
Abstract
Cell wall recycling and β-lactam antibiotic resistance are linked in Enterobacteriaceae and in Pseudomonas aeruginosa. This process involves a large number of murolytic enzymes, among them a cytoplasmic peptidoglycan amidase AmpD, which plays an essential role by cleaving the peptide stem from key intermediates en route to the β-lactamase production (a resistance mechanism) and cell wall recycling. Uniquely, P. aeruginosa has two additional paralogues of AmpD, designated AmpDh2 and AmpDh3, which are periplasmic enzymes. Despite the fact that AmpDh2 and AmpDh3 share a common motif for their respective catalytic domains, they are each comprised of multidomain architectures and exhibit distinct oligomerization properties. We review herein the structural and biochemical properties of orthologous and paralogous AmpD proteins and discuss their implication in cell wall recycling and antibiotic resistance processes.
Introduction
G
The bacterial cell wall is comprised of crosslinked strands of peptidoglycan (PG), which encase the entire cytoplasmatic membrane. A healthy cell wall is critical for survival of bacteria and homeostasis requires simultaneous biosynthetic and degradative processes. Gram-negative bacteria have evolved to recycle the cell wall that is degraded, as a means of existence. During a single doubling process, up to 60% of the PG is turned over in Gram-negative bacteria. Cell wall-active antibiotics such as β-lactams cause defect in the cell wall,1–3 which leads to the degradative events to generate some of the same recycling intermediates. This is the common link between the two events. A large number of enzymes are involved in these processes.
AmpD peptidoglycan amidases in cell wall recycling
Bacterial cell wall recycling is set in motion by degradation of PG, the major constituent of the cell wall, by the action of lytic transglycosylases4–6
(Fig. 1). These enzymes digest the peptidoglycan saccharide backbone to generate various fragmentation products, referred to as muropeptides.
7
The major end product is N-acetyl-β-
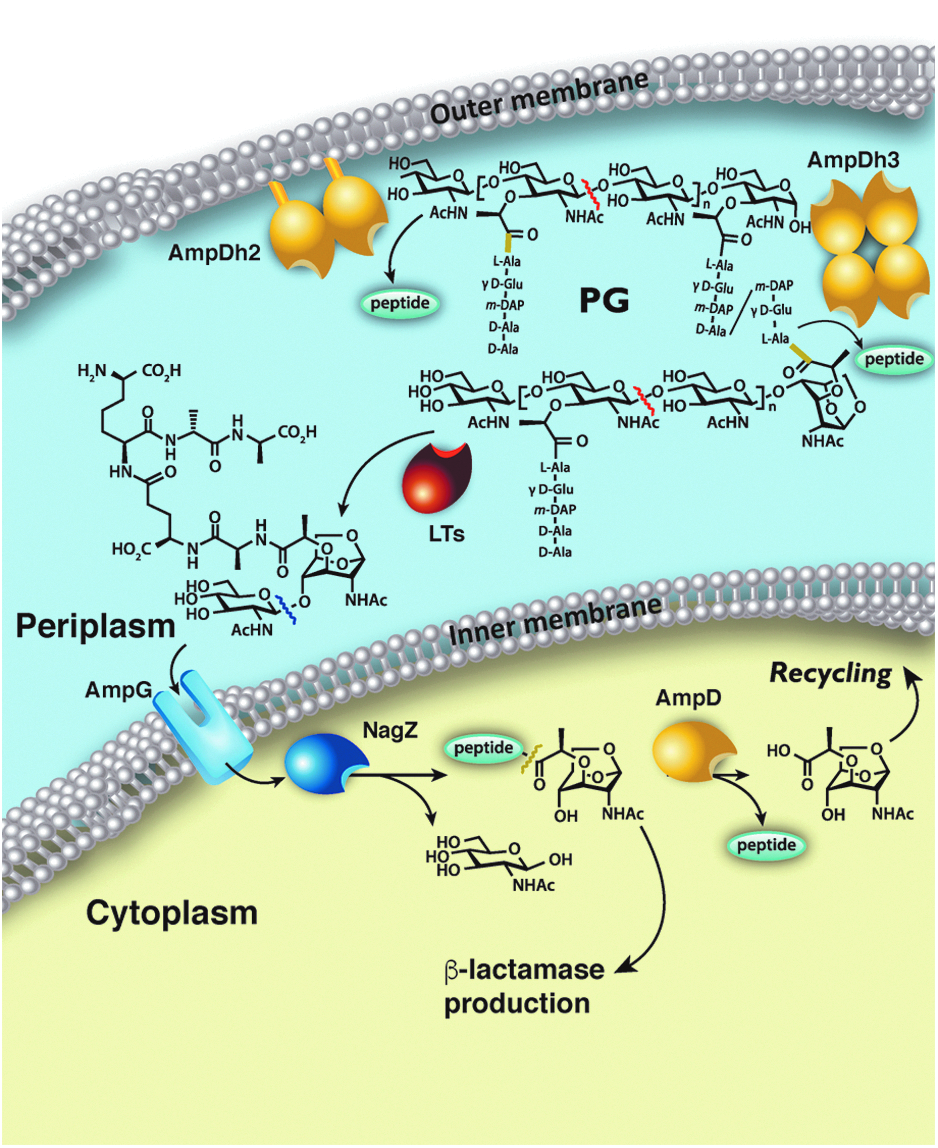
General cell wall recycling mechanism in Gram-negative bacteria. Figure adapted from Ref. 14 Yellow bond indicates where AmpD, AmpDh2, and AmpDh3 cleave. Red and blue lines indicate bonds cleaved by LTs and NagZ, respectively.
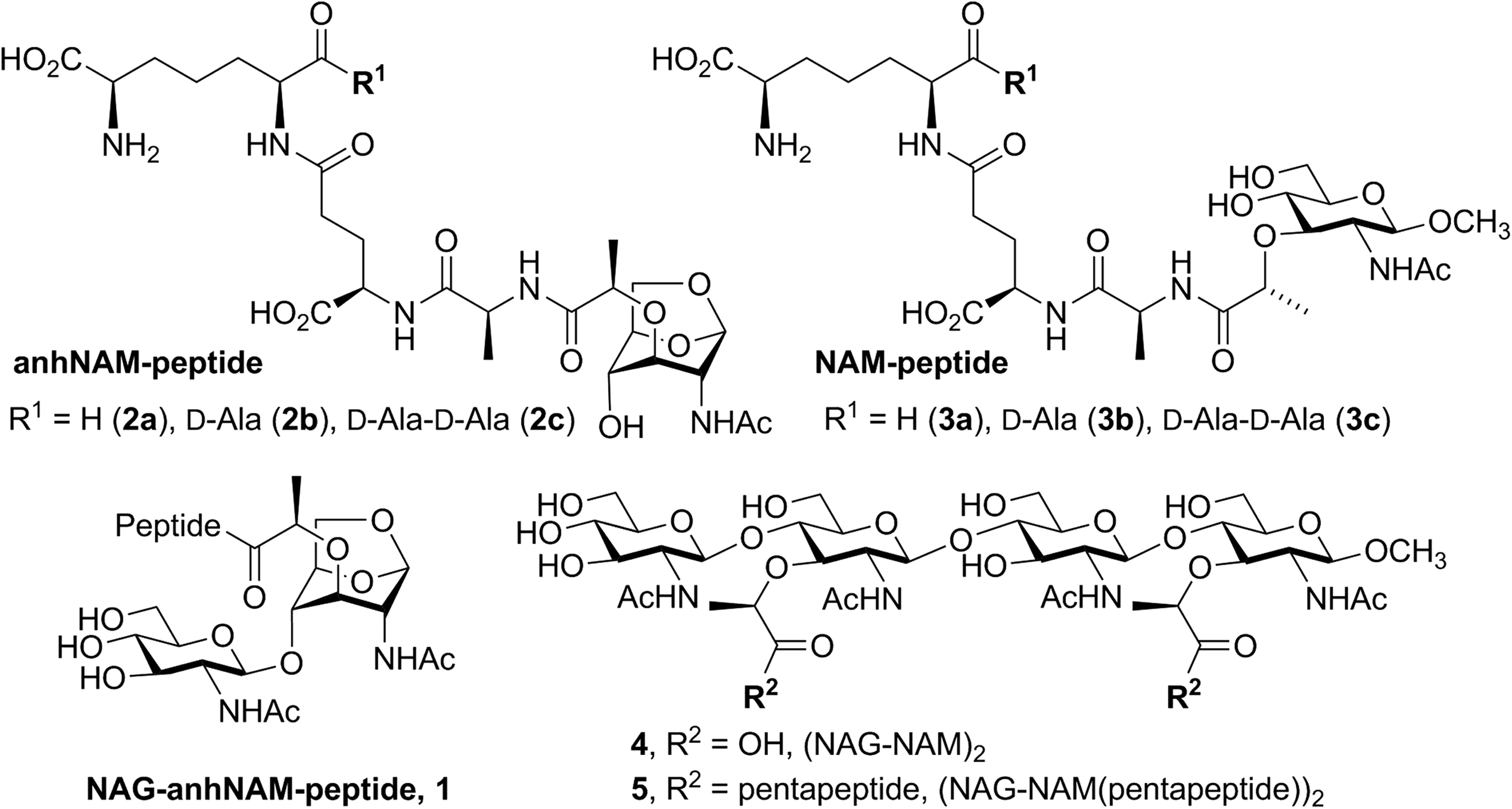
Chemical structures of synthetic substrates and product analog used for this study.
P. aeruginosa, in contrast to other organisms, has three closely related AmpD enzymes. Their roles in the link between the biochemical steps of recycling and inducible antibiotic resistance mechanism involving the AmpC β-lactamase have only been elucidated recently8,10 (Fig. 1). In contrast to other Gram-negative bacteria, peptidoglycan recycling in P. aeruginosa shows certain particularities inclusive of the PG amidases designated AmpD, AmpDh2, and AmpDh3.
11
The localization of these enzymes is critical for their functions. AmpD is clearly cytoplasmic, consistent with the observed localization of AmpD homologs in other species, as it turns over only the muropeptide substrate that is transported to the cytoplasm (such as compounds
AmpDh2 and AmpDh3 show 42% and 46% amino acid sequence identity (57% amino acid sequence similarity in both cases) to E. coli AmiD, respectively. The fact that both the P. aeruginosa AmpDh2 and the E. coli AmiD 10 are anchored to the inner leaflet of the outer membrane would suggest that AmpDh3 is an extra amidase in P. aeruginosa. Indeed, by infecting mice with the knockout strains of AmpD, AmpDh2, and AmpDh3, AmpDh3 would appear to be a bigger contributor than AmpDh2 to repression of ampC in an ampD mutant. Nonetheless, AmpDh2 and AmpDh3 also play a role in virulence, which is much less understood. 11
Catalytic mechanism(s) of AmpD, AmpDh2, and AmpDh3
Notwithstanding that both AmpDh2 and AmpDh3 turnover the peptidoglycan, they exhibit differences in their activities in that variations exist as in the degradation pattern of the peptidoglycan. For example, their specific activities are different, they exhibit differences in their choice of saccharides for substrates (compound
As indicated earlier, the kinetic parameters for turnover of synthetic substrates (compounds 1–3) of different AmpD enzymes from P. aeruginosa have been evaluated
8
using diverse muropeptides and peptidoglycan-based surrogates for the cell wall. As expected, the catalytic machineries of these proteins resemble those of typical zinc ion-dependent proteases. The X-ray structures show the typical active site ensconcing the zinc ion, which on incubation with the metal chelator EDTA leads to enzyme inactivation. Yet, the substrates that they turn over are distinct. The reactions with synthetic substrates reveal that AmpD shows activity with 1,6-anhydromuramyl derivatives (compounds
Three-dimensional structures of AmpD enzymes
AmpD peptidoglycan amidases are T7 lysozyme-type metallopeptidases and belong to the amidase 2 family (PF01510), according to the Pfam classification. 15 All members are zinc ion-dependent amidases with a single Zn+2 in the active site, 15 with a canonical folding constituted by a five-stranded parallel β-sheet surrounded by β-helices. The zinc ion at the active site is positioned on one side of the β-sheet and is tetrahedrally coordinated by a water molecule. 16 The amidase 2 family includes enzymes from different pathogenic species (e.g., Mycobacterium tuberculosis, Streptococcus pneumoniae, Staphylococcus aureus, Clostridium botulinum among others) and the peptidoglycan-recognition proteins (PGRPs) that are pattern-recognition proteins that bind and/or hydrolyze PG of the bacterial cell walls.17–19 PGRPs are found in both invertebrates and vertebrates, but have developed different functions from host signaling pathways20–22 to bactericidal functions.22–24 In general, peptidoglycan amidase genes code for proteins of 20–30 kDa and exhibit a minimum of 54% amino acid sequence similarity.
AmpD
The reported NMR structure of AmpD from C. freundii (no structural information has been reported for the P. aeruginosa ortholog) revealed, for the first time, similarities with bacteriophage T7 lysozyme and with eukaryotic PGRPs. 25 This structure presented a small active site incapable of accommodating its substrate. The crystal structures of AmpD in its apo- and holoenzyme forms at two different pH values, as well as in complex with the reaction products revealed a structure that, while preserving the general fold observed in the NMR structure, exhibited a larger active site around the catalytic zinc ion 14 (Fig. 3a). A large rmsd value of 3.9 Å (for all Cα atoms) was calculated from superimposing NMR and crystal structures, indicating that the highest changes were concentrated in four specific regions of the protein (r1–r4) surrounding the catalytic zinc ion (Fig. 3a). Since AmpD crystals in the presence of the substrate showed catalytic activity, the crystal structure conformation was proposed to be the enzyme active form, while the NMR structure was considered to be that of the inactive form by exhibiting a “closed” conformation. 14 The reason why this PG amidase would present such activation mechanism is still unknown; however it is likely that in the cytoplasm this enzyme would have fortuitous proteolytic activities that an activation mechanism would moderate. 14 In addition, sequence conservation of the regions involved in activation of all bacterial cytosolic AmpD enzymes and in some intracellular PGRP with peptidase activity (but not in periplasmic or extracellular enzymes) further supports this hypothesis.
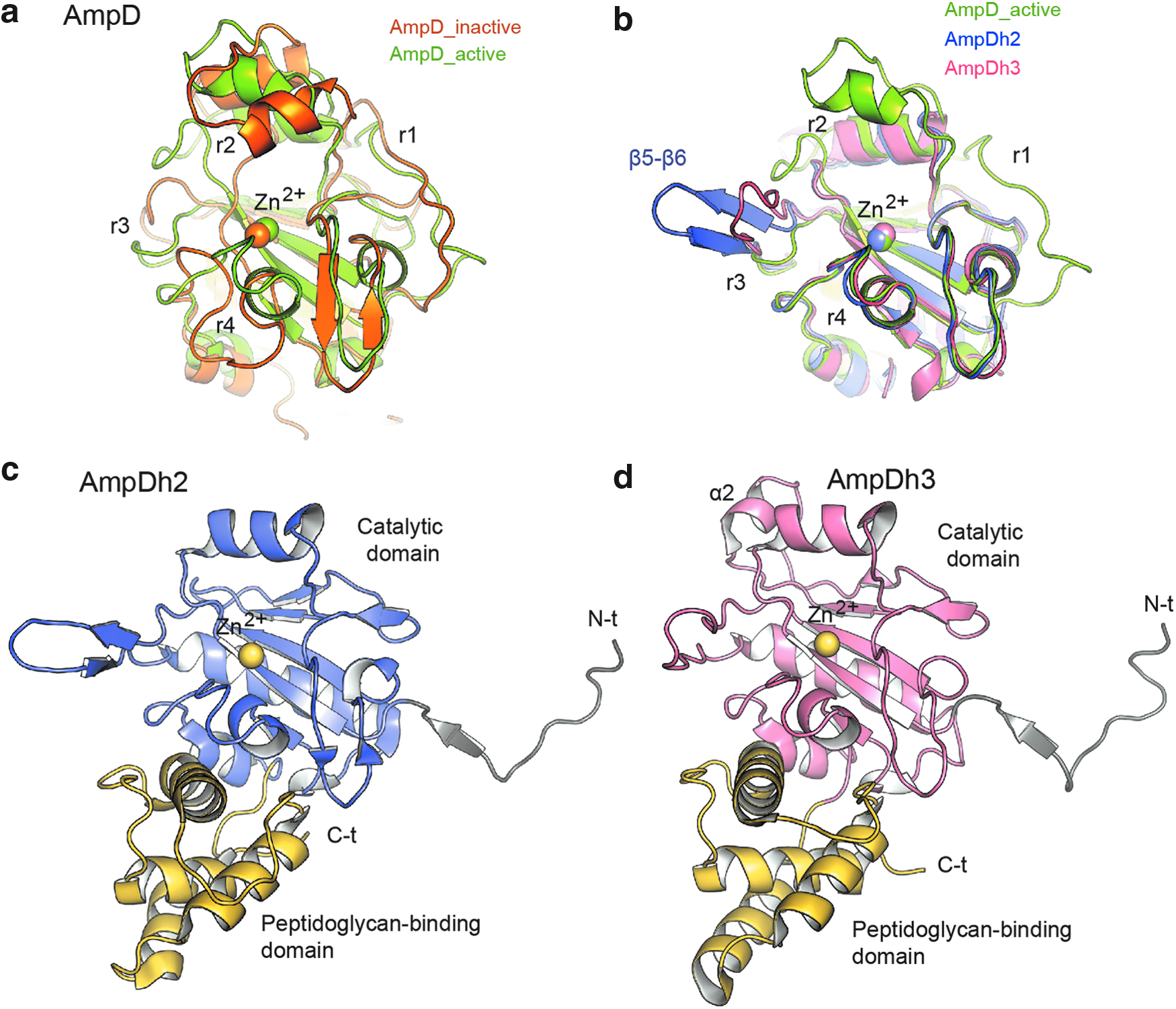
Three-dimensional structures of the different AmpDs in their monomeric forms.
AmpDh2 and AmpDh3
The crystal structures of AmpDh2 and AmpDh3 have been reported for the apo forms and in complexes with different substrate/product analogues.12,13 Both enzymes present similar multidomain structures (Fig. 3c, d) with an N-terminal coiled-coil/loop, a catalytic domain similar to that found in the cytoplasmic AmpD (Fig. 3b) and, finally, a globular C-terminal domain assigned as the peptidoglycan-binding domain based on Pfam annotation for this domain (PF01471). Both the peptidoglycan-binding domain and the N-terminal coiled-coil/loop, critical in the oligomeric state of AmpDh2 and AmpDh3 (see below), are not present in the cytoplasmic AmpD. Detailed structural comparison of the catalytic domains of the cytoplasmic and periplasmic paralogues reveals that while the r4 region, which is critical in substrate recognition of the cytosolic AmpD, is conserved in both AmpDh2 and AmpDh3 (Fig. 3b), regions r1, r2, and r3, key in the activation process of AmpD, are absent in the periplasmic enzymes. Sequence analysis reveals that the multidomain arrangement (Amidase 2 domain, followed by the PG-binding domain) observed in AmpDh2 and AmpDh3 is less common than the single-domain disposition of its cytoplasmic paralogue (177 sequences vs. 1728 sequences, as determined by Pfam). This modular architecture is found primarily (82%) in Gram-negative bacteria (Pseudomonas sp., Rhodopseudomonas sp., Rhizobium sp., Burkholderia sp., and Yersinia, among others) and in few Gram-positives (Clostridium sp., Mycobacterium sp., Streptomyces sp., among others). In contrast, the single-domain architecture having the Amidase_2 domain is largely distributed among various bacteria (as a part of the cell wall remodeling machinery), but also in insect and mammals (as a consequence of the incorporation of Amidase_2 domain in their immune system as pattern-recognition proteins), as assessed by the program Pfam.
Importantly, the oligomeric state is different for each AmpD paralogue, something that seems to be related to both their specificity for substrates and regulation. Cytoplasmic AmpD presents a globular monomer of around 40 × 49 Å dimensions for the active-site face (Fig. 4a). AmpDh2 is a large homodimer (60 × 69 Å) (Fig. 4b), both in the crystal and in solution. 13 The long N-terminal loop allows both the formation of the dimer and the anchoring of the oligomer to the inner leaflet of the bacterial outer membrane. Interestingly (as detailed below) the dimer also provides a more extensive surface for recognition of the cell wall. 13 On the other hand, the periplasmic AmpDh3 is soluble and is folded in a very large tetrameric arrangement of around 74 × 87 Å (Fig. 4c). The N-terminal coiled-coil/loop and an extra α helix (α2 in Fig. 3d), not present in homologous AmpDh2, provide a strong network of interactions that allow the formation of the tetramer in AmpDh3. This tetramer displays two active sites in one side of the oligomer and the other two in the back, allowing a multivalent binding of AmpDh3 onto the cell wall, which lends itself to its processive remodeling. 12

Oligomeric state and cell wall binding in AmpD enzymes. The enzyme structures are given as ribbon representation
PG recognition by AmpD enzymes
Each of the AmpD paralogues presents different substrate specificity. While cytoplasmic AmpD can recognize NAG-anhNAM(pentapeptide) (
The active site of AmpD presents both a glycan-binding site in which the 1,6-anhydro-N-acetylmuramyl moiety is accommodated and a peptide-binding site in which the peptide stem is bound (Fig. 4d). The PG-binding site of AmpD presents a similar topography to that of PGRPs.20,26–28 The groove is ∼26 Å long, with a narrow region near the active site and broader segments at the ends. The main differences between PG-binding cleft in AmpD and PGRPs are the entire r2 region and, to a less extent, the r1 region.
14
Different residues stabilize the ligand (
Crystal structures of AmpDh2 and AmpDh3 in complex with different ligands (compounds
The oligomeric arrangement of AmpDh3 displays two active sites on one side of the tetramer that could perform catalysis on two peptidoglycan strands simultaneously. A mechanism of action has been proposed in which on completion of the reaction with the two bound peptidoglycans, the tetramer would release the product strands and by rotating 60° the two active sites on the opposite side of the tetramer will be aligned to engage two neighboring strands for additional catalysis in a processive manner. 12
Conclusion
The AmpD enzymes of P. aeruginosa are key peptidoglycan amidases linking cell wall recycling processes and antibiotic resistance by repressing β-lactamase induction in Gram-negative bacteria. As discussed in this report, P. aeruginosa has evolved three paralogous genes for the AmpD proteases, designated as ampD, ampDh2, and ampDh3. Although these three enzymes share similar catalytic machineries and fold, their activities seem to be differentiated, whereas the cytosolic AmpD is the actual recycling enzyme, the periplasmic AmpDh2 and AmpDh3 are the enzymes involved in turn over of the bacterial cell wall itself and in its maturation. The additional AmpDh2 and AmpDh3 are primarily responsible for removing the peptides from both the exposed and the more sheltered peptidoglycan chains, respectively. From murine models of infection, while the inactivation of ampD was not sufficient to affect fitness or virulence of P. aeruginosa, double or triple mutants (loss of activities of AmpDh2 and AmpDh3) revealed to be defective in both. 10
The oligomerization states and differences in their three-dimensional structures are also related with their localization (membrane, periplasm, cytosol), which in turn influences binding and degradation of the intact cell wall chains or processed (by the actions of lytic transglycosylases and glucosaminidases) peptidoglycan units. The clear differences in specificity, structure, and oligomerization state found among the different AmpD paralogues point to specific functions in bacterial fitness that very likely will be also found in AmpD orthologues distributed in other bacteria. Whereas, as detailed above, clear functions have been assigned for these PG amidases, one cannot rule out additional functions that await discovery. For instance, as muropeptides are generated within the periplasm they are modified by AmpDh2 and AmpDh3. The resultant molecules could potentially be involved in signaling and regulatory events that govern bacterial physiology. This layer of complexity is underscored by the recent elucidation of the structures for at least 20 muropeptides in P. aeruginosa. 7
Footnotes
Acknowledgments
This work was supported by a grant from the NIH (GM61629) and by grants BFU2014-59389-P (the Spanish Ministry of Economy and Competitiveness) and S2010/BMD-2457 (the Government of Community of Madrid).
Disclosure Statement
No competing financial interests exist.