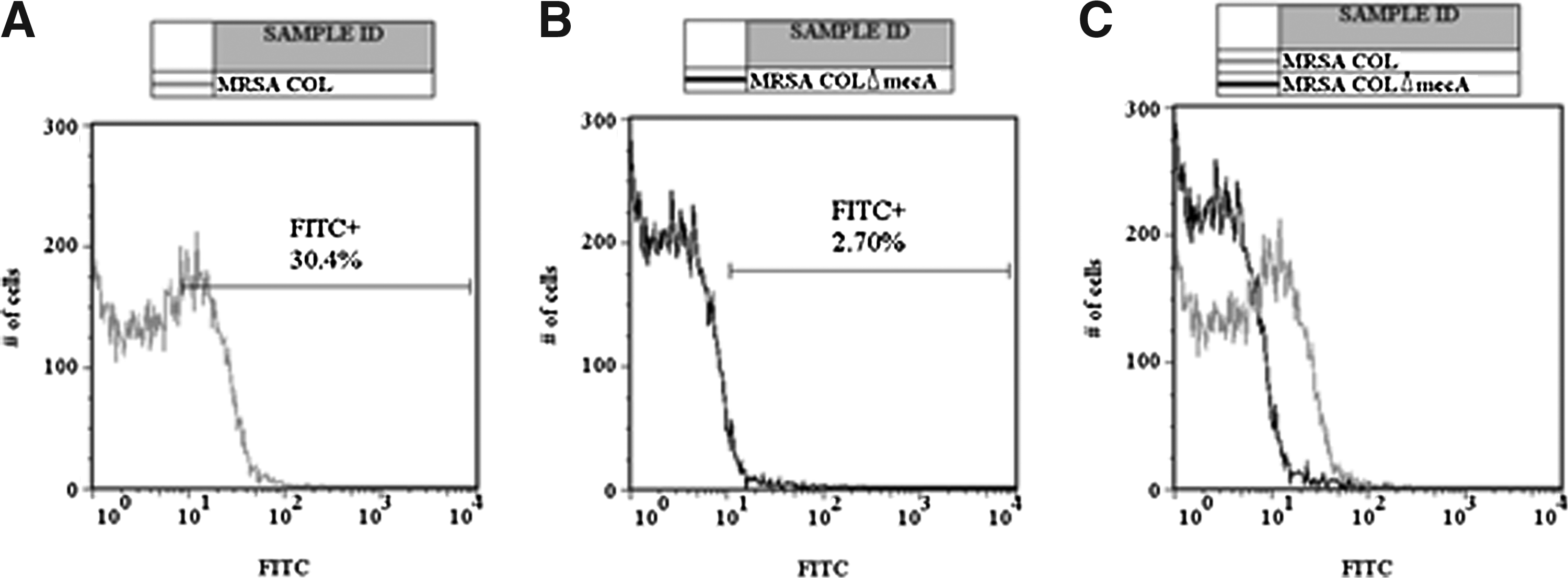
Flow cytometric evaluation of MRSA COL and MRSA COLΔmecA incubated with anti-PBP2a monoclonal antibody (clone 38) and stained with goat anti-mouse IgG-FITC conjugate. Histograms of FITC fluorescence (X axis) vs. number of cells (Y axis). (
Discussion
The emergence of infections due to bacteria multi-resistant to antimicrobials is presenting at an alarming rate. The prevalence of hospital infections caused by MRSA has increased worldwide and results in high morbidity and elevated cost due to the intensive use of antimicrobials and to a longer period of patient hospitalization. 21 Treatment based on chemotherapy has been showing signs of exhaustion, with the appearance of very few really novel and efficient drugs on the market. 22 In this context, passive immunotherapy (re)appears as a promising alternative, presenting a range of advantageous features compared to conventional chemotherapy, among them, lower toxicity, higher plasma half-life (which facilitates treatment by dose reduction and maybe a lower final treatment cost), and, especially in the case of the product presented here, selective toxicity as recommended by Ehrlich (in which the drug selectively eliminates the referred pathogen). 23 PBP2a is a unique feature of MRSA strains whose structure was elucidated in 2002. 24 Based on preliminary studies with a DNA vaccine in murine models13,14 and immunostructural analysis, an internal region coding the active center of the transpeptidase domain of PBP2a was chosen as antigen.
Two clones of MABs able to recognize PBP2a by ELISA were generated in this study. However, important differences among these clones were observed. Both MABs were able to recognize PBP2a in immunoassays, and the binding with denatured recombinant protein and cell lysates in Western blotting suggest that these antibodies are directed against linear epitopes.
Since PBP2a is an enzyme involved in cell wall biosynthesis, a process that happens predominantly during cell division, it is probably more exposed in the exponential phase of bacterial growth. In order to investigate if selected MABs were able to recognize PBP2a in MRSA cells in both the exponential and stationary phases, immunoblots with bacteria were performed, presenting positive results in both conditions. Furthermore, no reactions with methicillin-sensitive S. aureus (PBP2a negative) and E. coli (a gram negative bacteria) were seen, which is indicative that this antibody binds specifically to PBP2a.
Affinity assays with the selected MABs showed differences between these clones. In both tests (avidity and SPR), clone 38 was shown to be a better binder than clone 10. The higher association rate and lower dissociation rate of clone 38 (KD=57 picomolar) means that this antibody interacts strongly with the target remaining firmly bound during the period analyzed.
Flow cytometry assays were performed in order to demonstrate that this MAB is able to recognize PBP2a on the bacterial surface. PBPs are enzymes that participate actively in bacterial cell division, 12 and thus the exposure of these enzymes in principle would be greater during the exponential phase. However, in a heterogeneous population not all bacteria will be multiplying at the same time. The results of the flow cytometry are in agreement with these previous hypotheses, since about 30% of the analyzed cells showed a positive signal during the analysis.
In summary, we report the generation and extensive characterization of monoclonal antibodies that specifically recognize PBP2a of MRSA strains. Anti-PBP2a monoclonal antibodies have been used in in vitro diagnosis of MRSA strains25–27 ; however, none of these has been tested for the treatment of infections caused by this bacterium. The antibodies characterized here will be employed in future studies that will investigate their bactericidal activity and bestowed protection in animal models, with or without formulation or the simultaneous administration of conventional antibiotics.
Footnotes
Acknowledgments
We are grateful to Drs. Patrice Courvalin from Unité des Agents Antibactérien –Institut Pasteur, Agnes Figueiredo from Instituto de Microbiologia Paulo de Goés, Universidade Federal do Rio de Janeiro, and Juçara Steyer from Hospital de Pronto Socorro–Porto Alegre, who kindly furnished the bacterial strains used in this study. We are also grateful to Dr. Richard C. Garrat, who allowed us to use the BiaCore system in his laboratory (Center for Structural Molecular Biotechnology, Physics Institute, Universidade Federal de São Carlos) for this study and who kindly reviewed the final revision of this manuscript.
Author Disclosure Statement
The authors have no financial interests to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.