
Abstract
A series of siRNA duplexes containing cationic non-bridging 3′,5′-linked phosphoramidate (PN) linkages was designed and synthesized using a combination of phosphoramidite and H-phosphonate chemistries. Modified oligonucleotides were assayed for their thermal stability, helical structure, and ability to modulate the expression of firefly luciferase. We demonstrate that PN modifications of siRNAs are, in general, minimally destabilizing with respect to duplex thermal stability; destabilization can be mitigated through the incorporation of 2′-modified RNA-like residues or PN conjugates containing ionizable pendant moieties. We also demonstrate that single cationic dimethylethylenediamine PN linkages have little effect on siRNA potency, whether located in the passenger or guide strand of the duplex. Highly modified siRNA passenger strands were further modified with up to four cationic PN linkages, with little effect on duplex potency or helical structure. We envision that PN modifications could be useful in the production of therapeutic siRNAs with optimal biological properties.
Introduction
O
Chemical modifications of the sugar, nucleobase, and internucleotide linkage of oligonucleotides have been used to address many of the undesirable properties of oligonucleotides and to accelerate their clinical development [2–5]. In particular, modified internucleotide linkages have been extensively studied; some notable examples include phosphorothioates [6], methylphosphonates [7], boranophosphates [8], and amides [9]. Such backbone modifications have been used to alter nuclease resistance [8,10,11], biological activity [12], duplex thermal stability [10,12–14], and cellular uptake [15,16], leading to the development of a number of oligonucleotide therapeutics in clinical trials.
Phosphoramidate (PN) modifications of oligonucleotides, in which one of the non-bridging oxygen atoms of the phosphate linkage is replaced by an amino moiety (Fig. 1), were first reported in 1986 [17]. Since then, these modifications have been explored in the context of both antigene [18–20] and antisense [21–24] approaches; in each of these strategies, the active agent is a single strand of DNA [25]. In general, PN-modified oligonucleotides exhibit improved nuclease resistance relative to unmodified DNA; in addition, the introduction of cationic linkages within the oligonucleotide backbone could potentially facilitate cellular uptake due to the reduction of the net negative charge of the phosphodiester backbone [25]. Other biological properties of PNs vary depending on the structure and biophysical properties of the amine conjugate [26]; as the incorporation of PN linkages creates chirality at the phosphorus center, the stereochemistry of the phosphorus atom also plays a role [27,28]. Cationic alkylamine conjugates have been widely used in the exploration of PN oligonucleotides [19,27,29–31].
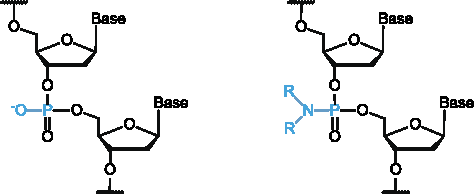
General structures of phosphodiester (left) and non-bridging phosphoramidate (right) linkages. Color images available online at www.liebertpub.com/nat
The study of PN linkages in RNA is more challenging. Interribonucleotide PN linkages are unstable in both aqueous and anhydrous acidic, alkaline, and neutral solutions due to the existence of the 2′-OH group in the cis-alpha position [32–35]. In acidic and alkaline solutions, PN-linked oligoribonucleotides are rapidly degraded, both with chain cleavage (to a mixture of ribonucleotide 2′,3′-cyclic phosphates and nucleosides) and without (to ribonucleotide isomers) [34]. As a result of this instability, the study of PN linkages has been limited to DNA-based applications; these modifications have not, to our knowledge, been systematically studied in RNA.
In this study, we examine the use of PN modifications in small interfering RNA duplexes (siRNAs). PN-modified linkages were incorporated in siRNA passenger and guide strands using 2′-deoxy-, 2′-fluoro-, and 2′-O-methyl-3′-H-phosphonate monomers to circumvent stability issues prevalent with PN-modified ribonucleotides. The PN modifications investigated herein were found to have a mild destabilizing effect on duplex thermal stability, with little variance observed with respect to the different amine conjugate structures. Negative impacts on biological properties of siRNAs were minimized when modified linkages were incorporated using RNA-like sugars. Notably, single PN modifications of RNA guide strands resulted in duplexes with comparable potency to unmodified siRNAs.
Materials and Methods
Oligonucleotide synthesis and purification
Standard phosphoramidite solid-phase synthesis conditions were used for the synthesis of all modified and unmodified oligonucleotides containing phosphodiester linkages on an ABI 3400 DNA synthesizer. Each oligonucleotide was synthesized at the 1 μmol scale, using UnyLinker CPG (ChemGenes) as solid support. Phosphoramidites were prepared as 0.1 M (DNA) and 0.15 M (RNA) solutions in acetonitrile. 5-Ethylthiotetrazole was used as activator, 3% trichloroacetic acid in dichloromethane was used to detritylate, acetic anhydride in tetrahydrofuran (THF) and 16% N-methylimidazole in THF were used to cap, and 0.1 M I2 in 1:2:10 pyridine/water/THF was used for oxidation of standard phosphite triester linkages. DNA amidites were coupled for 110 s (270 s for dG); all other phosphoramidites were coupled for 600 s (900 s for G).
Deprotection and cleavage from the solid support were achieved by treatment with 40% aqueous methylamine for 12 min at 65 C. After decanting the methylamine solution, the CPG was washed with a mixture of 3:1:1 ethanol/acetonitrile/water. The washes were combined and dried under vacuum in a SpeedVac evaporator. For oligonucleotides containing RNA residues, desilylation was achieved in a solution of 6:4:3 TREAT-HF/Et3N/N-methylpyrrolidone (300 μL) for 90 min at 65°C, followed by quenching with 3 M NaOAc buffer (50 μL) and precipitation of the crude oligonucleotide from cold 1-butanol (1 mL).
Purifications were performed by HPLC, using an Agilent PL-SAX analytical anion-exchange column. A mobile phase of 1 M LiClO4 in milli-Q water (18.2 MΩ resistivity) was used for analysis and purification using a gradient of 0%–30% over 30 min. Following purification, excess LiClO4 salts were removed using Gel Pak 2.5 size-exclusion columns (Glen Research). Purified oligonucleotides were then lyophilized to dryness, characterized by electrospray ionization-mass spectrometry, and quantitated by UV spectroscopy. Extinction coefficients were determined using the IDT OligoAnalyzer tool (www.idtdna.com/analyzer/Applications/OligoAnalyzer).
Synthesis of PN-modified linkages
To form modified linkages, CPG derivatized with an oligonucleotide of the desired length was detritylated on an automated synthesizer, and then transferred into a 10 mL glass peptide synthesis vessel. The solid support was washed with acetonitrile, and then dried under vacuum and flushed with argon. Coupling was performed manually by addition of a suitably protected 3′-H-phosphonate monomer (125 mM in 300 μL 3:1 ACN/pyridine), followed by addition of pivaloyl chloride (400 mM in 300 μL 3:1 ACN/pyridine); the vessel was then shaken for 5–12 min. Capping was omitted as H-phosphonate coupling reactions are self-capping. Oxidation was performed by addition of a solution of the desired amine in carbon tetrachloride (10% v/v, 2 mL) to form the corresponding phosphoramidate linkage; the vessel was shaken for 25 min. The solid support was washed with pyridine and dichloromethane, dried under vacuum, and flushed with argon between each step. Finally, the solid support was transferred back into a synthesis column and returned to the automated synthesizer for detritylation and further oligonucleotide synthesis using standard phosphoramidite chemistry as previously described.
Thermal denaturation experiments
Melting curves were recorded on a Cary 300 spectrophotometer (Varian). Equimolar amounts (1.5 nmol) of modified oligomers and their targets were combined, dried, and resuspended in 1 mL of 5 mM sodium phosphate buffer (pH 7.2) containing 140 mM KCl and 1 mM MgCl2. The samples were preheated to 90°C, slowly cooled to 5°C, and then slowly heated to 90°C. Cooling and heating rates were ramped at 0.5°C/min. The cell compartment was flushed with dry nitrogen for temperatures below room temperature. Melting temperature values were determined as the maxima of the first derivative plots of absorbance versus temperature; each value is an average of at least three experiments.
Circular dichroism experiments
CD spectra were recorded on an Applied Photophysics instrument. Equimolar amounts (1 nmol) of modified oligonucleotides and their targets (or 2 nmol of single strands) were combined, dried, and resuspended in 200 μL of 5 mM sodium phosphate buffer (pH 7.2) containing 140 mM KCl and 1 mM MgCl2 to give a final concentration of 10 mM. Circular dichroism was monitored scanning from 300 to 200 nm. Each spectrum was averaged over five experiments, with four degrees of smoothing applied.
Luciferase assays
Luciferase knockdown assays were performed as described in Deleavey et al. [36] with a few modifications. Typically, HeLa cells were counted and seeded at a density of 10,000 cells/well in a 96-well plate. Cells were allowed to recover for 24 h at 37°C with 5% CO2. Subsequently, cells were washed once with serum-free DMEM media and then 80 μL of serum-free DMEM media was added. siRNA and control nucleic acid preparations were diluted up to 20 μL with serum-free media and transfection reagent (Oligofectamine, Invitrogen) and added to the appropriate well (for a total of 100 μL) at increasing concentrations (0.032, 0.16, 0.8, 4, 20, and 100 nM). Cells were incubated overnight (for a total of 24 h after oligonucleotide addition). Then, 50 μL of ONE-Glo luciferase reagent (Promega) was added to each well and luminescence was measured and normalized to protein levels using a Biotek Synergy HT plate reader. Data were acquired with the Gen5 software suite and manipulated and plotted using GraphPad Prism software suite.
Cell viability assays
To text the toxicity of siRNA duplexes, we proceeded to measure cell viability using the Cell Titer Blue Assay (Promega). Briefly, HeLa cells were seeded at 5,000 cells/well in a 96-well plate. The next day, samples were prepared as in the Luciferase Assay (100 nM final concentration) and incubated with HeLa cells for 24 h. After overnight incubation, the Cell-Titer Blue reagent was added and sample fluorescence was measured using a Biotek Synergy Plate Reader (Ex. 560 nm and Em. 590 nm). Analysis was performed using GraphPad Prism software and samples were normalized to the Oligofectamine control.
Results and Discussion
Experiment design
Dinucleotides containing alkylamino PN linkages are positively charged at neutral pH, which can markedly impact their hybridization properties [27]. Preliminary modeling experiments and a previous study by Deglane et al. [37] suggested the potential for electrostatic attraction across the minor groove of an oligonucleotide duplex, between phosphate oxygen anions and the pendant amine function of a PN linkage.
To further explore this interaction, four different conjugate structures were selected, as shown in Fig. 2. Three of these conjugates contain tertiary amine moieties that were expected to be positively charged at physiological pH, having pKaH ∼ 10 [38]; these structures vary only in the length of the carbon spacer linking the two amine functions. The fourth conjugate contained a neutral isopropyl group at its terminus, to verify whether the presence of a pendant ionizable moiety affects duplex thermal stability and biological activity.
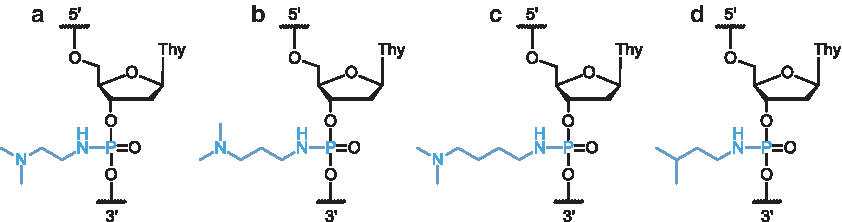
Phosphoramidate conjugate structures used in this study.
To examine the positional effects of these modifications, three positions within the passenger strand of a duplex targeting firefly luciferase mRNA (from position +1818 to +1836) were selected for the incorporation of modified linkages, as shown in Fig. 3. Each of these positions was modified with each of the four conjugates. To probe the effects of duplex type as they relate to PN modifications, modified passenger strands incorporating thymidine residues at the site of modification (sequences d,r[x,y,z]) were synthesized using both DNA and RNA residues.
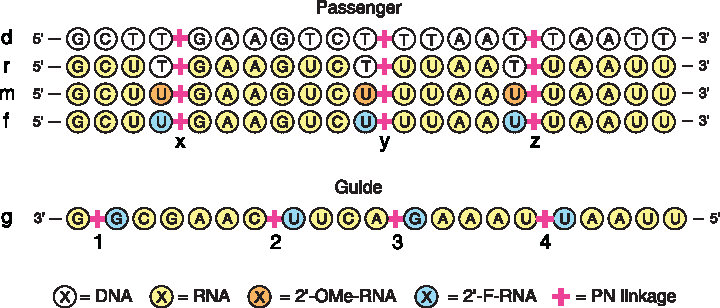
Schematic of singly modified sequences studied. Sequences are either passenger (d, r, m, f) or guide (g) strands of a 21mer siRNA targeting firefly luciferase mRNA from position +1818 to +1836. Positions modified in passenger strand: x, y, and z. Positions modified in guide strand: 1, 2, 3, and 4. Color images available online at www.liebertpub.com/nat
To examine the difference between DNA-like and RNA-like residues at the site of modified linkages with respect to duplex structure and biological properties, 2′-F- and 2′-OMe-uridine-3′-H-phosphonate residues were also incorporated into RNA passenger strands. This permitted the investigation of sequences that were entirely RNA like in nature, as analogous sequences incorporating modifications using thymidine residues would likely contain destabilizing A-B junctions [39]. Finally, modification of siRNA guide strands was examined through the incorporation of PN modifications at four different positions, as shown in Fig. 3. A complete list of the sequences investigated herein can be found in Supplementary Table S1 of the Supplementary Data; mass spectrometry data for all sequences is presented in Supplementary Table S2 (Supplementary Data are available online at www.liebertpub.com/nat).
Preparation of PN-modified oligonucleotides
To circumvent stability issues relating to the presence of 2′-OH groups adjacent to PN-modified linkages, 2′-deoxy-, 2′-fluoro-, and 2′-O-methyl-3′-H-phosphonate residues were used herein to incorporate modified linkages in both DNA and RNA sequences.
2′-Blocked residues can be incorporated within siRNAs as the 2′-OH function is not required for RNAi activity [40]. As shown in Supplementary Fig. S1, these monomers were coupled to the growing oligonucleotide as suitably protected 3′-H-phosphonate triethylamine salts, resulting in the formation of H-phosphonate diester linkages. The diester linkages were then transformed into the desired PN linkages through an Atherton-Todd oxidative amination [41,42] using a 10% solution (v/v) of the corresponding amine in carbon tetrachloride.
While the synthesis of PN-modified DNA sequences is relatively straightforward, the incorporation of modified linkages within RNA sequences necessitated some modifications to standard synthetic protocols. In particular, PN linkages are known to be unstable in both acidic and alkaline conditions [34], and as a result, care must be taken to select conditions that will facilitate the retention of modified linkages. To determine the specific stability of the PN linkage, dimethylethylenediamine (DMEDA) PN dinucleotides were synthesized and subjected to treatment with a wide range of reagents and conditions commonly used throughout oligonucleotide synthesis, including detritylation, activation, oxidation, ammonolysis, and desilylation.
In the synthesis of RNA, triethylamine trihydrofluoride (TREAT-HF) is a commonly used, reliable reagent for the removal of 2′-O-TBDMS protecting groups [43]. As shown in Supplementary Fig. S2, treatment of a DMEDA PN dinucleotide with TREAT-HF resulted in cleavage of the PN linkage after only 1 h (complete desilylation of a 21mer oligonucleotide would require 48 h). The PN peak present at ∼10 ppm was replaced by three peaks at -5, -10, and -15 ppm (J = 998 Hz); interestingly, this pattern of peaks corresponds well with the expected 31P NMR spectra of phosphorofluoridate dinucleotides [44,45].
To avoid the use of neat TREAT-HF, a 6:4:3 (v/v) mixture of TREAT-HF/triethylamine/N-methylpyrrolidone was used to desilylate modified oligonucleotides. Compared with TREAT-HF, this condition resulted in only minimal chain cleavage, permitting isolation of the desired full-length oligonucleotide.
Thermal stability of modified oligonucleotides with DNA and RNA complementary strands
In general, RNA:RNA duplexes are more stable than either DNA:DNA or hybrid DNA:RNA duplexes, although melting temperature values vary widely with sequence [46,47].
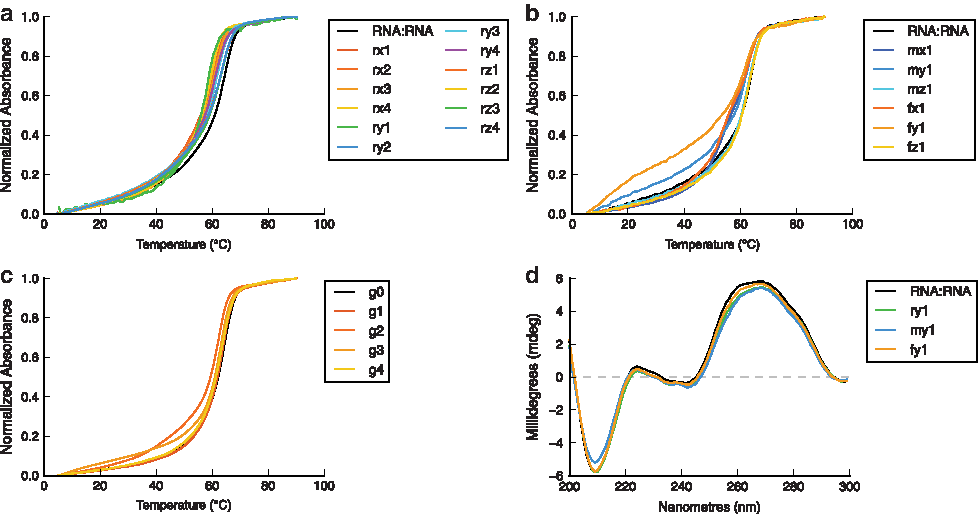
In addition, PN-modified DNA sequences generally form stronger duplexes with complementary DNA than with RNA, since RNA is much more sensitive to distortions in structure resulting from modified linkages [31]. To examine this further, our PN-modified DNA and RNA passenger strands were each hybridized with both DNA and RNA guide strands. The results of thermal denaturation experiments are summarized in Table 1. Tm curves of the RNA:RNA duplexes are shown in Fig. 4a; Tm curves of the DNA:DNA, DNA:RNA, and RNA:DNA duplexes are shown in Supplementary Fig. S3.
Thermal melting
Sequences: 5′-gct t(x)ga agt ct(y)t taa t(z)ta att-3′ (dx, dy, dz); 5′-GCU t(x)GA AGU Ct(y)U UAA t(z)UA AUU-3′ (rx, ry, rz). Complements: 3′-ggc gaa ctt cag aaa tta att-5′ (DNA); 3′-GGC GAA CUU CAG AAA UUA AUU-5′ (RNA). DMEDA, dimethylethylenediamine; DMPDA, dimethylpropanediamine; DMBDA, dimethylbutanediamine; isoPA, isopentylamine. Buffer: 10 mM sodium phosphate, 140 mM KCl, 1 mM MgCl2, pH 7.2.
DNA:DNA duplexes were the least sensitive to modification, while those duplexes comprising RNA in either or both strands were much more destabilized by the inclusion of modified linkages. On average, RNA and hybrid duplexes were destabilized by 2.1°C–2.6°C per modification, compared to an average destabilization of only 1.0°C per modification in DNA duplexes. We hypothesize that this could be due, in part, to the formation of destabilizing A-B junctions in the duplex resulting from the presence of DNA residues within the RNA strand.
The length of the spacer between amine moieties within the conjugate (Fig. 2, structures a-c) did not have any apparent effect on hybridization; average destabilization from each of the three cationic conjugates containing pendant amine functions was similar, ranging from 1.7°C to 2.0°C per modification. However, the neutral conjugate (Fig. 2, structure d) was slightly more destabilizing, with an average ΔTm of 2.9°C per modification; it is possible that this additional destabilization resulted from the absence of electronic effects provided by the pendant tertiary amine moiety present in the diamine structures DMEDA, DMPDA, and DMBDA [37].
Upon calculating the mean destabilization for all modified duplexes, it was observed that, in general, modified DNA passenger strands were most sensitive to modifications incorporated mid-strand, while RNA passenger strands were most sensitive to modifications incorporated at the 3′-end. This discrepancy could be a result of duplex type or a sequence-dependent effect [47].
To assess the difference between the thermal stabilities of sequences modified using DNA-like and RNA-like sugars, passenger strands containing 2′-F- and 2′-OMe-uridine at the site of modification were hybridized with complementary RNA. Results of thermal denaturation experiments are shown in Table 2; Tm curves are shown in Fig. 4b. With respect to the duplexes modified with thymidine residues (average ΔTm = −2.2°C per modification), these duplexes were much less destabilized, with ΔTm values of −1.0°C and −1.2°C per modification for 2′-OMe and 2′-F residues, respectively. This is perhaps due in part to the absence of any destabilizing A-B junctions within these duplexes [36,39].
Sequences: 5′-GCU U(x)GA AGU CU(y)U UAA U(z)UA AUU-3′ (mx, my, mz, fx, fy, fz); 3′-G(1)GC GAA C(2)UU CA(3)G AAA U(4)UA AUU-5′ (g1, g2, g3, g4). Complements: 3′-GGC GAA CUU CAG AAA UUA AUU-5′ (for modified passenger strands); 5′-GCU UGA AGU CUU UAA UUA AUU-3′ (for modified guide strands). Buffer: 5 mM sodium phosphate, 140 mM KCl, 1 mM MgCl2, pH 7.2. ΔTm values calculated relative to an unmodified RNA:RNA duplex.
Finally, modified RNA guide strands containing DMEDA-modified linkages incorporated using 2′-F-2′-uridine- and 2′-F-2′-deoxyriboguanosine-3′-H-phosphonate monomers were hybridized with complementary RNA and assayed for their thermal stability. Results of these experiments are shown in Table 2; Tm curves are shown in Fig. 4c. It was determined that modification of the guide strand at either the 3′-end or in the seed region was neutral, while modification of the two central positions of the guide strand was slightly destabilizing. These values compared well with those obtained for duplexes containing 2′-F-modified residues in the passenger strand, for which ΔTm values ranged from −0.3°C to −2.1°C, with more destabilization observed when modifications were positioned centrally within the sequence.
Circular dichroism
CD spectra of duplexes comprising DMEDA-modified linkages incorporated with T, 2′-OMe-rU, and 2′-F-U residues were contrasted with an unmodified RNA:RNA duplex; averaged spectra are shown in Fig. 4d. It was observed that single PN modifications had little effect on the global helical structure, regardless of the sugar type used to incorporate them. If distortions were to exist, they would likely be minimal and localized at or near the site of modification.
Luciferase activity of modified oligonucleotides
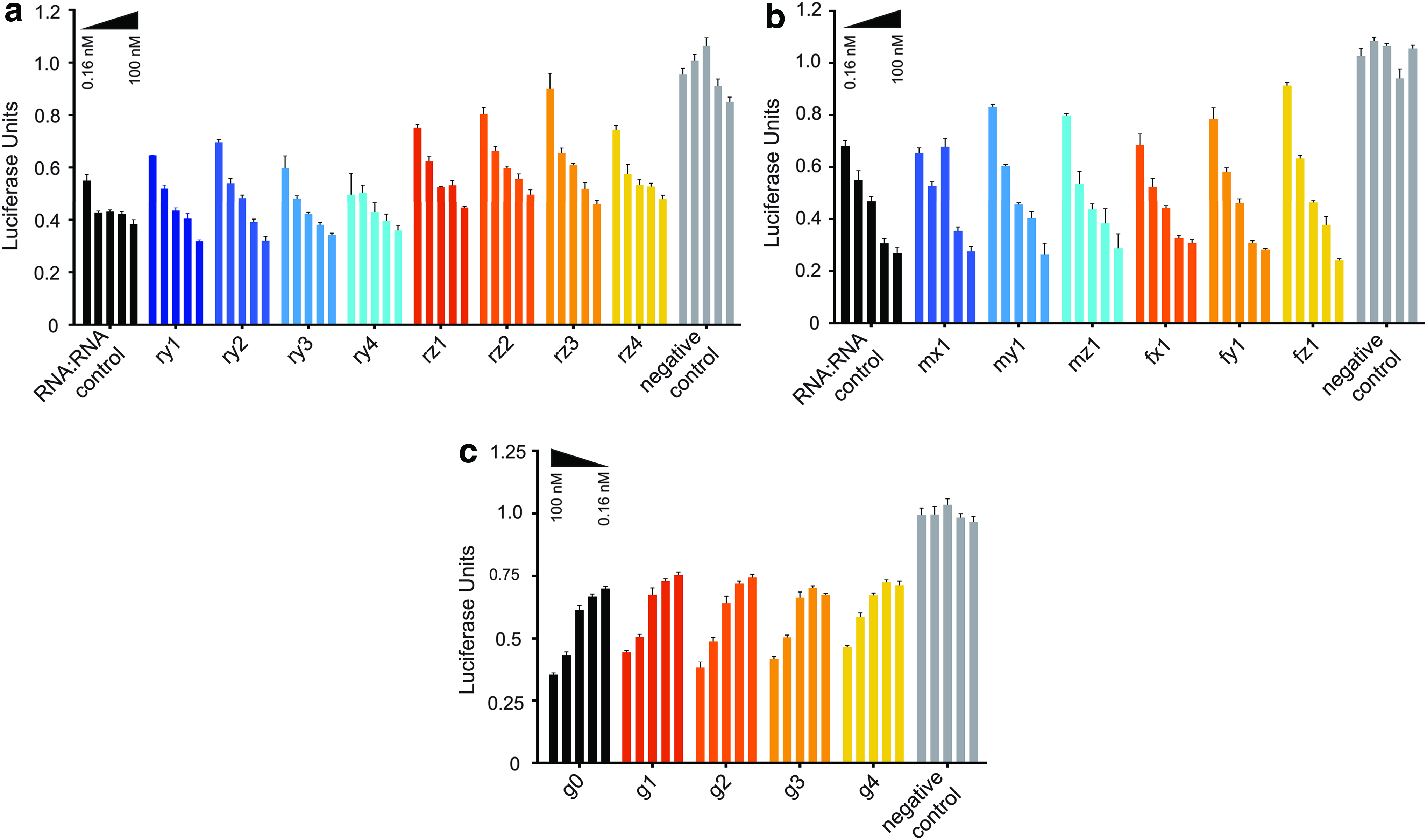
First, modified DNA and RNA passenger strands containing the DMEDA conjugate in each of the three positions (x, y, and z) were hybridized with complementary RNA and assayed for their ability to knock down expression of firefly luciferase. Results are shown in Supplementary Fig. S4. The potency of the modified duplexes was compared with that of the analogous unmodified DNA:RNA and RNA:RNA duplexes. It was determined that single PN modifications within the passenger strand had a minimal detrimental effect on potency. Furthermore, this effect appeared to correlate with the position of the modification. For instance, modifications within the middle of the passenger strand resulted in duplexes with potency similar to that of unmodified siRNA, while duplexes modified near the 5′-end of the passenger strand were least potent.
Next, modified RNA passenger strands containing each of the four conjugate structures (dimethylethylenediamine, DMEDA; dimethylpropanediamine, DMPDA; dimethylbutanediamine, DMBDA; and isopentylamine, isoPA) in one of two positions (y,z) were hybridized with complementary RNA and assayed for their activity against luciferase. Results are shown in Fig. 5a. In this study, it was observed that, among the various conjugate structures, duplexes containing DMPDA modifications were least potent, while duplexes containing isoPA modifications were most potent. This was particularly interesting given that the isoPA modification was the most destabilizing in terms of thermal stability; we envision that, with the appropriate choice of position, these qualities could be exploited in the design of siRNA scaffolds with optimal biological properties.
Luciferase assay results for siRNA duplexes containing single DMEDA phosphoramidate linkages.
Modified RNA passenger strands containing modified linkages incorporated using 2′-F- and 2′-OMe-uridine-3′-H-phosphonate monomers were then hybridized with complementary RNA strands and assayed in comparison with unmodified siRNA. Results are shown in Fig. 5b.
It was observed that siRNA duplexes containing these modified sugars in combination with the cationic PN linkages had similar potency relative to unmodified controls, regardless of the location of modification within the passenger strand.
Finally, siRNA duplexes comprising guide strands modified with single DMEDA PN linkages hybridized with unmodified RNA were assayed for their ability to knock down luciferase expression in comparison with unmodified siRNA; results are shown in Fig. 5c. In general, the entirety of the passenger strand and the 3′-tail region of the guide strand are known to be relatively tolerant to modification, while the 5′-end and central regions of the guide strand are quite sensitive to structural changes [40,48,49]. This is unsurprising as human argonaute2 (hAgo2) binds the guide strand during RISC loading; interaction with the phosphates of the guide strand permits efficient Ago2-mediated cleavage of the mRNA target [48].
Interestingly, it was observed that all duplexes containing modified guide strands had very similar potency compared with native siRNA, regardless of the position of modification. Perhaps most strikingly, this was true even when the modified linkage was located at the cleavage site of the duplex.
Synthesis and study of highly modified siRNA architectures
Recent work by several groups [36,50–52] has suggested that extensive modification can result in siRNAs with improved affinity for target sequences, enhanced in vivo stability, reduced off-target effects, and more efficient selection of the intended guide strand by RISC. To this end, a number of common highly modified scaffolds have emerged, with many incorporating 2′-fluoro- and 2′-O-methyl-modified residues in various patterns [50,53].
Given our promising results from the incorporation of PN linkages using 2′-F- and 2′-OMe-modified sugars, we aimed to incorporate multiple PN modifications within a siRNA passenger strand fully modified with 2′-F- and 2′-OMe-RNA residues in an alternating manner. This motif has been shown to produce duplexes with improved thermal stability and in vitro potency when hybridized with unmodified RNA [50]. In addition, removing all 2′-OH residues from the sequence allowed us to avoid the issues previously experienced with the desilylation of PN-modified oligonucleotides containing 2′-O-TBDMS protecting groups.
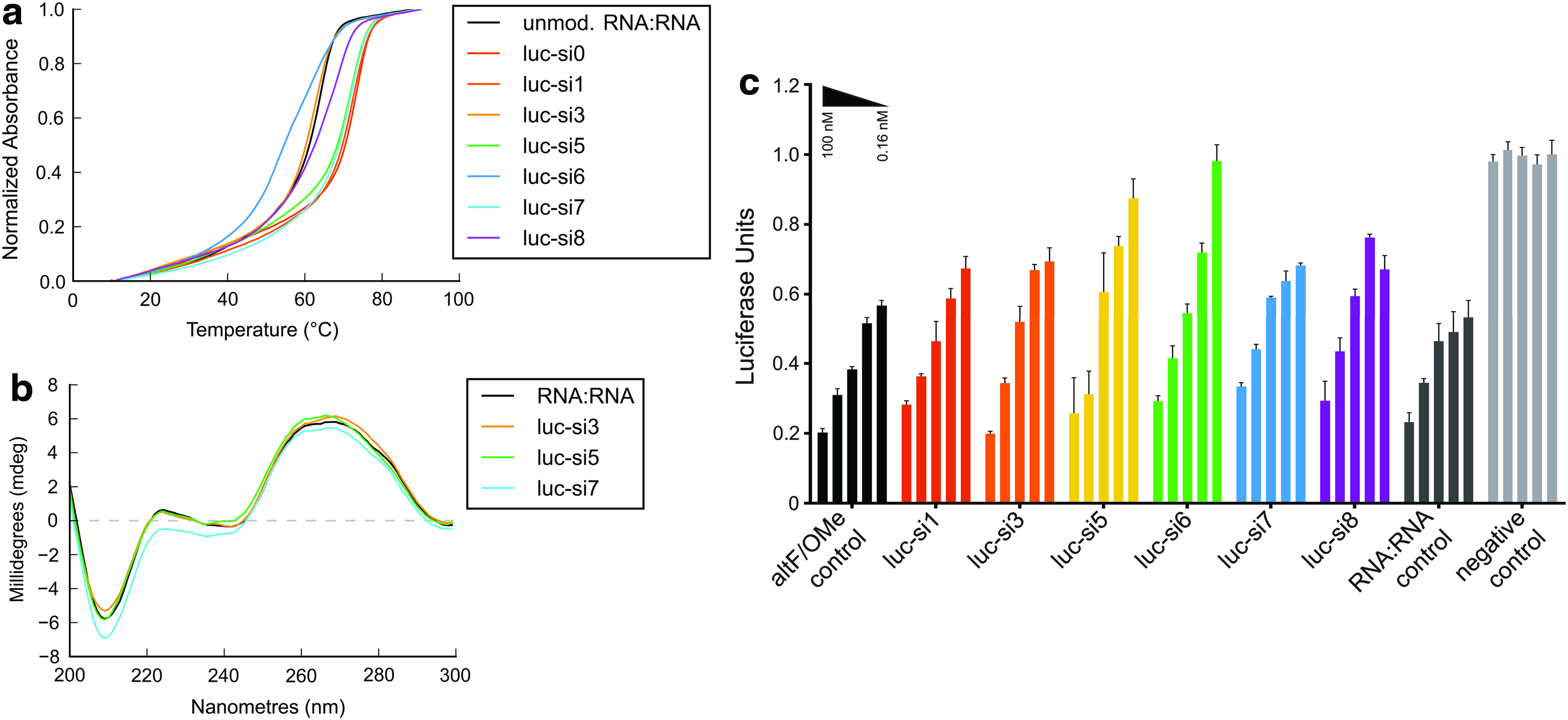
A series of modified passenger strands targeting the same region of luciferase mRNA was designed, incorporating multiple DMEDA PN linkages at either the 3′- or 5′-end of the sequence in a consecutive, alternating, or spaced motif. Sequences are shown in Table 3. Synthesis of these sequences was accomplished using an automated process combining H-phosphonate and phosphoramidite chemistry, to be described in detail in a future publication. Sequences were hybridized with complementary unmodified RNA to form siRNA duplexes with 2nt 3′-overhangs; duplexes were then assessed to determine their thermal stability, helical structure, and ability to knock down luciferase expression. These results are presented in Fig. 6.
Stability, structure, and activity of duplexes containing highly modified passenger strands having multiple phosphoramidate modifications.
Legend: RNA; 2'-OMe-RNA;
With respect to thermal stability, the use of the alternating 2′-F/2′-OMe motif markedly increased thermal stability, as expected, with an improvement of 9.1°C relative to the unmodified siRNA duplex. Tm values for all sequences are shown in Table 3.
Interestingly, for those sequences having modifications at the 3′-end, the average ΔTm per modification (−0.4°C) was much less than the average value observed for singly modified siRNAs (average ΔTm/mod = −1.0°C). Destabilization resulting from DMEDA PN modifications was observed to be more intense when modifications were clustered at the 5′-end of the passenger strand (average ΔTm/mod = −3.8°C). Since these results were consistent across all three motifs, this additional destabilization was hypothesized to be sequence dependent, rather than motif dependent.
As shown in Fig. 6b, CD spectra of three of the modified duplexes (si3, si5, and si7) compared with an unmodified RNA:RNA duplex showed that multiple modifications had little effect on the global helical structure, regardless of their number and the pattern in which they occurred. Spectra for all compounds, including the unmodified duplex, showed a strong negative band at 210 nm, indicating an A-form helical structure.
With respect to potency, the activity of PN-modified duplexes compared well with the positive control, with only a minimal decrease in potency observed across all modified sequences. Results are shown in Fig. 6c. This was of particular interest in light of our thermal stability results, which demonstrated a large difference in thermal stability based on the position of the modifications. Based on these results, one might expect that those duplexes having modifications at the 5′-end of the passenger strand would be much less potent in comparison with the other sequences, given the thermal bias demonstrated by the RISC complex [54]; however, this was not observed. It is thus likely that multiple factors affect the activity of these duplexes in addition to thermal asymmetry.
To examine the toxicity of the modified duplexes in HeLa cells, cell viability assays were performed using a wide range of modified and unmodified duplexes, at the highest concentration employed in our luciferase assays (100 nM). Cellular viability of cells treated with modified and unmodified duplexes was compared with that of untreated cells. Results of this assay are shown in Supplementary Fig. S5. While some variability among samples was apparent, no significant detriment to cellular viability was observed following treatment with modified duplexes.
Conclusions
PN modifications of oligonucleotides have been well studied in DNA, but have not been evaluated in RNA due to stability issues stemming from the presence of the 2′-OH group. We have demonstrated that PN-modified linkages can be incorporated within siRNA duplexes using a variety of 2′-blocked 3′-H-phosphonate monomers. Single modifications incorporated using 2′-deoxy residues generally had a slightly destabilizing effect (1–2°C/mod) on the thermal stability of DNA, RNA, and hybrid duplexes; this destabilizing effect was most pronounced in duplexes containing RNA and when modifications were incorporated within the central region of the passenger strand. Single modifications incorporated in RNA passenger strands using RNA-like 2′-F and 2′-OMe residues were even less destabilizing, possibly owing to the entirely RNA-like character of the resulting duplexes.
The activity of siRNA duplexes containing singly modified DNA and RNA passenger strands compared favorably with that of unmodified siRNA duplexes, resulting in a minimal loss of potency, particularly when those modifications were incorporated using RNA-like sugars. Notably, single PN modifications of RNA guide strands resulted in duplexes with comparable potency to unmodified siRNAs. Initial investigations of highly modified siRNA passenger strands containing multiple PN modifications also exhibited favorable results with respect to thermal stability, duplex structure, and potency.
Footnotes
Acknowledgments
The authors wish to acknowledge financial support provided by the Natural Sciences and Engineering Council of Canada (NSERC DG to MJD and NSERC CREATE to DV). We also thank Prof. H. Sleiman for providing cell culture facilities (luciferase screening assays) and Dr. A. Wahba for mass spectrometry support.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.