
Abstract
Osteoarthritis (OA) has become a major public health problem not only because of its increasing prevalence worldwide but also because of its frequent association with cardiovascular disease, the leading cause of death in industrialized countries. There is growing evidence that OA is not simply a disease related to aging or mechanical stress of joints but rather a “metabolic disorder” in which various interrelated lipid, metabolic, and humoral mediators contribute to the initiation and progression of the disease process. Indeed, OA has been linked not only to obesity but also to other cardiovascular risk factors, namely, diabetes, dyslipidemia, hypertension, and insulin resistance.
Introduction
Epidemiology of Oa and Its Association with the Metabolic Syndrome
Data from the Third National Health and Nutrition Examination Survey (NHANES III) have shown that the prevalence of OA among U.S. adults ages >35 years is approximately 21%. 1 Of the 115.9 million U.S. adults surveyed, 24.3 million have OA. Recent data from the National Arthritis Data Workgroup have shown that nearly 27 million U.S. adults have clinical OA, indicating that its prevalence is increasing. 2 Furthermore, the prevalence of OA increases with advancing age. 1 –4 In particular, nearly all persons aged 65 or older have radiographic evidence of OA. A more recent analysis of NHANES III data comprising a representative sample of 7,714 subjects (of whom 975 subjects had OA and 6,739 had no OA) showed that metabolic syndrome was prevalent in 59% of the OA population, but only 23% of the population without OA. 5 In addition, each of the five cardiovascular risk factors that comprise metabolic syndrome was more prevalent in the OA population versus the population without OA: Hypertension (75% vs. 38%), abdominal obesity (63% vs. 38%), elevated triglycerides (47% vs. 32%), low high-density lipoprotein cholesterol (44% vs. 38%), and hyperglycemia (30% vs. 13%). Metabolic syndrome was more prevalent in subjects with OA regardless of sex or race. The association between OA and metabolic syndrome was greater in younger subjects and diminished with increasing age. Having OA at age 43.8 years (mean age of the general population) was associated with a 5.26-fold increased risk of metabolic syndrome. A high prevalence of metabolic syndrome in OA patients was also observed in a cohort of 1,460 patients (of different ethnicities) undergoing total knee arthroplasty for end-stage OA. 6 Among the 1,334 white patients, 114 (8.5%) had metabolic syndrome (defined as body mass index [BMI] >30 kg/m2, diabetes, hypertension, and hypercholesterolemia) as compared with 18 of 90 (20%) Asians, and only 3 of 36 (8.3%) blacks. Further analysis showed that patients of Asian ethnicity had a two times greater odds of metabolic syndrome as compared with those of other ethnicity.
There is also epidemiologic evidence of increased mortality among individuals with OA compared with the general population. 7,8 In a cohort study of 296 women aged 42–76 years, an increasing prevalence of full-body radio-graphically defined OA in women was associated with decreased survival, which was independent of age and other co-morbid conditions, such as diabetes, smoking, alcohol use, and BMI. 7 Similarly, a large Finnish study has shown that the presence or identification of OA in any finger joint (a marker for generalized OA) predicts cardiovascular death in men. 9
Finally, there are several studies showing that serum concentrations of high-sensitivity C-reactive protein (hsCRP), a marker of systemic inflammation and a strong predictor of cardiovascular disease events, are higher in individuals with OA 10,11 and are associated with OA severity 12 as well as prevalent and incident knee OA. 13 Levels of hsCRP were also shown to correlate with waist circumference and BMI, 13,14 suggesting that CRP elevations in OA may be related in part to increased fat mass associated with obesity. CRP has also been shown to predict OA progression over several years. 15,16
Major Risk Factors of Oa
OA may be viewed as a heterogeneous disorder that is thought to result from a complex interplay of genetic, metabolic, hemodynamic, nutritional, and environmental factors that interact with each other and contribute to the development and progression of OA (Fig. 1).
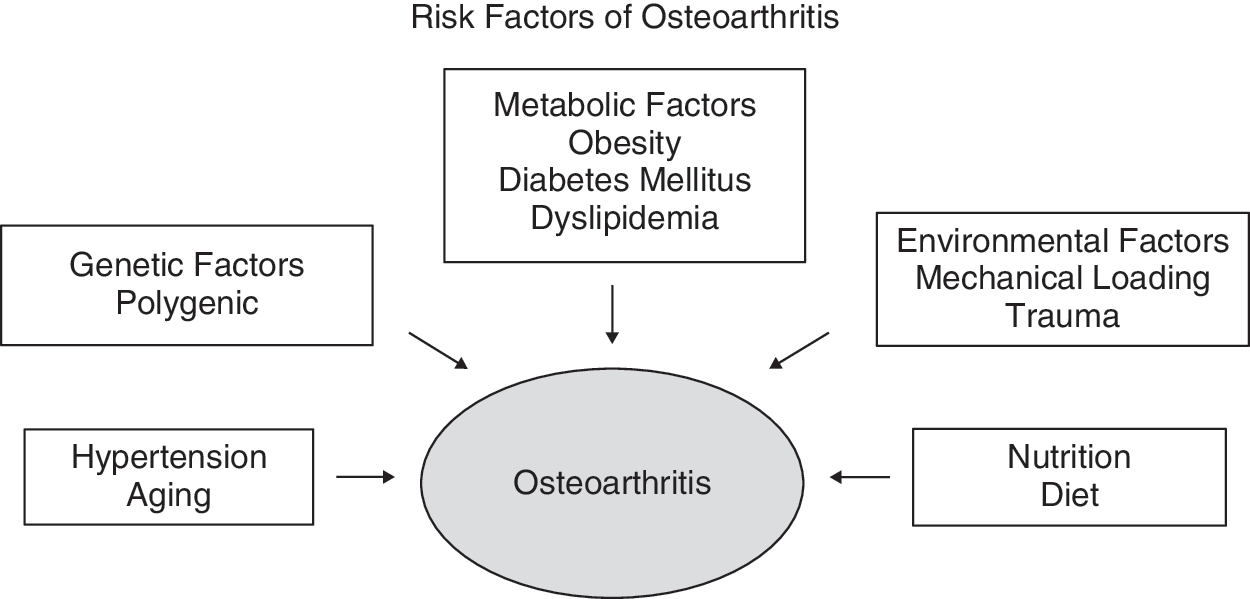
Risk factors of osteoarthritis.
Among the metabolic risk factors, obesity has long been recognized as a dominant factor for developing OA. 17 The impact of obesity on OA is observed particularly in weight-bearing joints, such as the knee and hip, and has been attributed in part to mechanical trauma associated with excess body weight. However, there is also evidence of an association between obesity and OA in non-weight–bearing joints. 17,18 In a recent cross-sectional study of 3,585 patients (age ≥55 years), Dahaghin et al. 19 have shown that overweight (defined as BMI >27.4 kg/m2) was significantly associated with radiographic evidence of hand OA, confirming an earlier longitudinal study that showed an association of relative body weight with subsequent severity of hand OA. 18 Moreover, the concurrent presence of overweight, diabetes, and hypertension showed an even higher prevalence of hand OA, and this prevalence increased further in the younger age groups. These findings suggest that the association between overweight and OA in non-weight–bearing joints might be mediated through factors linked to a metabolic disorder. The metabolic syndrome itself has been suggested as an important risk factor for the development of OA. 20 Positive correlations of OA with diabetes mellitus and the metabolic syndrome have been documented in several studies. 21 –24 In a recent report, the presence of the metabolic syndrome was shown to be associated with significantly increased risk of knee OA, an association that is largely related to increased BMI. 24
The association of OA with the metabolic syndrome is further supported by the recent observation that individuals with genetic predisposition to diabetes mellitus and hypertension (if they are obese) are likely to develop early diffuse idiopathic skeletal hyperostosis (DISH), a variant of OA. 25 DISH is but one example of a variety of disorders associated with OA that similarly manifest a metabolic component (Table 1). A close association of primary OA with insulin resistance and dyslipidemia is further exemplified in the STR/Ort mouse, a murine model that spontaneously develops osteoarthritis of the knees along with several features of the metabolic syndrome. 26 These include hyperglycemia, hypertriglyceridemia, hyperinsulinemia, insulin resistance, as well as low levels of serum adiponectin and abnormalities in nonesterified fatty acid (NEFA) metabolism. Collectively, these observations strongly support the concept that OA is part of a multisystem disorder with a metabolic component in its pathogenesis.
The influence of heredity on the development of OA is well documented in a variety of studies, including epidemiological studies of familial clustering, twin studies, gene association studies using single nucleotide polymorphisms (SNPs), and genome-wide scans. 27 –30 In several familial and twin studies, genetic factors were estimated to account for 39%–65% of both radiographic hand and knee OA. 27 –29 Genetic association studies have identified an increasing number of SNPs that are associated with pathology of OA. These include, among others, SNP rs143383 of the growth differentiation SNP of the ADAM12 gene, 30 interleukin-1 (IL-1) gene cluster polymorphisms, 31 ANP 32A gene, 32 and frizzled-related protein (FRZB) gene polymorphisms. 33 Given the heterogeneity of OA pathophysiology, these SNPs can contribute to only a small genetic component of OA. Recently, a more global approach that combines microRNA gene profiling with proteomic analysis has identified other proteins associated with OA pathobiology that are also involved in lipid metabolism and inflammatory pathways. 34 For example, the proteins SOX11, CCR3, and WWOX were found to be differentially expressed in OA cartilage, and their expression correlated with BMI, indicating that these metabolism-related proteins are involved in cartilage destruction associated with OA.
Mediators Linking Oa and the Metabolic Syndrome
OA is characterized by the progressive breakdown of articular cartilage and by reactive changes around joints as well as in subchondral bone. The disease is marked by ischemia, inflammation, cartilage loss, and osteophyte formation that together results in pain, deformity, and decreased function of affected joints (Fig. 2). The mechanisms involved in the pathogenesis of OA are incompletely understood. Several mediators, including glucose, fatty acids, hormones, certain growth factors, transcription factors, nitric oxide (NO), cytokines, and reactive oxygen species (ROS), which are key players in the pathogenesis of metabolic syndrome, have also been suggested to play a role in the development and progression of OA (Fig. 2).
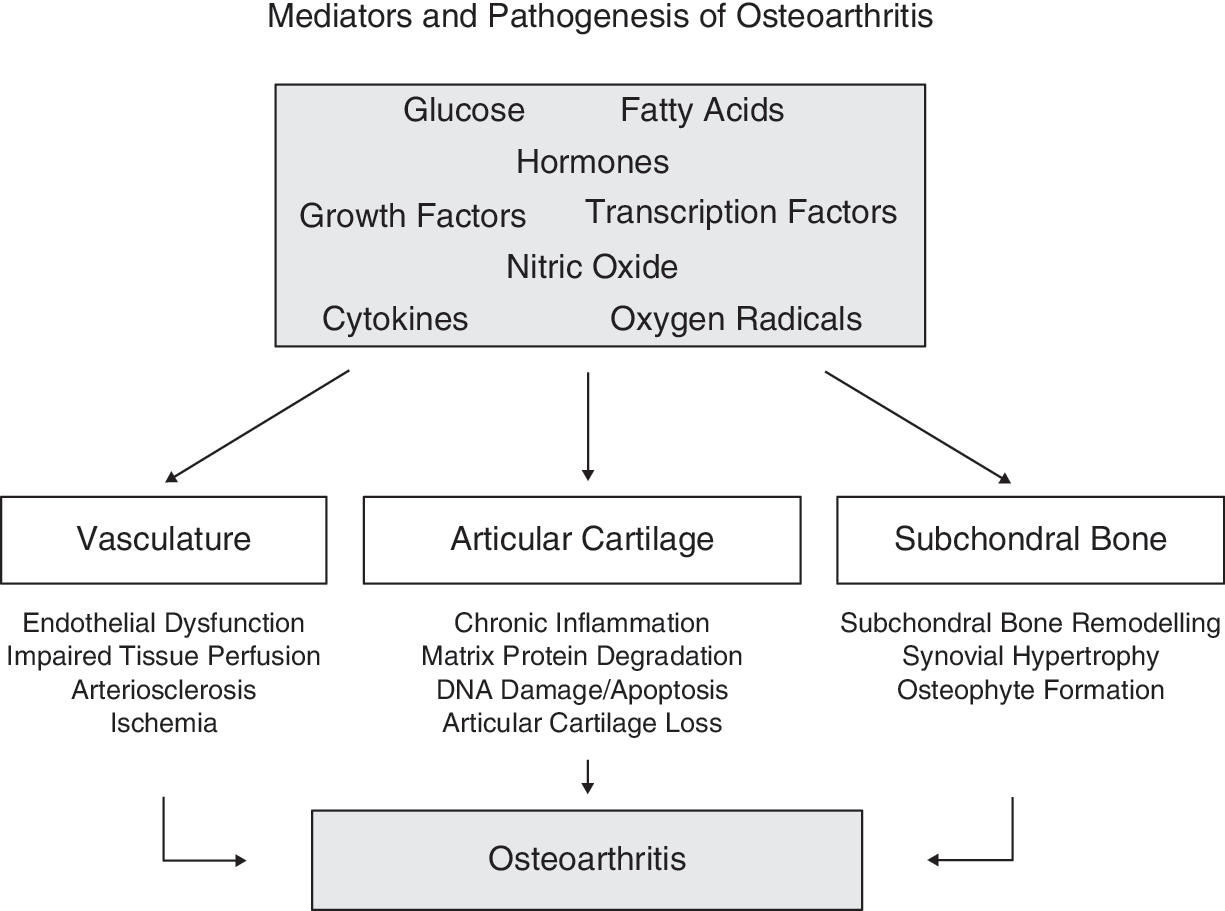
Mediators and pathogenesis of osteoarthritis.
Role of glucose
It is well known that chondrocytes take up and use glucose as a primary substrate for energy production, cell homeostasis, and extracellular matrix (ECM) synthesis. 35 Human articular chondrocytes express several isoforms of facilitative glucose transporters—the GLUT/SLC2A transporters—among which glucose transporter-1 (GLUT-1) is particularly relevant as this transporter is regulated by anabolic and catabolic stimuli that affect cartilage synthesis and degradation. 35 Recent in vitro studies by Rosa et al. 36 have shown that exposure of both normal and OA chondrocytes to low extracellular concentrations of glucose induced an increase in glucose uptake and GLUT-1 protein expression, whereas exposure to high glucose produced the opposite effects only in normal, but not in OA, chondrocytes. In addition, high glucose was shown to induce ROS production, an effect that lasted longer in OA than in normal chondrocytes. These findings suggest that OA chondrocytes, unlike normal chondrocytes, have impaired ability to down regulate the GLUT-1 content and glucose transport when exposed to high ambient glucose, which leads to the intracellular accumulation of glucose and increased oxidative stress. Such impaired GLUT-1 downregulation may be one mechanism whereby hyperglycemia seen in humans with diabetes mellitus and other conditions associated with impaired glucose metabolism can promote chondrocyte degeneration.
Prolonged hyperglycemia in diabetes results in the production and accumulation of advanced glycation end products (AGEs). In low-turnover tissues, AGEs also accumulate with age and may contribute to the pathophysiology of aging and long-term complications of diabetes. 37 Accumulation of AGEs in cartilage has also been proposed as a mechanism for the age-related development of OA. 38,39 Articular cartilage from patients with a focal degenerative cartilage lesion showed higher AGE levels than healthy cartilage from donor subjects in which there are no signs of OA. 39 In addition, articular chondrocytes also express the receptor for AGEs (RAGE), and RAGE levels are increased in OA cartilage, 40,41 suggesting that AGEs contribute to cartilage damage in OA via activation of RAGE. Investigating the relationships between AGEs, RAGE, and inflammation may be a fruitful area for elucidating the common mechanisms between OA and metabolic syndrome.
Role of fatty acids
Free fatty acids (FFAs) are the major forms of lipids found in plasma and provide the major substrates for energy production, membrane fluidity, and lipid storage in tissues. FFAs are well documented to play a central role in the pathogenesis of insulin-resistant states, including type 2 diabetes, obesity, and the metabolic syndrome itself. 42,43 Acute physiological elevations of plasma FFA have been shown to increase insulin resistance in experimental animals as well as in normal and diabetic individuals. 42,43 In addition, changes in the type or composition of FAs in the diet exert an influence on insulin resistance. For example, a high-fat diet rich in saturated fatty acids (SFAs), or polyunsaturated (omega-6) fatty acids (n-6 PUFAs) induces insulin resistance, whereas a diet rich in polyunsaturated (omega-3) fatty acids (n-3 PUFAs) improves insulin sensitivity in rats. 44 Similarly, in a controlled study of healthy human subjects, substitution of dietary SFA for monounsaturated fatty acid (MUFA) in an isoenergetic diet for 3 months reduced insulin sensitivity. 45
Evidence is emerging that suggests that lipids and FFAs also play a role in the pathogenesis of OA. 46 Elevated levels of fat and n-6 PUFAs have been observed in osteoarthritic bone. 47 A higher dietary intake of SFAs, but not MUFAs or PUFAs, was shown to increase the risk of developing bone marrow lesions (BMLs), an effect that was independent of age, gender, and BMI. 48 The independent impact of dietary fat on BMLs is worth noting because the presence of BMLs predicts the progression of cartilage defects and loss of cartilage volume associated with OA. 49
In a prospective study of 148 asymptomatic middle-aged women (aged 40–67 years) who underwent magnetic resonance imaging (MRI) of their dominant knee at baseline and at 2.2 years later, serum cholesterol and triglyceride levels but not high-density lipoprotein (HDL), low-density lipoprotein (LDL) (P = 0.20), or total cholesterol/HDL ratio were shown to be associated with the incidence of BMLs over 2 years. 50,51 Whether treatments aimed at reducing serum lipids may have a role in reducing the burden of knee OA remains to be determined.
More recently, Soran et al. evaluated lipid parameters along with enzymes related to oxidant stress and lipid peroxidation in 36 patients with knee OA and 30 healthy individuals, and found that that serum HDL-C, total thiol (total free sulfhydryl groups, –SH) levels, paraoxonase, and arylesterase activities were significantly lower in OA group than those in the controls, wheras lipid hydroperoxide LOOH and LDLC levels were significantly higher. 52 Lower serum paraoxonase-1 activity and lower levels of HDL-C seem to be related to increased oxidative stress and inflammatory condition in these patients. It is known that paraoxonases reduce oxidative stress in serum and tissues, thereby protecting against cardiovascular disease, particularly atherosclerosis. Thus, decreased paraoxonase and arylesterase activities play a role in the pathogenesis of atherosclerosis through increased susceptibility to lipid peroxidation in patients with osteoarthritis.
The precise mechanisms whereby FFAs influence cartilage degradation and bone lesions associated with OA are unknown. In vitro studies of chondrocytes isolated from OA patients have shown that supplementation of certain FAs, namely conjugated linoleic acids, induce a reduction in the production of the eicosanoid prostaglandin E2 (PGE2) and NO, which are known mediators involved in the pathogenesis of OA. 53 Additionally, exposure of cultured bovine chondrocytes to varying amounts n-3 PUFAs was shown to reduce the mRNA levels for several proteins known to be involved in the pathology of OA, including ADAMTS-4, ADAMTS-5, metalloproteinase-3 (MMP-3), MMP-13, and cyclo-oxygenase 2 (COX-2). 54 However, the n-6 PUFA arachidonic acid (AA) had no effect on the gene expressions of these proteins. These studies suggest that diets rich in SFAs and n-6 PUFAs have adverse effects on OA progression, whereas dietary n-3 PUFAs have beneficial effects on OA lesions, in addition to their well-known effects on atherosclerosis. Nutritional intervention studies are needed to determine whether dietary n-3 PUFAs can prevent or slow disease progression in patients with OA. Because SFAs are known to accelerate generalized atherosclerosis, the deleterious effects of SFAs on articular cartilage and bone lesions in OA may also be related in part to ischemia resulting from atheromatous vascular disease of subchondral bone. Indeed, a causal link between the progression of OA and atheromatous vascular disease has recently been proposed. 55,56 Reduced blood flow in small vessels in the subchondral bone may accelerate OA by altering the cartilage nutritional milieu as well as by virtue of direct ischemic effects on bone.
Hormones
Hyperinsulinemia and insulin resistance are considered the key factors linking the cluster of abnormalities in metabolic syndrome (e.g., glucose intolerance, obesity, dyslipidemia, and hypertension). Evidence also exists for the presence of insulin resistance in patients with OA as well as in animal models. 57,58 For example, serum insulin levels have been reported to be significantly higher in overweight patients with knee OA compared to subjects without OA. 57 Insulin sensitivity, as determined from the Quantitative Insulin Sensitivity Check Index (QUICKI), was observed to be decreased in patients with OA, a finding similar to that observed in patients with rheumatoid arthritis. 58 Both hyperinsulinemia and insulin resistance are also characteristic features of the spontaneous OA model, the STR/Ort mouse. 26
Leptin, the 16-kD protein product of the obese (ob) gene is primarily produced and secreted by the white adipose tissue and acts on specific receptors (Ob-R) to regulate food intake and energy balance. 59,60 Plasma levels of leptin correlate closely with body fat mass, 61 and leptin levels are known to be elevated in human obesity. 62 Because of the wide distribution of leptin receptors in peripheral tissues, 63 leptin has been considered a pleiotrophic hormone involved in other physiologic processes, including lipid metabolism, 64 insulin secretion, 59 thermogenesis, 60 and immune functions. 65 Leptin has also been shown to regulate bone growth by inducing osteoblast proliferation, collagen synthesis, and bone mineralization. 66 Leptin also enhances bone formation by stimulating endochondral ossification. 67 Leptin also has been shown to have a direct stimulatory effect on human chondrocytes 1n vitro by increasing chondrocyte proliferation and synthesis of proteoglycans and collagen. 68 Thus, leptin not only enhances skeletal growth and endochondral ossification but also affects cartilage generation, which are key events in the development of OA.
Dumond et al. showed that leptin was detected in synovial fluid (SF) obtained from patients with OA, and that levels of leptin correlated with the BMI. 69 Moreover, leptin was strongly overexpressed in human OA cartilage and in osteophytes. Human OA chondrocytes also produced growth factors, the topographic localization and staining intensity of which varied according to the histology grade of cartilage destruction and paralleled levels of leptin. Results of animal studies indicated that this adipocytokine stimulated anabolic functions of chondrocytes and induced the synthesis of growth factors in cartilage. Taken together, these data provide evidence for a key role of leptin in the pathophysiology of OA.
Simopoulou et al. showed that leptin levels were significantly increased in SF than serum samples from patients with knee OA. 70 Moreover, these investigators also showed that mRNA expression and protein levels of leptin and leptin's receptor isoform (Ob-Rb) were significantly increased in advanced OA knee cartilage compared to minimally affected OA cartilage. Furthermore, leptin's mRNA expression in advanced OA cartilage was significantly correlated with BMI of the patients, suggesting that leptin may be a metabolic link between obesity and the BMI of OA patients suggests that leptin may be a metabolic link between obesity and OA.
Adiponectin is another hormone secreted by white adipose tissue and exists as the most abundant adipokine in the human plasma. 71 Circulating levels of adiponectin are decreased in obesity and type 2 diabetes in close association with hyperinsulinemia and insulin resistance. 72,73 Adiponectin increases insulin sensitivity in the liver and skeletal muscle and reduces atherosclerosis. 73 In addition to these effects, adiponectin has also vasculoprotective effects mediated via an increase in endothelial NO production or modulation of expression of adhesion molecules and scavenger receptors. 73 Recently, Chen et al. have shown that adiponectin is present in synovial fluid of OA patients and that adiponectin levels in OA synovial fluid were considerably lower compared to levels in OA plasma. 74 In addition, the expression of adiponectin receptors 1 and 2 (AdipoR1 and AdipoR2) differed in human OA tissues in that AdipoR1 was abundantly expressed in cartilage, bone, and synovial tissues, whereas AdipoR2 was rarely detected in these tissues. Pretreatment of primary chondrocytes with recombinant adiponectin was shown to upregulate TIMP-2 expression and partially abolish IL-1β–induced MMP-13 expression. These findings indicate that adiponectin may play a protective role in the development and/or progression of OA.
Growth factors
Insulin-like growth factor-1 (IGF-1) is a peptide hormone with structural and functional similarities to insulin that is produced mainly by the liver and plays an essential role in glucose and lipid metabolism. IGF-1 has been implicated in the pathogenesis of insulin resistance and cardiovascular disease. 75 Circulating levels of IGF-1 have been shown to correlate inversely with markers of insulin resistance, such as serum leptin, waist-to-hip ratio, and BMI. 76 Low serum IGF-1 levels are independently associated with glucose intolerance, diabetes, abdominal obesity, and dyslipidemia. 77–78 Moreover, administration of recombinant human IGF-1 to diabetic patients reduces insulin dose requirement and serum glucose levels 79 and improves glucose tolerance, hyperinsulinemia, and hypertriglyceridemia. 80,81 These data suggest a protective role of IGF-1 in the development of the metabolic syndrome. There is evidence that IGF-1 also has a protective effect against endothelial dysfunction, atherosclerotic plaque formation, and ischemic myocardial damage. 81
The IGF-I system has also been implicated in OA pathogenesis. 82 IGF-1 receptors are known to be abundantly expressed in human cartilage and bone. 83,84 In vitro work has shown that both chondrocytes and osteoblasts produce and respond to IGFs I and II, suggesting that locally produced IGF acts in an autocrine fashion to regulate cartilage metabolism and bone turnover. IGF-1 expression is increased in chondrocytes isolated from OA cartilage. 85,86 In addition, production of IGF-binding proteins (IGFBPs) are also increased in OA cartilage. 86,87 However, OA chondrocytes, in contrast to normal chondrocytes, are hyporesponsive to IGF-1 stimulation, a phenomenon attributed to increased levels of IGFBPs, which have a high affinity for IGF. 88 The elevated expression of IGFBPs suggests that these proteins may be acting as competitors for IGF-I in OA cartilage, thus reducing the anabolic action of IGF-I in this tissue, which results in the net loss of cartilage. IGF-I was shown to stimulate the production of cell-associated matrix (CAM) compounds, namely aggrecan and type II collagen by human cartilage cells, indicating that it also regulates cartilage ECM homeostasis. 89 It has also been suggested that local activation of IGF-1 by bone cells may contribute to the abnormal subchondral bone remodeling and sclerosis seen in OA. 84,90
Although normal adult cartilage is an avascular tissue, physiological angiogenesis can be observed during endochondral bone development. 91 However, in pathological states, such as inflammation and hypoxia, a number angiogenic factors, including vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), and transforming growth factor-β (TGF-β), are expressed in growth plate cartilage frequently invaded by the granulation tissue. These findings suggest that angiogenic factors may involved in the pathogenesis of OA. 91 Indeed, increased expression of VEGF and its receptors has been demonstrated in OA cartilage. 92–93 Experimental studies in vitro also have shown that VEGF stimulates the proliferation and chemotactic migration of primary human osteoblasts, 94 suggesting a functional role for this growth factor in bone formation and remodeling.
Endogenous TGF- β, a pivotal growth factor required for cartilage integrity and repair, has been shown to be involved in osteophyte formation and synovial thickening in OA. 95,96 Expression of the three TGF-β isoforms is increased in cartilage sampled from patients with OA. 97 Osteoarthritis-like changes have been observed in the murine knee joint after intraarticular injections of TGF-β. 98 In addition, inhibition of TGF-β activity by a scavenging soluble TGF-β-RII not only prevented osteophyte formation but also markedly enhanced cartilage proteoglycan loss and reduced articular cartilage repair. 99
Transcription factors
The transcription factor complex hypoxia-inducible factor 1 (HIF-1) is well recognized as the central regulator that mediates systemic and cellular homeostatic responses to hypoxia in various tissues. 100 Under hypoxic conditions, HIF-1 expression is increased, which in turn activates the transcription of genes involved in oxygen homeostasis—energy metabolism, glucose transport, angiogenesis, vasomotor control, apoptosis, proliferation, and ECM production. 102,103 Recent studies indicate that HIF-1 plays a pivotal role in cartilage development and homeostasis. 104,105 Furthermore, HIF-1 α is known to be expressed in human normal and osteoarthritic chondrocytes. 106 Overexpression of HIF-1α during hypoxia also induces transcriptional activation of VEGF isoforms by epiphyseal chondrocytes, a mechanism that may contribute to the formation of blood vessels in long bone development. 107 In addition to hypoxia, catabolic stress and inflammatory induce the expression of HIF-1α in articular chondrocytes, further suggesting an involvement of HIF-1α in the pathogenesis of osteoarthritis. 108 These observations suggest that HIF-1 activation in OA chondrocytes may serve as a compensatory mechanism to protect chondrocytes from injury during hypoxia and catabolic stress.
Nuclear factor kappa B (NF-βB) is a pleiotrophic transcription factor that regulates the expression of a number of genes that are involved in inflammation, immunity, apoptosis, as well as cell proliferation and differentiation. 109 –111 (These include genes encoding adhesion molecules, inflammatory cytokines, antiapoptotic genes, and cell cycle regulatory genes. NF-κB is activated in a wide variety of pathological disorders associated with chronic inflammation and is also implicated in obesity and diet-induced insulin resistance, 111 atherosclerosis, 112 and bone loss. 113 There are reports that NF-κB is also activated in the osteoarthritic chondrocytes. 114,115 Furthermore, in a rat model of experimental OA, in vivo delivery of the adenoviral vector-mediated NF-κBp65-specific small interfering (si) RNA inhibited NF-κB activation and the expression of NF-κBp65 in cartilage and synovium of the knee and reduced cartilage degradation in the early phase of OA. 116
Serum amyloid A–activating factor 1 (SAF-1) is another transcription factor identified in human chondrocytes. 117 Low levels of SAF-1 protein are expressed in normal cartilage. By contrast, increased SAF-1 protein expression has been demonstrated in degenerated cartilage matrix and in osteophytes in OA tissues. Activation of SAF-1 in OA chondrocytes was also shown to increase the expression of the matrix metalloproteinase-1 (MMP-1) promoter, whereas interference of endogenous SAF-1 activity by antisense SAF-1 messenger (m) RNA inhibited IL-1–mediated MMP-1 promoter activity. 118 These findings indicate that SAF-1 is involved in the regulation of MMP-1 gene expression and is highly abundant in the articular cartilage of OA patients.
Nitric oxide
NO is a highly reactive ubiquitous molecule produced by the enzyme nitric oxide synthase (NOS) present in virtually all mammalian cells via the oxidation of the amino acid substrate L-arginine to L-citrulline. NO is well recognized as the “multifunctional mediator” with diverse physiological and pathological roles, for example, in vascular homeostasis, metabolism, neurotransmission, cell growth, inflammation, and atherosclerosis. In the vascular endothelium, endothelium-derived NO is produced from L-arginine by endothelial NOS (eNOS) and acts to relax vascular smooth muscle. NO produced in other tissues interacts with growth factors and cytokines to modulate cell proliferation, ECM production, and inflammation. Impaired release or reduced bioavailability of NO is one of the mechanisms implicated in endothelial dysfunction associated with hypertension, 119 diabetes, 120 obesity, 121 hyperlipidemia, 122 and atherosclerosis. 123
There is experimental evidence that NO is involved in the pathogenesis of OA. 124 –126 Elevated levels of markers of NO production are found in OA cartilage. For example, in vivo expression of inducible NOS (iNOS) is increased in chondrocytes harvested from patients with OA, 127,128 suggesting that NO production is enhanced in human OA. Explants of knee menisci from OA patients were shown to produce NO and PGE2, which were further increased in response to proinflammatory cytokines IL-1, TNF-α, and IL-17. 129 NO has also been shown to inhibit cytokine-induced proteoglycan synthesis in rabbit cartilage cultures. 130 Similarly, elevation of the NO via induction of iNOS expression by IL-1 was shown to increase the production of MMPs, which are proteases involved in the degradation of ECM in OA. 131 These findings suggest that NO has a catabolic role in OA. On the other hand, there is also evidence that NO, under certain conditions, may exert a protective action on chondrocytes. 125,132 –134 In cultured bovine chondrocytes, the addition of an exogenous NO donor inhibited IL-1-induced NF-κB activation and NOS II expression. 133 Similarly, exogenous NO donor can stimulate collagen synthesis in cultured human tendon cells. 133 Taken collectively, these observations suggest that NO activation in cartilage may have different effects on chondrocyte metabolism, either by inhibiting the synthesis of both collagen and proteoglycans or by modulating the inflammatory response of chondrocytes.
Cytokines
The role of cytokines in the pathophysiology of OA is well-documented. 134 Among the various cytokines, IL-1 and TNF are important mediators of the osteoarthritis process. 135 These cytokines are known to modulate chondrocyte expression of proteases, (such as aggrecanases and collagenases), that are ultimately involved in matrix breakdown resulting in the formation of OA lesions.
Recent work by Benito and colleagues 136 has shown that expression of TNF and IL-β was associated with increased expression of the NF-κB1 and COX-2 in synovial tissue from patients with OA. In addition, synovial tissue from patients with early OA demonstrated significantly greater CD4+ and CD68+ cell infiltration, blood vessel formation, VEGF, and intercellular adhesion molecule-1 (ICAM-1) expression. These inflammatory changes appear to persist in late OA.
Radical oxygen species
The role of oxidative stress as a common mechanism in obesity-associated metabolic syndrome, diabetes, hyperlipidemia, hypertension, and the development of cardiovascular complications has been extensively investigated in both animals and humans 137 –144 and described in detail in several reviews. 144 –146 In both animal and humans with obesity, fat accumulation closely correlates closely with the markers of systemic oxidative stress. 137,138,142 In addition, increased production of ROS in accumulated fat resulting from increased nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and decreased antioxidative enzymes also leads to increased oxidative stress in peripheral blood, affecting other organs including the liver, skeletal muscle, and aorta. 139 Additionally, ROS participates in the various stages of inflammation and have been shown to activate multiple intracellular signaling molecules and transcription factors associated with inflammatory responses, namely NF-κB and activator protein-1 (AP-1).
In like manner, oxidative stress may play a role in the generation of OA changes in cartilage. 147 Oxidative stress may adversely impact not only the senescent chondrocytes but the extracellular joint matrix as well and contribute to cartilage degradation seen in OA. 148 Finally, increased oxidative stress or ROS has also been suggested as contributing to sarcopenia, 149,150 a condition of skeletal muscle loss that is not only seen with aging but also a characteristic feature associated with early OA changes in women. 151
Therapeutic Implications
There is little data in the literature about the effects of therapeutic interventions on metabolic alterations in OA patients with the metabolic syndrome and how such interventions affect or ameliorate metabolic markers. However, it is known that weight reduction in insulin-resistant obese subjects improves insulin sensitivity. Similarly, treatment of obesity with gastric bypass surgery has been shown to induce rapid and prolonged improvements in insulin sensitivity. 152 Along with these reports, there are observational studies describing the fact that weight loss after bariatric surgery results in improvements in many obesity-related co-morbid conditions. 153,154 In these studies, most obese patients experience improvement or even resolution of diabetes as well as significant improvement of hyperlipidemia, hypertension, OA, and obstructive sleep apnea. 153,154 One prospective study conducted in 64 obese patients with radiological evidence of knee OA showed significant improvements in knee function and joint space narrowing. 155
Conclusions
In summary, a large body of evidence indicates that OA is part of a generalized metabolic disorder in which various interrelated metabolic and hemodynamic factors contribute to the progressive degradation of articular cartilage, which is the pathological hallmark of OA. This is derived from numerous epidemiological studies as well as experimental studies in humans and animals showing a strong and independent association of the occurrence of OA with several components of the metabolic syndrome, namely obesity, diabetes mellitus, hypertension, hyperlipidemia, and atherosclerosis. Moreover, in vivo and in vitro studies suggest that several biochemical, cellular, and molecular mediators, such as glucose, fatty acids, hormones, growth factors, transcription factors, nitric oxide, cytokines, and oxygen radicals (known to be involved in the development of the metabolic syndrome), may also participate in the chondrocyte damage associated with OA, thereby pointing to a common biochemical and pathophysiological milieu shared by OA and the metabolic syndrome.
Footnotes
Author Disclosure Statement
The authors have no disclosures relevant to the content of this article.