
Abstract
Antisense oligonucleotides (ASOs) regulate gene expression by binding to complementary target RNA, and ASOs can be designed to take advantage of a growing array of post RNA binding molecular mechanisms. Intracellular trafficking of ASOs influences their efficacy. We have identified a number of membrane-less structures in the nucleus, nucleolus, and cytoplasm where phosphorothioate-modified ASOs (PS-ASOs) accumulate and have shown that PS-ASOs can induce the formation of new nuclear structures such as PS-bodies and paraspeckle-like structures. In this study, we report that PS-ASOs can localize to cytoplasmic processing bodies (P-bodies) and increase the number of P-bodies in cells. The antisense activity of PS-ASOs was not affected by the absence of essential P-body assembly proteins DDX6 and LSm14A. Moreover, the effects of PS-ASOs on P-body assembly were independent of their antisense activities. The phosphorothioate modification stabilizes the association between ASOs and cellular proteins and is essential for the P-body localization of ASOs. Since PS-ASOs bind to major P-body components, PS-ASOs may serve as scaffolds for P-body formation. Taken together, these results indicate that interactions of PS-ASO with proteins, rather than antisense activities, are essential for the dynamic interplay between PS-ASOs and P-bodies.
Introduction
Antisense oligonucleotides (ASOs) bind to complementary RNA. They are usually designed to mediate transcript degradation through the recruitment and activation of endonuclease RNase H1, but can be designed to exploit other mechanisms as well [1–5]. ASOs are useful research tools for controlling gene expression and have broad therapeutic applicability. Chemical modification has been widely used in antisense technology to increase the stability, to enhance target affinity, or to facilitate uptake by particular cell types. Increasing the number of phosphorothioate (PS) backbone modification facilitates ASO uptake by cells and protects ASOs against nuclease degradation but does not prevent the activity of RNase H1. Modifications at the 2′ ribose such as 2′-O-methoxyethyl (MOE), 2′-O-methyl (OMe), 2′-fluoro (F), 2′4′-locked nucleic acid (LNA), and 2′,4′-constrained 2′-O-ethyl (cEt) are also commonly incorporated to enhance the stability and binding affinity of ASOs to their target RNAs [2,3,6,7]. To stimulate the sequence specific cleavage activities of RNase H1, ASOs were commonly designed as “gapmer” oligonucleotides with a central deoxynucleotide gap flanked at each end (wing) by 2′-ribose-modified nucleotides [2,7].
In previous work we identified the major cellular proteins that interact with PS-ASOs and characterized how these interactions influence cellular uptake and efficacy of PS-ASOs [8–12]. For example, the uptake of chemically modified PS-ASOs into cells is facilitated through binding with cell surface receptors such as asialoglycoprotein receptor [13,14] and epidermal growth factor receptor [15]. The release of PS-ASOs from endocytic organelles is mediated through association with different proteins. We recently showed that early endosome protein annexin A2 and endoplasmic reticulum-Golgi transport protein STX5 bind PS-ASOs during endocytic transport and promote the productive release of PS-ASOs [8,12,16].
Protein binding contributes to the subcellular distribution of PS-ASOs to various cytoplasmic and nuclear structures, including stress granules, paraspeckles, nucleoplasm, nucleoli, and perinucleolar caps [10,17]. Interestingly, when high concentrations of PS-ASOs are delivered inside cells through transfection, PS-ASOs seed the formation of nonmembranous organelles. Fluorescently labeled PS-ASOs are observed as distinct foci in the nucleus, termed PS bodies [18]. The formation of PS-bodies is at least partially due to the association between PS-ASOs and the β subunit of T-complex protein 1 [9]. We also found that PS-ASOs can replace the long noncoding RNA NEAT1 as a scaffold for the formation of nuclear paraspeckles. PS-ASOs appear to seed the assembly of paraspeckle-like structures by recruiting major paraspeckle proteins such as P54nrb and PSF [10]. Since cellular uptake, subcellular trafficking, and functional release of PS-ASOs are influenced by their association with different cellular proteins, characterization of protein/PS-ASO interaction will lead to a better understanding of the pharmacokinetics of PS-ASO-based drugs.
Through affinity-selection approaches, our group recently showed that many ASO binding proteins are known nucleic acid binding proteins found in cytoplasmic ribonucleoprotein (RNP) granules [16]. Interestingly, several key components of cellular processing bodies (P-bodies), such as DDX6, UPF1, and MOV10, were also found to be strongly associated with PS-ASOs [4,16]. We hypothesized that PS-ASOs are localized to P-bodies through their association with major P-body assembly factor such as DDX6 [19–22].
P-bodies are microscopically detectable cytoplasmic foci formed by the association of mRNAs and multiple proteins involved in translational inhibition. Enzymes involved in different stages of mRNA degradation such as GW182 and argonaute proteins, which are components of the RNA interference (RNAi) machinery, decapping enzymes DCP1/2, deadenylating complex factors PAN2-PAN3 and CCR4-NOT1, and 5′-3′ exonuclease XRN1, are detected in P-bodies [23]. Moreover, nonsense-mediated decay (NMD) factor UPF1 [24,25] and AU-rich element decay pathway proteins TTP and BRF1 are also localized to P-bodies [26,27]. Due to the presence of mRNA decay-mediating proteins in P-bodies, it has been speculated that P-bodies serve as centers for mRNA decay. However, recent evidence clearly indicates that the physical presence of P-bodies is not required for mRNA degradation mediated by small interfering RNA (siRNA), NMD, or microRNA (miRNA) [28–30] and that siRNA-mediated gene silencing nucleates P-bodies [31]. Thus, the formation of P-bodies may be the result rather than the cause of RNA decay activities.
In this study, we found that after delivery by transfection, PS-ASOs bind to core P-body proteins and induced formation of cytoplasmic P-bodies. The pharmacological activity of PS-ASOs was not affected even in the absence of essential P-body assembly factors DDX6 and LSm14A. The number of P-bodies was partially restored by PS-ASO transfection. Mechanistically, PS-ASO-mediated P-body formation was dependent on their protein association rather than RNase H1-based RNA degradation activities. Taken together, our data support the notion that the recruitment of P-body assembly factors affects the subcellular localization of PS-ASOs. Moreover, we also determined that the interaction between PS-ASOs and P-body components influences the dynamics of cellular P-bodies in a gene silencing-independent manner.
Materials and Methods
ASOs, siRNAs, antibodies, and primer probe sets used for quantitative real time-polymerase chain reaction (qRT-PCR) are listed in Supplementary Data.
Plasmid construction and primers
PCMV6-DDX6-turbo green fluorsecent protein (tGFP) plasmid was purchased from Origene. DDX6-tGFP mutants were generated using Site-Directed Mutagenesis Kit (Thermo Fisher) following manufacturer's protocols. Primer sets used for PCR are listed in Supplementary Data.
Cell culture and transfection
HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 0.1 μg/mL streptomycin, and 100 U/mL penicillin. Cells were seeded at ∼70% confluency and grown for 16 h before transfection. siRNA was transfected into cells using Lipofectamine 2000 (Thermo Fisher) at 10 nM final concentration following manufacturer's protocols. For overexpression experiments, plasmids were transfected into cells using FuGENE 6 at 1 μg/mL following manufacturer's protocols. For activity assay of ASOs, HeLa cells were treated with control siRNA or siRNAs against different RNA targets for 24 h. Cells were then reseeded in 96-well plates at ∼5,000 cells per well. After 24 h, ASOs of different concentrations were transfected into cells for 4 h using Lipofectamine 2000 at 8 μL/mL.
RNA preparation and qRT-PCR analysis
RNA preparation and qRT-PCR assay using TaqMan primer probe sets were performed as described previously [32]. Briefly, total RNA was prepared using RNeasy Mini Kit (Qiagen). qRT-PCR was performed in triplicate using StepOne Real-Time PCR system and TaqMan primer probe sets with AgPath-ID™ One-step RT-PCR Kit (Applied Biosystems). qRT-PCR in 20 μL reactions was performed using the following program: 48°C for 10 min, 94°C for 10 min, and 40 cycles of 45 s each at 94°C and 60°C. The qRT-PCR results were quantified using StepOne Software V2.3, calculated and plotted using Excel. Target RNA levels were normalized to the levels of total RNA measured by Quant-iT RNA Assay Kit (Thermo Fisher).
Western analysis
Cells were harvested directly using radioimmunoprecipitation assay buffer (Thermo Fisher) after being washed once with 1× phosphate-buffered saline (PBS). Cell lysate was cleared by centrifugation at 10,000g for 10 min at 4°C. Proteins (10–20 μg/lane) were separated by 4%–12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to membranes using iBlot transfer system (Thermo Fisher), and proteins were detected with specific antibodies and visualized using Forte Western HRP Substrate Kit (Millipore).
Immunofluorescent staining
Cells grown in glass bottom microscopic dishes (MatTek) were washed with 1× PBS, fixed with 4% paraformaldehyde for 10 min at room temperature, permeabilized, and blocked for 20 min in blocking buffer (0.1% Triton, 3% bovine serum albumin in 1× PBS). Cells were incubated with primary antibodies at 1:200–1:1,000 dilution in blocking buffer overnight at 4°C. Next morning, dishes were washed thrice by PBS and incubated for 1 h at room temperature with secondary antibody conjugated with fluorophores at 1:1,000 dilution (Thermo Fisher) in blocking buffer. After washing thrice, dishes were mounted and sealed with Fluoromount-G containing 4′,6-diamidino-2-phenylindole (SouthernBiotech). Images were acquired using confocal microscope (Olympus FV-1000) and processed using FV-10 ASW 3.0 Viewer software (Olympus). Quantification of P-body numbers was performed by manually counting EDC4 positive foci in at least 30 cells from 5 to 10 random selected fields. Quantification of colocalization events was performed manually from ∼20 cells, by counting overlapping foci where images from both channels could be clearly distinguished from the background without saturation. Statistics analysis was performed based on unpaired t-test using prism.
Affinity selection of ASO-associated proteins
Affinity selection using biotinylated ASO IONIS 386652 was performed as described previously [9]. Briefly, 25 μL streptavidin beads (Thermo Fisher) were incubated with 25 μL of 100-μM biotinylated PS-ASOs with different numbers of PS linkages at 4°C for 2 h in immunoprecipitation (IP) buffer (Thermo Fisher). Beads were then washed thrice with IP buffer. ASO-coated beads were incubated at 4°C overnight with ∼300–500 μg HeLa cell extract prepared in IP buffer plus cocktail protease inhibitors (Roche). After three washes with IP buffer, ASO associated proteins were eluted by competition with 50 μL of 50 μM ASOs as indicated in figures. The eluted proteins were separated on 4%–12% SDS-PAGE, analyzed either by silver staining or by western analyses. For direct elution of isolated proteins using biotinylated ASOs, coisolated proteins were eluted by boiling in 2× lithium dodecyl sulfate (LDS) loading buffer (Thermo Fisher) for 10 min. For RNase A treatment, equal amount of ASO-coated beads/protein complex was resuspended into 1 mL PBS with or without 100 μg/mL of RNase A (Thermo Fisher) for 1 h at 4°C. The complexes were then washed thrice with IP buffer, and ASO conjugated proteins were eluted as described before.
Results
PS-ASOs associate with P-bodies
Using an affinity selection assay, we previously observed that proteins known to associate with P-bodies, for example, DDX6 and UPF1, bound to PS-ASOs [16]. We first tested whether PS-ASOs associated with other P-body components. Affinity selection was performed to isolate PS-ASO associated proteins using a biotinylated, 5-10-5 PS-MOE gapmer ASO incubated with cell lysate, and bound proteins were eluted by competition using 5-10-5 gapmer PS-ASOs containing different 2′ modifications at the wings or by uniform PS-DNA or phosphodiester (PO) DNA ASOs without 2′ modifications. These ASOs have the same sequence but different chemical modifications. We found that LSm14A, eIF4e-T, and PAT1B bound to PS-DNA-ASOs, as well as gapmer 2′-MOE (IONIS 116847) and gapmer 2′F PS-ASOs (IONIS 404130) (Fig. 1A). P-body proteins were barely eluted from the beads by ASOs with PO linkages. Previously, we have shown that PO-backbone ASOs remain stable during this elution process [9]. These results suggest that the above P-body proteins can bind PS-ASOs, similar to many other ASO-binding proteins identified previously [16].
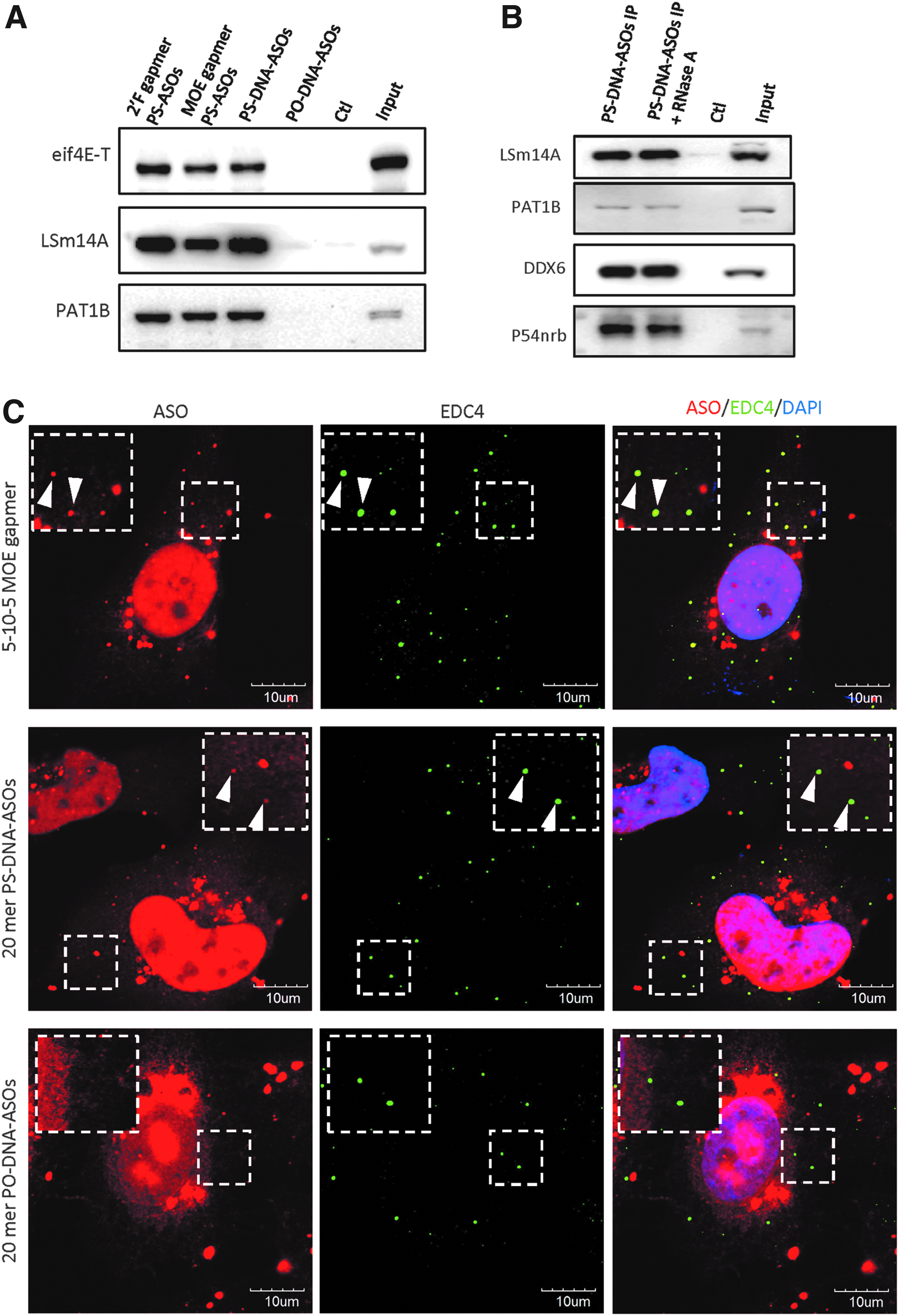
PS-ASOs associate with P-body components and are localized in P-bodies.
Since P-bodies are composed of both RNA and RNA binding proteins, the presence of RNA may serve as a scaffold and mediate the aggregation of different P-body components in the absence of a physical association of these proteins. In support of this hypothesis, P-body proteins UPF1 and MOV10 have been coimmunoprecipitated with DDX6 only in the presence of RNA [20]. To exclude the possibility that the association between PS-ASOs and different P-body components is mediated indirectly through RNA molecules, we treated the beads after affinity selection and washing steps with RNase A. After RNase A treatment and washing, the levels of DDX6, LSm14A, and eIF4E-T proteins associated with the beads were similar with or without RNase A treatment (Fig. 1B), implying that the interactions between PS-ASOs and P-body proteins are not mediated by RNA.
We next investigated whether the strong associations between PS-ASOs and P-body proteins result in their colocalization in cells. We first transfected HeLa cells with 50 nM Cy3-conjugated 5-10-5 MOE gapmer PS-ASO (IONIS 446654), which has the same sequence as the PS-ASO used in the affinity selection assay. At 4 h after transfection, cells were fixed and stained with P-body marker EDC4 (Fig. 1C). As expected, PS-ASOs were mainly localized in the nucleus and formed distinct punctate structures known as PS-bodies. Cytoplasmic PS-ASOs were also observed in aggregates that may be liposomes formed by transfection reagents or lysosomes loaded with ASOs. The PS-ASOs were also observed within cytoplasmic foci marked by P-body protein EDC4 (Fig. 1C). Although P-body localized PS-ASOs accounted for a small fraction of intracellular PS-ASOs, PS-ASOs were observed in almost all EDC4-positive foci.
Since the formation and composition of P-bodies are highly dynamic, we also stained PS-ASO-transfected HeLa cells with a spectrum of antibodies against known P-body components DCP1B, DDX6, LSm14A, EDC3, and eIF4E-T (Supplementary Fig. S1). Strong colocalization was observed between PS-ASOs with each of these P-body proteins, and all staining patterns were similar. The colocalization between PS-ASOs and P-bodies is independent of 2′ modification as MOE-, or cEt-, F-modified gapmer PS-ASOs (Supplementary Fig. S2A), and PS-DNA ASOs without 2′ modification (Fig. 1C, middle panel) were observed in cytoplasmic P-bodies. We also confirmed that P-body localization was sequence independent, as MOE gapmer PS-ASOs containing different sequences were also detected in EDC4-labeled foci 4 h after transfection (Supplementary Fig. S2B, C). PO-ASOs of the same sequence as the MOE gapmer PS-ASO did not associate with any P-body proteins (Fig. 1A); thus, this Cy3-labeled PO-ASO was used as a negative control for potential artifacts caused by transfection or channel leakage during macroscopic visualization. PO-ASOs were condensed in the perinuclear region of the cytoplasm and were rarely colocalized with P-bodies (Fig. 1C). Taken together, our data indicated that PS-ASOs bind directly to P-body proteins and localize to P-bodies independent of 2′ modification and sequence.
PS-ASOs induce formation of P-bodies
P-bodies are composed of mRNAs associated with nonribosomal proteins. The numbers and sizes of P-bodies are highly dynamic [28]. Interestingly, it has been shown that robust RNA silencing activities mediated by siRNAs, double-stranded RNA, or miRNA result in increased numbers of P-bodies [23,33–35]. Since transfection of cells with PS-ASOs leads to target mRNA degradation within hours, we asked whether PS-ASOs trigger the assembly of P-bodies. An increase in the number of P-bodies was observed in HeLa cells transfected with 50 nM PS-ASO (Fig. 2A). By quantifying the number of EDC4 foci in each cell, we found that PS-ASOs stimulated the formation of P-bodies in a dose-dependent manner (Fig. 2B). The average number of P-bodies increased from ∼5 to 15 per cell when the concentration of PS-ASO was increased from 1 to 200 nM. It appears that increase in P-body numbers is saturable, as no further increase was observed when ASO concentration was higher than 50 nM. At low concentration (1 nM), the majority of PS-ASOs were found in the cytoplasm. Nevertheless, their colocalization with P-bodies was readily detected, confirming that P-bodies are very active sites for accumulation of PS-ASOs even at concentrations that PS-body was not detected [9,10]. At all concentrations, PS-ASOs were colocalized with almost all detected P-bodies (Fig. 2A). Levels of P-body proteins DDX6, LSm14A, PAT1B, eIF4E-T, DCP1B, EDC4, and XRN1 were comparable in cells not transfected with PS-ASO and those transfected with PS-ASOs (Fig. 2C), indicating that ASOs likely promote the formation of P-body through the recruitment of preexisting P-body proteins rather than directly modulating their expression levels.
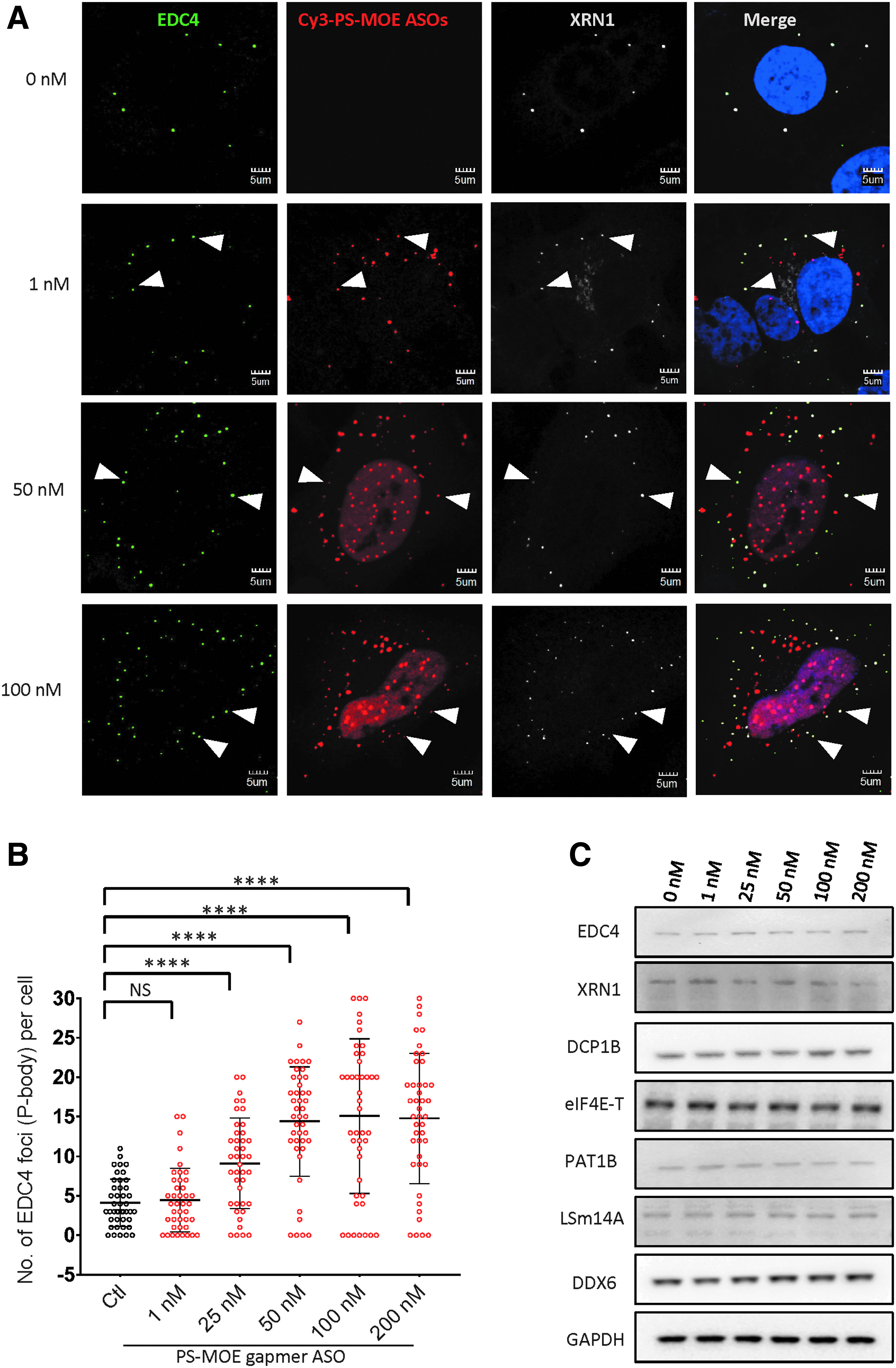
PS-ASOs increased the number of P-body in a dose-dependent manner.
Under stress conditions, nontranslating mRNAs and their associated proteins can aggregate to form stress granules [17,36]. Although P-bodies and stress granules are generally considered to be two distinct cytoplasmic structures, they share several protein components and are often juxtaposed or interlinked [36,37]. We previously reported that a cEt gapmer PS-ASO was localized to stress granules under stress conditions [17]. To confirm that the structures induced by PS-ASOs were indeed P-bodies, we stained PS-ASO-transfected HeLa cells with stress granule marker G3BP1 and found that none of the foci containing PS-ASOs was G3BP1-positive stress granules (Supplementary Fig. S3). Taken together, our data suggest that PS-ASO treatment results in a significant induction of P-bodies without the formation of stress granules.
The intracellular trafficking of PS-ASOs has been shown by us and others to be a time-dependent process involving cytosol and nuclear diffusion, docking with different cell organelles such as endosomes and lysosomes, and formation of new structures such as PS-bodies [4,18]. To determine the temporal interplay between P-bodies and transfected PS-ASOs, we monitored the accumulation of P-bodies and their association with PS-ASOs through a 48-h time course. P-body localization of PS-ASOs was observed at 0.5 h after transfection and at subsequent time points (Fig. 3A). The PS-ASOs were detected in the nucleus at the 0.5 h time point; however, no nuclear PS-bodies were detected (Supplementary Fig. S4). At 4 h after transfection, induction of P-body formation and the presence of nuclear PS-bodies were apparent (Fig. 3A and Supplementary Fig. S4). From 24 to 48 h, cytoplasmic PS-ASOs were gradually concentrated in the perinuclear regions (Fig. 3A). The number of P-bodies in PS-ASO-transfected cells increased over time and reached maximum within 4 h (Fig. 3B). The protein level of major P-body components DDX6, LSm14A, eIF4E-T, EDC4, XRN1, EDC3, and DCP1B did not increase during the 48-h time course after transfection (Fig. 3C), excluding the possibility that the increase in the numbers of P-bodies was due to an induction in P-body protein expression.
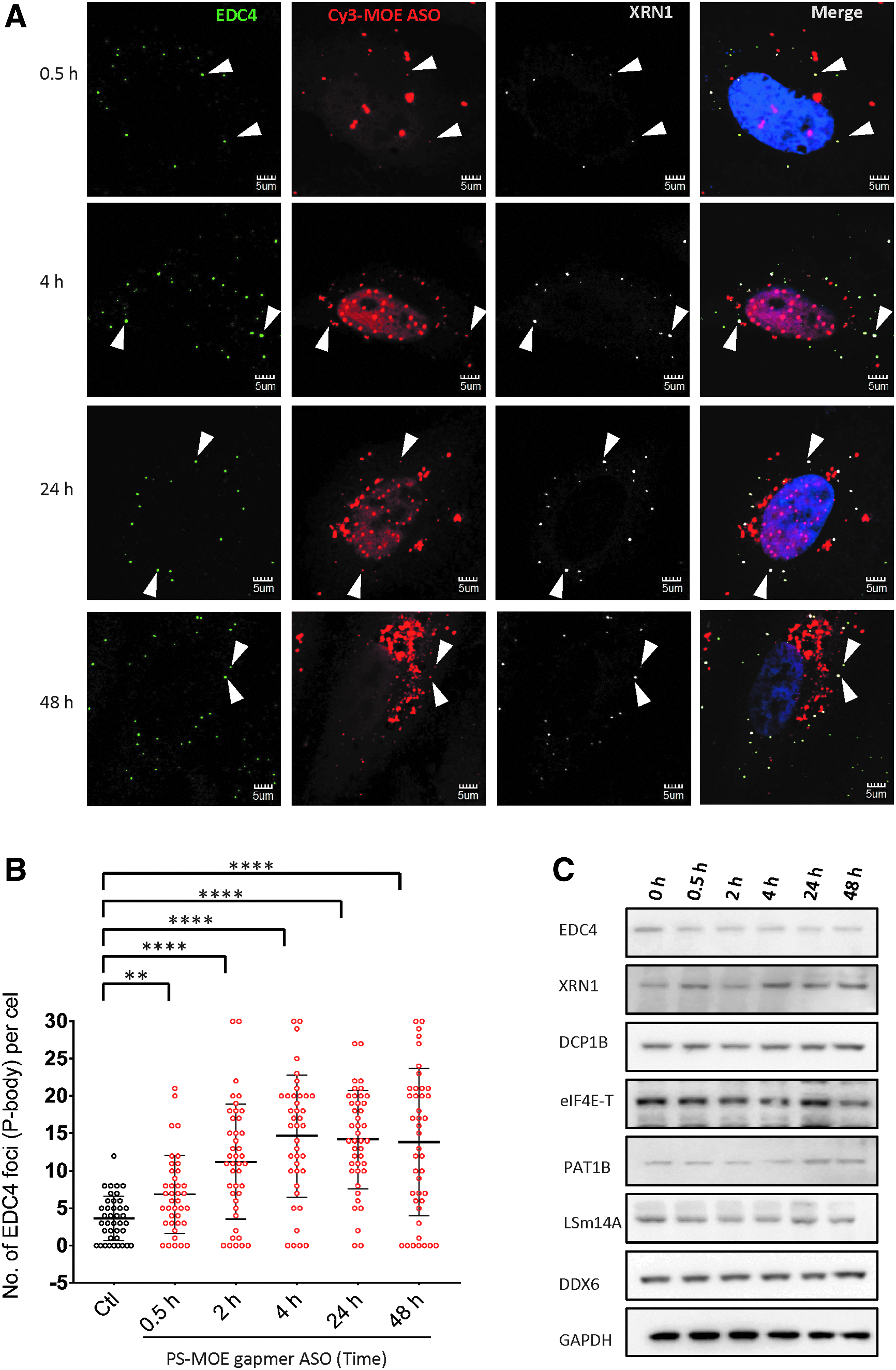
Transfection of PS-ASOs increased the number of P-bodies in a time-dependent manner.
To determine whether the observed increase in the number of P-bodies depends on the PS-ASO sequence, we transfected HeLa cells with MOE gapmer PS-ASOs complementary to Malat1 RNA (IONIS 713841) or mouse Sod mRNA (IONIS 651106). As expected, both colocalized with P-bodies (Supplementary Fig. S2B, C) and induced similar increases in P-body numbers as did the PTEN mRNA targeted MOE PS-ASOs at 4 h after transfection (Supplementary Fig. S5), suggesting that the induction is sequence independent. P-bodies are highly organized cytoplasmic nonmembraned structures that composed of individual nontranslating messenger RNP complexes. Thus, the presence of nontranslating RNA pool is indispensable for the formation of P-bodies [37,38]. To test whether PS-ASO induced EDC4 positive foci are indeed P-bodies rather than the aggregation of P-body proteins, we treated PS-ASO transfected cells with cycloheximide for 1 h to trap mRNA in polysomes and deplete the availability of cytoplasmic mRNA pool. Cycloheximide significantly disrupted PS-ASO induced EDC4 foci, further confirming that PS-ASO-induced EDC4 positive foci are newly assembled P-bodies (Supplementary Fig. S6). In summary, our results suggest that PS-ASOs rapidly localize to P-bodies after transfection and induce the formation of P-bodies without affecting the expression levels of major P-body proteins.
Deletion of P-body assembly factors does not alter the antisense activities of PS-ASOs
The relationship between P-bodies and the RNAi pathway is controversial; conclusions depend on methods used to eliminate P-bodies [28,31,33,35]. For example, the depletion of GW182, which is essential for the formation of P-bodies and miRNA functions, eliminated P-bodies and gene silencing mediated by both miRNAs and siRNAs [22,33,39]. In contrast, P-body depletion achieved by depletion of P-body assembly factors DDX6 and LSm14A was not associated with decreased siRNA-mediated RNA silencing activities [22,28,34,40]. Interestingly, RNase H1-dependent PS-ASOs were capable of mediating RNA degradation in the cytoplasm [41,42]. Since after transfection PS-ASOs preferentially localized to P-bodies, we asked whether a decrease in the number of P-bodies would reduce activities of these PS-ASOs. To decrease the number of P-bodies, we used siRNAs to deplete cells of DDX6 or LSm14A, which are essential for P-body assembly [21,34]. At 48 h after transfection of cells with siRNA, PS-ASOs were transfected into cells, and protein and RNA were analyzed after an additional 4 h. Levels of DDX6 and LSm14A were significantly reduced after siRNA treatment (Fig. 4A, B), and P-body numbers were accordingly decreased (Fig. 5A); however, the activities of MOE gapmer PS-ASOs targeting PTEN, NCL, or Malat1 RNAs were not affected (Fig. 4A, B, right panels). We detected strong colocalization between cEt- and F-modified gapmer PS-ASOs with P-bodies (Supplementary Fig. S2), but the activities of these PS-ASOs were not altered when cells were depleted of DDX6 or LSm14A (Supplementary Fig. S7A, B).
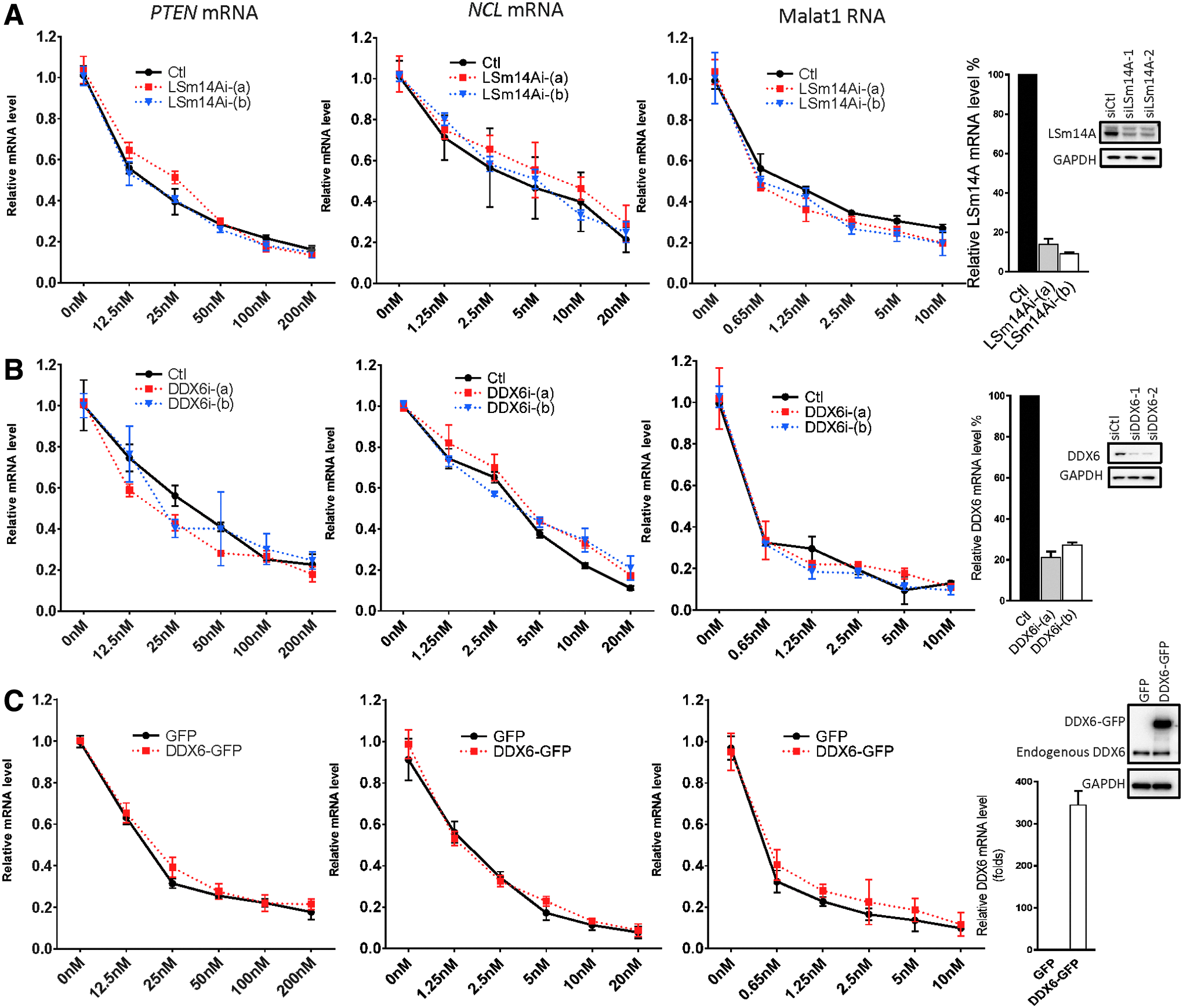
Depletion or overexpression of P-body assembly factors had no significant effect on the activities of ASOs.
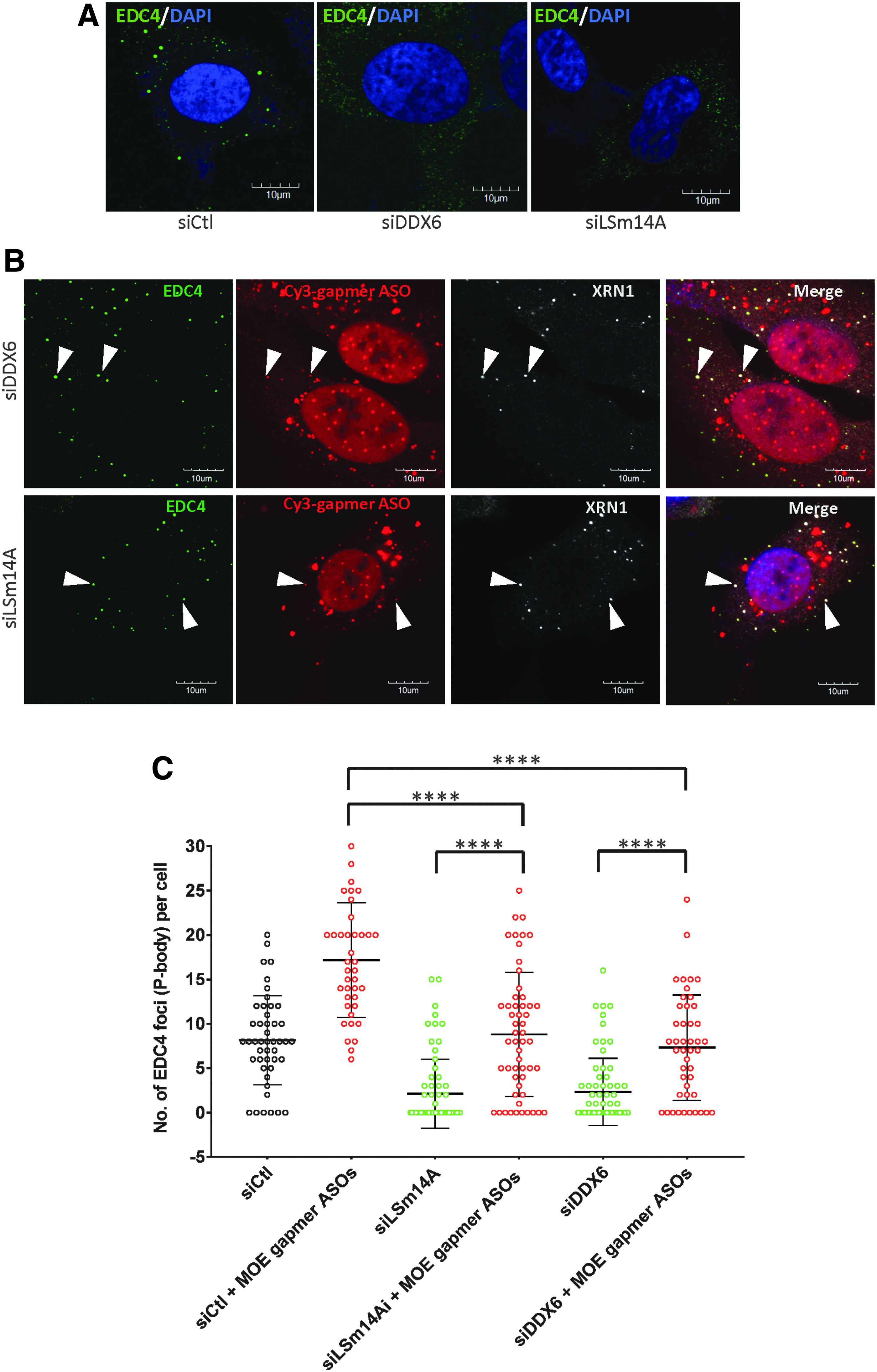
Transfection of PS-ASOs restored the formation of P-bodies in the absence of P-body assembly factors.
Depletion of other P-body proteins such as eIF4E-T, PAT1B, and CNOT1 has also been shown to disrupt P-body formation [21,43,44]. To further confirm that antisense activities of ASOs are independent of the presence of P-bodies, we used siRNAs to deplete cells of these factors. Consistent with DDX6 and LSm14A depletion, reductions in expression of these additional P-body proteins had no notable effects on antisense activities of MOE gapmer PS-ASOs against different targets (Supplementary Fig. S8). Reversely, antisense activities of PS-ASOs were also not affected when P-body component DDX6 was overexpressed (Fig. 4C). Taken together, these results indicate that PS-ASO-mediated RNA cleavage is independent of the formation of P-bodies.
Introduction of PS-ASOs induces P-body assembly in DDX6- and LSm14A-depleted cells
RNAi mediated by siRNAs has been reported to stimulate the formation of P-bodies [28,34]. Interestingly, reduction in expression of P-body assembly factors such as LSm14A and DDX6 does not prevent the nucleation of P-bodies stimulated by treatment of cells with siRNAs [34]. Since PS-ASOs elicited RNA cleavage in cells depleted of DDX6 and LSm14A, we asked whether PS-ASOs induced formation of P-bodies in these cells. The number of P-bodies, identified as large EDC4-positive foci, was decreased in cells treated with siRNAs targeting DDX6 or LSm14A as expected (Fig. 5A). Transfection with PS-ASOs in these cells restored the number of P-bodies to the levels observed in control cells (Fig. 5B), as confirmed by quantitative analysis of more than 40 cells in each group (Fig. 5C). It should be noted that the extent of P-body induction by PS-ASOs was less in cells depleted of DDX6 and LSm14A than in cells treated with a control siRNA (Fig. 5C), implying that the presence of intact P-body components is important, although not completely required, for PS-ASO-mediated assembly of P-bodies. To validate that EDC4-positive foci in cells depleted of DDX6 were indeed P-bodies, we stained these cells with additional P-body markers, including eIF4E-T, PAT1B, and DCP1B. These foci were stained for each of these P-body markers (Supplementary Fig. S9), confirming that PS-ASOs were able to mediate P-body assembly even in the absence of a major P-body assembly factor. We tested two additional MOE gapmer PS-ASOs of different sequences in cells depleted of LSm14A, and the data indicate that PS-ASO mediated P-body restoration is independent of sequence (Supplementary Fig. S10).
Disruption of P-bodies can also be achieved by depletion of other P-body components [21,43,44]. Treatment of HeLa cells with siRNAs against CNOT1, PAT1B, or eIF4E-T led to the abolishment of P-bodies (Supplementary Fig. S11A, C). Transfection of these P-body component-depleted cells with PS-ASOs restored the assembly of P-bodies (Supplementary Fig. S11B, C). In summary, our results demonstrate that transfection of PS-ASOs induced P-body assembly in cells depleted of P-body components that are necessary for P-body formation in the absence of PS-ASOs.
Antisense activities are not required for PS-ASO–induced P-body assembly
It was previously shown that gene silencing induced by siRNA was necessary for siRNA-mediated P-body induction and reassembly in the absence of DDX6 [34,45]. As evidences, RNAi machinery proteins AGO2 and GW182 are required for siRNA-induced increase of P-body numbers [34]. To distinguish the difference between PS-ASOs and siRNA-mediated P-body induction, we tested whether PS-ASOs promoted P-body assembly in cells depleted of AGO2 and GW182. The number of P-body was significantly increased by PS-ASOs even in the absence of AGO2 (Supplementary Fig. S12) or GW182 (Supplementary Fig. S13), suggesting that RNAi machinery is not required for PS-ASO–mediated P-body induction. It should also be noted that the colocalization between PS-ASOs and P-body was not inhibited in AGO2 depleted cells (Supplementary Fig. S12D), while P-body localization of siRNA relied on the presence of AGO2 [45].
Since the mechanisms of gene silencing induced by siRNAs and gapmer PS-ASOs are different, we asked whether PS-ASO-induced RNA degradation is required for PS-ASO-mediated P-body induction. We generated mismatch mutation in the core region of a MOE gapmer PS-ASO against Malat1 RNA (IONIS 713841) to disrupt target specific gene silencing activities and compared the level of P-body induction between parental and mutated PS-ASOs. As shown in Supplementary Fig. S14A and B, P-body was similarly induced by both parental and mutated PS-ASOs. After transfection, mutated PS-ASOs were also preferentially localized in P-bodies (Supplementary Fig. S14A), although the mutated PS-ASO showed dramatically reduced activity compared with the parental PS-ASO (Supplementary Fig. S14C). Furthermore, PS-ASOs induced P-body formation even when PS-ASO-mediated target RNA degradation was compromised by RNase H1 knockdown (Supplementary Fig. S15).
To exclude the potential interference from the off-target antisense activity by PS-ASOs, we transfected cells with PS-ASOs modified at all positions with 2′OMe or MOE. These PS-ASOs cannot mediate RNase H1 cleavage of the target mRNA [1,6]. As expected, these PTEN-targeted OMe or MOE uniformly modified PS-ASOs did not induce RNA degradation, whereas the MOE gapmer PS-ASO did (Fig. 6A). Like the MOE gapmer PS-ASO, uniform OMe and MOE PS-ASOs preferentially localized in EDC4–positive P-bodies (Fig. 6B). The number of P-bodies was also increased in cells transfected with these uniformly modified PS-ASOs (Fig. 6B, C), and uniformly modified PS-ASOs restored P-body formation in cells depleted of DDX6 (Fig. 6B) or LSm14A (Supplementary Fig. S16). The visual observation was confirmed by quantification of the numbers of P-bodies (Fig. 6C). Taken together, these data indicate that PS-ASOs stimulate the assembly of P-bodies through a mechanism different from siRNA-mediated target-dependent P-body induction and that antisense activities of PS-ASOs may not be required for their interplay with P-body.
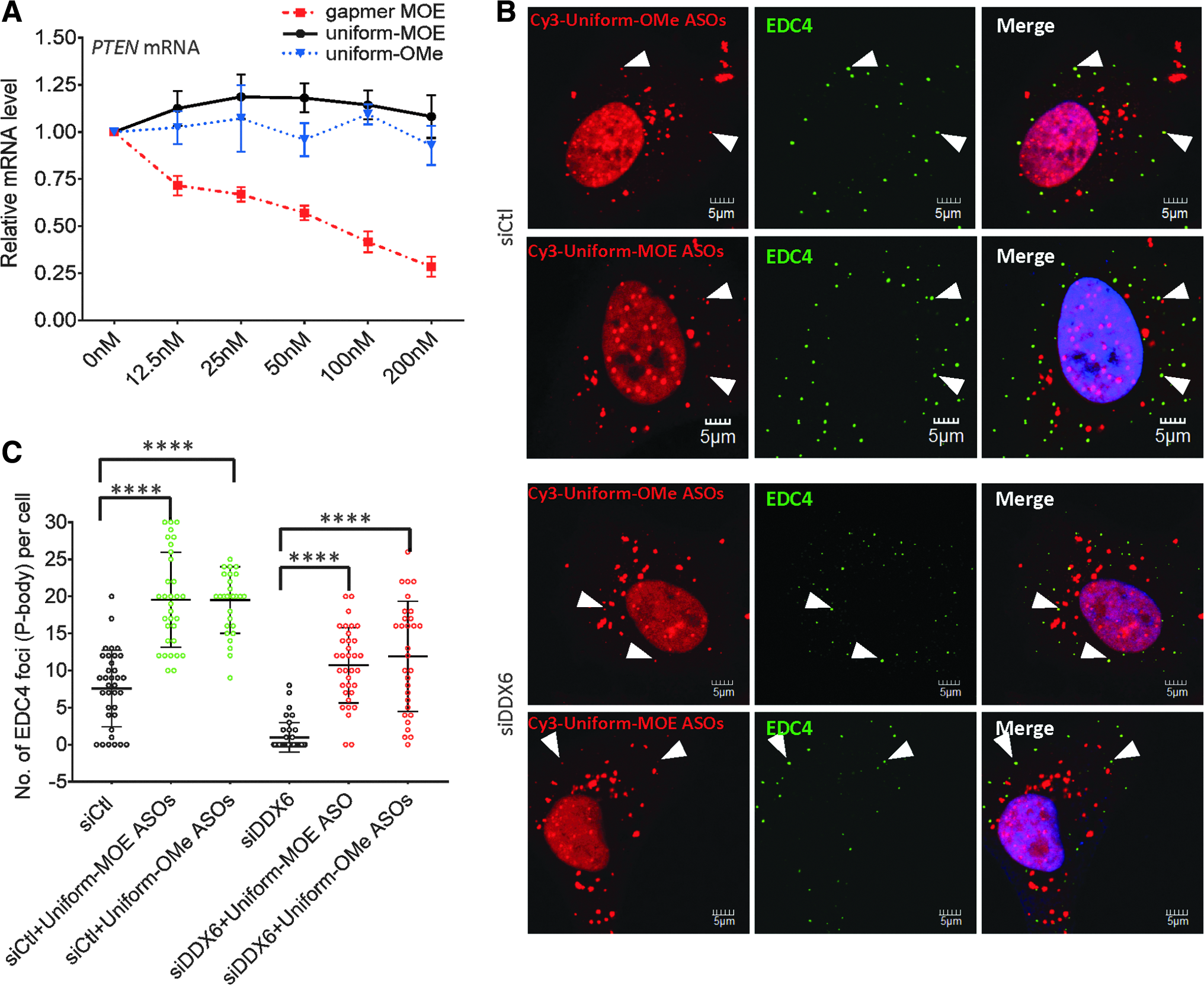
Antisense activity is not required for PS-ASO-mediated P-body formation.
PS-ASOs associate with the C-terminal domain of DDX6 to scaffold P-body assembly
We next sought to determine the properties of PS-ASOs responsible for their colocalization with P-bodies and their induction of P-body formation. As PS-ASOs were directly associated with multiple P-body components (Fig. 1B, C), this raised the possibility that these proteins govern the interplay between PS-ASOs and P-bodies. Protein binding properties of ASOs depend on the number of PS modifications in the backbone [3,11,46]. We therefore evaluated a set of 2′ unmodified, 20-mer PS-DNA-ASOs with decreasing numbers of PS moieties (PS19 to PS5). These ASOs have the same sequence as the Malat1-targeted MOE PS-ASO. A sequence-matched PO-ASO was used as a negative control. Affinity selection was performed using a biotinylated PS-MOE ASO as described above, and coisolated proteins were eluted with the DNA ASOs containing different numbers of PS backbones. As expected, reduction in the number of PS linkages led to an overall decrease in protein binding (Fig. 7A, top panel). More specifically, the association between ASOs and P-body proteins was also significantly diminished when ASOs had 10 or fewer PS linkages (Fig. 7A, lower panel).

P-body induction by PS-ASOs depends on their affinity for proteins.
To test the effect of reduced affinity for proteins on ASO P-body localization, ASOs with various numbers of PS linkages labeled with Cy3 were transfected into HeLa cells. EDC4 staining of these cells revealed that the colocalization of ASOs and P-bodies, as well as the presence of nuclear PS-bodies, was proportionally reduced with decreasing PS numbers (Fig. 7B, C). As previously described, the localization pattern of the PO-ASO was distinct from those of ASOs with PS linkages, and PO-ASOs did not colocalize with EDC4-labeled foci (Fig. 1). ASOs with fewer than 10 PS linkages did not induce formation of P-bodies in untreated cells (Fig. 7D) and in cells depleted of LSm14A (Supplementary Fig. S17). Taken together, these data suggest that interactions of PS-ASOs with P-body component proteins can contribute to the formation of P-bodies.
Since DDX6 is a core P-body assembly factor with high affinity for PS-ASOs (Fig. 1A, B), we generated a set of tGFP-conjugated DDX6 mutants with deletions of different functional domains and investigated cellular localization and interaction with MOE gapmer PS-ASOs (Fig. 8A). The expression of these mutants in HeLa cells was confirmed by western analyses (Fig. 8B). The localization of these DDX6 mutants was determined by confocal imaging. P-body component XRN1 was used to confirm P-body localization. Motif Q (amino acids 96–108) and Motif I (amino acids 140–147) deletions did not affect the P-body localization of DDX6 (Fig. 8C, left panel). As expected, transfected PS-ASOs colocalized with Motif Q- or Motif I-deleted DDX6 mutants (Fig. 8C, right panel). When the core DEAD domain (amino acids 246–249) was deleted, the P-body localization of DDX6 was impaired (Fig. 8C, D). Interestingly, PS-ASOs partially restored the P-body localization of DDX6 mutant missing the DEAD domain (Fig. 8C, D), and PS-ASOs were detected in these P-bodies. This suggested that the PS-ASO can seed the formation of P-bodies, likely by recruiting other factors essential for P-body reassembly. Consistent with previous reports [19,47], deletion of C-terminal domain (CTD) completely abolished P-body localization of DDX6 (Fig. 8C, D). The even cytoplasmic distribution pattern of DDX6 mutant lacking the CTD domain was not reversed by the introduction of PS-ASOs (Fig. 8C, D). An analysis of proteins precipitated with a biotinylated PS-ASO confirmed that the deletion of the CTD domain completely abolished the binding of DDX6 to PS-ASOs, whereas deletions of Motif Q, Motif I, or DEAD domains did not affect the association of DDX6 with PS-ASOs (Fig. 8B). In summary, PS-ASOs may interact with the CTD of DDX6 to recruit DDX6 to the PS-ASO–seeded P-bodies.
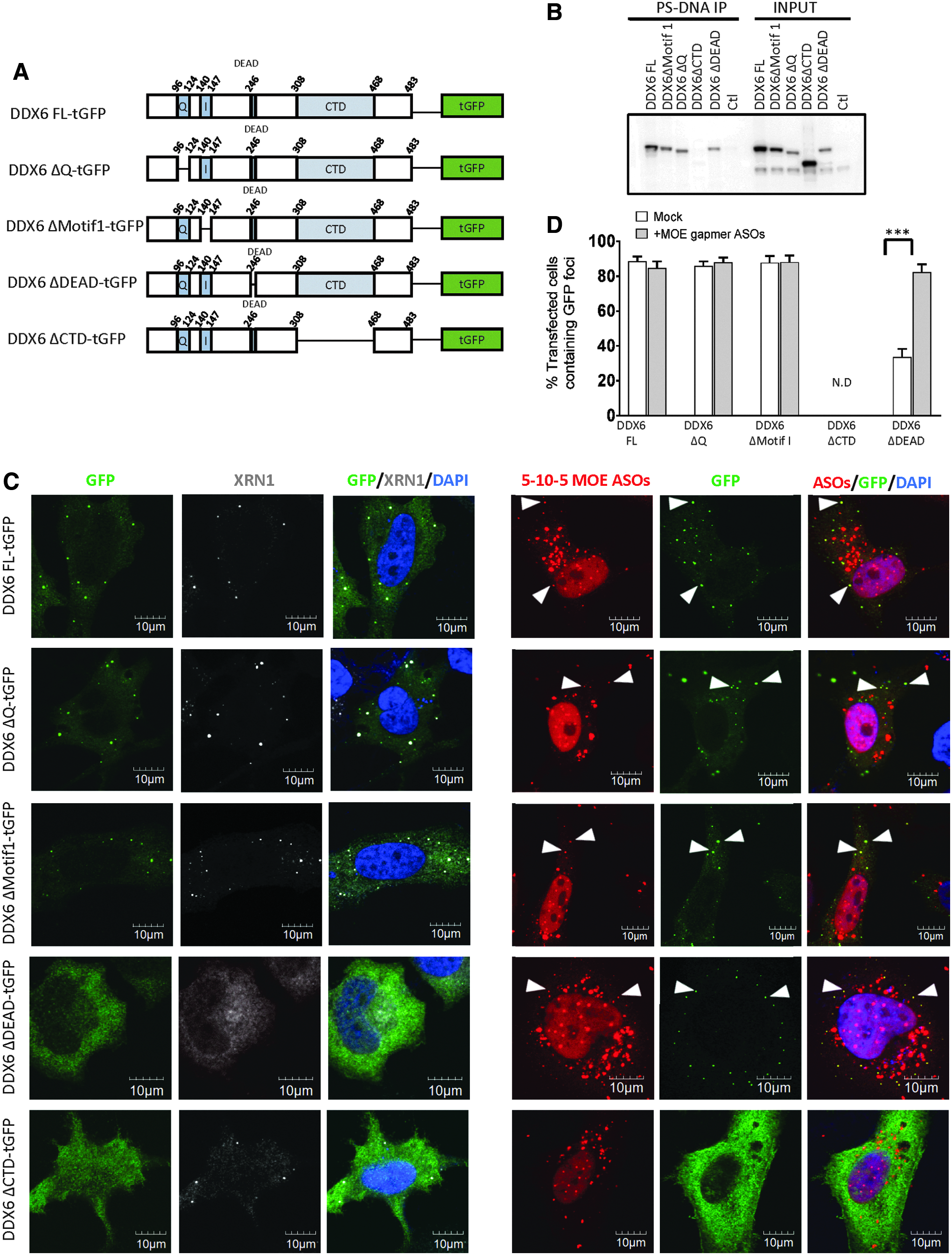
PS-ASOs associate with the C-terminal domain of DDX6 to scaffold P-body assembly.
Discussion
In this report, we examined the interplay between PS-ASOs and P-bodies. Cytoplasmic P-bodies are formed by the aggregation of translationally repressed mRNAs and their associated proteins [48]. Proteins involved in RNA degradation, translational repression, and RNA-mediated gene silencing accumulate in P-bodies [27,28]. Moreover, siRNAs and mRNA decay intermediates are detected in P-bodies [33,34,45]. In this study, we accordingly found that cytoplasmic PS-ASOs localized in P-bodies after transfection into cells.
Since proteins involved in siRNA- and miRNA-mediated mRNA degradation are detected in P-bodies, it has been proposed that P-bodies are essential for gene silencing through the RNAi pathway. P-body numbers have virtually no effect on siRNA-, miRNA-, or NMD-mediated RNA degradation [28,29]. However, treatment of cells with siRNA or miRNA does increase the number of P-bodies [34,40]. Similarly, we observed that PS-ASO–mediated mRNA degradation was not affected in cells depleted of P-body assembly factors and that the transfection of ASOs significantly induced the number of P-bodies.
Although siRNAs and PS-ASOs can both promote the formation of P-bodies, several lines of evidence suggest that the underlying mechanisms are different. First, siRNA-mediated P-body assembly is target dependent: siRNAs with no complementarity to expressed transcripts do not cause P-body formation [34,45]. In contrast, PS-ASOs stimulated P-body formation even when they had no complementarity to the transcriptome. Second, siRNA-mediated P-body induction requires functional RNAi machinery, as inhibition of expression of either GW182 or AGO2 completely abolished induction of P-body formation by siRNAs [34]. In contrast, inhibition of RNase H1 expression had no effect on the number of P-bodies formed in the presence of PS-ASOs (Supplementary Fig. S15). Moreover, PS-ASOs uniformly modified at the 2′ position of the sugar that do not support RNase H1 cleavage of RNAs nucleated P-body formation to a level comparable to that of gapmer PS-ASOs. Third, siRNA gradually stimulates the assembly of P-bodies, as the numbers and the sizes of P-bodies increase for 3 days after transfection of cells with siRNA [34]. In contrast, the number of P-bodies was maximal at around 4 h after transfection of cells with PS-ASOs. These discrepancies clearly suggest that P-body formation induced by PS-ASOs and siRNAs occurs through different mechanisms.
One of the key differences between PS-ASOs and siRNAs lies in their affinity for proteins. The PS moiety used in the design of most ASOs significantly increases protein association and plays an essential role for the uptake, distribution, and efficacy [4,7]. The strong association between PS-ASOs with a variety of P-body proteins and the findings that uniformly 2′-modified PS-ASOs induce P-body formation sequence independently led us to postulate that protein interactions, rather than antisense activities, result in PS-ASO–mediated P-body assembly. Our data provided strong evidence to support this hypothesis. First, protein binding affinity and induction of P-body formation were correlated with the number of PS linkages in the ASO. Second, the depletion of P-body assembly factors DDX6 and LSm14A reduced PS-ASO–mediated P-body induction, although P-bodies were observed at near-control levels in cells depleted of DDX6 or LSm14A upon treatment of PS-ASOs. Third, PS-ASOs only restored the P-body localization of DDX6 mutants that contained the PS-ASO interaction domain. Based on these observations, we deduce that the interactions between PS-ASOs and various P-body components are important for PS-ASO–mediated P-body assembly.
The rapid increase in sizes and numbers of P-bodies after PS-ASO transfection suggests that PS-ASOs stimulate a late stage of P-body assembly rather than the initiation stage. It is possible that PS-ASOs bind to preexisting RNP granules that contain translationally repressed mRNAs or to early stage, submicroscopic P-bodies and recruit additional components to form large, microscopically visible P-bodies. In contrast, siRNAs may cause the de novo formation of P-bodies, as there is a minimal increase in numbers of P-bodies at 24 h after siRNA treatment, a time when the gene silencing activity of these agents is observed [34].
Although our data demonstrate that protein interactions with PS-ASOs are essential for PS-ASO mediated P-body induction, we cannot exclude the potential regulatory effects of RNA in this process, as P-bodies are basically aggregates of multiple translationally repressed mRNAs and associated proteins [28]. Previous studies have shown that P-bodies disperse when mRNAs are degraded by RNase A or when protein synthesis is inhibited by cycloheximide treatment [38]. We observed a similar disruption of P-bodies in PS-ASO treated cells shortly after cycloheximide treatment (Supplementary Fig. S6), suggesting that PS-ASOs and proteins are not sufficient for formation or maintenance of P-bodies.
It should also be noted that we observed colocalization of PS-ASOs with P-bodies when PS-ASOs were delivered by transfection but not when HeLa or A431 cells (data not shown) were treated with PS-ASOs without transfection reagents [49]. However, in the absence of transfection reagent, PS-ASOs with LNA modification reportedly localized to GW182-positive foci in Huh7 cells [41,50]. This discrepancy could be explained by subtle differences between the composition of P-bodies and GW182-positive bodies in different cell lines. In Huh7 cells, the LNA-modified gapmer PS-ASOs colocalized with GW182-positive foci rather than DCP1B positive P-bodies [41]. In addition, the mechanism by which Huh7 cells internalize PS-ASOs by free uptake or the mechanism of release from endosomes may differ in Huh7 and HeLa cells. Arguably, an alternative explanation may be that the accumulation of PS-ASOs in P-body may be dependent on the peak cytosolic PS-ASO concentrations as transfection results in a much more rapid accumulation of PS-ASOs in the cytoplasm than free uptake. Additional studies will be required to better understand the cytosolic trafficking of PS-ASOs when delivered by the cellular free-uptake approach.
In conclusion, we determined that PS-ASOs colocalized with P-bodies, stimulated P-body assembly, and restored P-body formation in cells depleted of core P-body proteins DDX6 and LSm14A. The interactions of PS-ASOs with proteins, rather than antisense activities, underlie this colocalization. Our findings increase our mechanic understanding of P-body dynamics and present a unique case in which the association between PS-ASO and protein not only mediates intracellular trafficking of PS-ASOs but also substantially remodels cellular organelles. Finally, it is important to note that most siRNAs used for therapeutic purpose contain a meaningful number of PS moieties. Thus, these siRNAs might accumulate in P-bodies through the similar process as reported here.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.