
Abstract
Cognitive impairment is common following traumatic brain injury (TBI), and neuroinflammatory mechanisms may predispose to the development of neurodegenerative disease. Apolipoprotein E (apoE) polymorphisms modify neuroinflammatory responses, and influence both outcome from acute brain injury and the risk of developing neurodegenerative disease. We demonstrate that TBI accelerates neurodegenerative pathology in double-transgenic animals expressing the common human apoE alleles and mutated amyloid precursor protein, and that pathology is exacerbated in the presence of the apoE4 allele. The administration of an apoE-mimetic peptide markedly reduced the development of neurodegenerative pathology in mice homozygous for apoE3 as well as apoE3/E4 heterozygotes. These results demonstrate that TBI accelerates the cardinal neuropathological features of neurodegenerative disease, and establishes the potential for apoE mimetic therapies in reducing pathology associated with neurodegeneration.
Introduction
In addition to these acute neurocognitive deficits, TBI has been suggested to predispose to the later development of neurodegenerative disorders such as Alzheimer's disease (AD). In fact, there are many pathological features common to both acute brain injury and AD, including Aβ deposition, tau phosphorylation, neurite degeneration, synapse loss, and microgliosis (Ikonomovic et al., 2004; Uryu et al., 2007). Neuroinflammatory responses may serve as a common denominator between these two entities, and play a central role in mediating secondary neuronal injury in both chronic neurodegenerative diseases and acute brain injury. In the acute setting, glial activation results in an upregulation of amyloid precursor protein (APP), as well as other inflammatory mediators. This increase in APP expression and the neuroinflammatory response following injury may contribute to a cycle of Aβ deposition and microglial activation that ultimately results in chronic neuropathology.
Given the important role of glial activation in acute brain injury and in the pathogenesis of neurodegenerative disease, it is of interest that genetic modifiers of neuroinflammation, such as APOE (APOE: gene; apoE: protein) polymorphisms, play an important role in both (Corder et al., 1993; Zhou et al., 2008). There are three common polymorphisms of APOE, designated APOE2, APOE3, and APOE4, which differ by cysteine to arginine substitutions at positions 112 and 158. APOE4 is associated with increased susceptibility of developing late-onset familial and sporadic AD (Corder et al., 1993), and is also associated with increased deposition of Aβ in the setting of acute brain injury (Nicoll et al., 1995) and neurodegenerative disease (Schmechel et al., 1993). Although the mechanisms by which the APOE4 allele increases susceptibility to AD remain poorly defined, there is accumulating evidence demonstrating that apoE modifies neuroinflammatory responses in an isoform-specific fashion (Barger and Harmon, 1997; Laskowitz et al., 1997, 2001), an effect that is likely receptor-mediated (Herz and Beffert, 2000; Hoe et al., 2005; Misra et al., 2001; Ophir et al., 2005). Although a great deal of research has focused on the mechanisms by which apoE may selectively modulate amyloid metabolism and fibrillogenesis in an isoform-specific manner (Gallo et al., 1994), it is also plausible that the association between apoE4 and increased amyloid deposition is related to its effect on glial activation and neuroinflammatory responses. This is consistent with the evolving understanding of the role of glial activation and oxidative stress in AD, and would accommodate the multiple clinical and preclinical observations associating apoE4 with poor outcome after acute brain injury (Zhou et al., 2008). A more complete understanding of the role that apoE plays in modifying glial activation might also have therapeutic implications. For example, a series of apoE-mimetic peptides have recently been developed that suppress glial activation in vitro (Laskowitz et al., 2002, 2006), and improve functional recovery in murine models of TBI (Hoane et al., 2007; Kaufman et al., 2010; Lynch et al., 2005; Laskowitz et al., 2007). These findings raise the potential of developing novel therapeutic approaches that harness the anti-inflammatory properties of endogenous apoE (Laskowitz et al., 2007).
To assess the role of APOE polymorphisms in the neurodegenerative pathology seen following TBI, we utilized double-transgenic animals expressing both the human APOE3 or APOE4 polymorphism in the correct genetic locus (APOE-targeted replacement), and the mutant human APP (V717P), in a minigene driven by a platelet-derived growth factor (PDAPP; Games et al., 1995). The use of double-transgenic animals allows examination of the interaction between mutated APP overexpression and the common humanized apoE isoforms seen following brain injury. We establish that systemic administration of a rationally derived apoE-mimetic peptide reduces many of the features of post-traumatic brain injury, including Aβ deposition, microgliosis, tau phosphorylation, and neuronal degeneration. We also demonstrate a pharmacogenomic interaction between the humanized APOE genetic background and the therapeutic effect of the apoE peptide.
Methods
PDAPP/APOETR mice
All animal care and experimental procedures were approved by the Duke University Animal Care and Use Committee. PDAPP mice expressing a mutated form of human amyloid precursor protein (APPV7l7F), under the control of the platelet-derived growth factor promoter (PDAPP+/+) were bred with APOE-targeted replacement (APOETR) mice, which were created by replacing only the coding sequences of mouse apoE with human APOE allele-specific coding sequences, knocking out the endogenous mouse APOE gene without disturbing any known regulatory sequences. This results in animals that express human apoE at physiological levels similar to those of primates and humans in both temporal and spatial patterns (Sullivan et al., 1997, 2004). The experimental mice used in this project were PDAPP+/+/APOE3TR+/+(PDAPP/APOE33TR), and PDAPP+/+/APOE4TR+/+(PDAPP/APOE44TR). PDAPP+/+/APOE3TR+/APOE4TR+(PDAPP/APOE3TR/APOE4TR) mice were generated by mating PDAPP/APOE33TR mice to PDAPP/APOE44TR mice. Littermates were used for controls.
Traumatic brain injury
Our murine TBI model has been previously described as a model of diffuse closed head injury (Lynch et al., 2005; Wang et al., 2006). Twelve- to 14-week-old male PDAPP/APOE33TR and PDAPP/APOE44TR mice were pre-randomized to treatment or vehicle groups prior to injury. The mice were anesthetized with 4.3% isoflurane in oxygen at an F
Drug administration
ApoE mimetic peptide COG1410 (acetyl-AS-Aib-LRKL-Aib-KR-amide) was derived from apoE residues 138–149 with aminoisobutyric acid (Aib) substitutions at positions 140 and 145. These modifications of the receptor binding region of apoE have been previously demonstrated to enhance their anti-inflammatory properties (Laskowitz et al., 2006). Twelve-week-old PDAPP/APOE33TR and PDAPP/APOE44TR mice received a single intravenous injection of 0.6 mg/kg COG1410 or vehicle 30 min following TBI, with subsequent daily injections for the first week after TBI, followed by weekly injections for the ensuing 8 weeks. The peptides were synthesized by NeoMPS (San Diego, CA) to a purity of 95%.
Testing of motor deficits
The vestibulomotor function was assessed using an automated rotarod (Ugo Basile, Comerio, Italy). On the day prior to TBI, mice underwent two consecutive conditioning trials at a set rotational speed (16 rpm) for 60 sec, and then two additional trials with an accelerating rotational speed. The average time to fall from the rotating cylinder in the latter two trials was recorded as baseline latency. From day 1 to day 5 post-TBI, the mice were given two consecutive daily trials with accelerating rotational speed (intertrial interval = 15 min). The average latency to fall from the rod was recorded. The latency value for mice unable to grasp the rotating rod was 0 sec (n = 11 animals in the treated group; n = 10 animals in the vehicle group).
Tissue processing and histological methods
After anesthesia and intracardial perfusion with phosphate-buffered saline (PBS), the brains were rapidly removed and dissected in the mid-sagittal plane. One hemisphere was flash-frozen in liquid nitrogen and stored at −80°C, and the other hemisphere was immersion fixed in 10% formaldehyde for 24 h, transferred to 1 × PBS, and stored at 4°C. Frozen coronal sections (40 μm) were collected on a sliding microtome. For immunostaining, tissue was incubated in 1% hydrogen peroxide, permeabilized by 0.1% saponin, and blocked with 10% goat serum. When possible to minimize background resulting from the use of monoclonal antibodies and for antigen retrieval purposes, the brain sections were first microwaved in SSC and antigen retrieval buffer (Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions. The sections used for immunostaining of Aβ were first pretreated with 90% formic acid for 30 min. Then primary antibodies were diluted to the final working concentration in 1% goat serum, and then incubated overnight at 4°C. The Vectastain Elite ABC kit/DAB (diaminobenzidine) staining kit (Vector Laboratories) was used according to the manufacturer's instructions. Brain sections were mounted on slides, dehydrated, cleared, and cover-slipped using DPX mounting media (Fluka, Milwaukee, WI). Thioflavine-S and Fluoro-Jade B staining were done using standard protocols (Wang et al., 2007). For immunohistochemistry staining, the following antibodies were used: anti-mouse F4/80 antibody (1:10,000 rat monoclonal; Serotec, Raleigh, NC), anti-neuronal-specific nuclear protein (NeuN) antibody (1:30,000 mouse monoclonal; Chemicon, Temecula, CA), anti-phosphorylated-tau antibodies (AT8, 1:300 mouse monoclonal; Pierce Endogen, Rockford, IL; AT-180, 1:3000 mouse monoclonal; Pierce; and AD-2, 1:2000 mouse monoclonal; [phosphorylated residues Ser396 and Ser404, a gift from Dr. Delacourte, INSERM Research Center, Lille, France] (Buee et al., 2000), and anti-mouse synaptotagmin antibody (1:1200 mouse monoclonal; Stressgen, Ann Arbor, MI). For Aβ immunohistochemistry, a biotinlyated mouse anti-human Aβ monoclonal antibody (4G8, 1:1000; Signet, Dedham, MA) was used and detected with the biotin-avidin-peroxidase method. Antibodies binding APP were also used, including anti-C-terminal APP antibody (1:1000 rabbit polyclonal; Invitrogen, Eugene, OR), and anti-N-terminal APP antibody (1:5000 goat polyclonal; Abcam, Cambridge, MA), which binds to N-terminal amino acids 44–63 of human APP. The bound antibody against synaptotagmin was detected by anti-mouse fluorescence secondary antibody (1:1500; Invitrogen). The rest of the antibodies were detected with the biotin-avidin-peroxidase method as described previously (Wang et al., 2007). In all immunohistochemical studies except those using biotinylated primaries, control studies were performed to evaluate non-specific staining by omitting the primary antibody. Double-immunofluorescence staining was performed using 4G8 and GRP78 BiP antibody (1:250, an ER marker, rabbit polyclonal; Abcam). The appropriate Alexa Fluor-conjugated secondary antibodies (Invitrogen) were used. The hippocampal pyramidal cells were imaged using a Leica (Nussloch, Germany) TCS SL confocal system set up on a Leica DMIRE2 inverted microscope equipped with epifluorescence illumination and a 60 × oil-immersion objective (numerical aperture 1.4). Images were captured using 10 × optical zoom.
Western blotting
Protein was extracted from frozen hemispheres in 1 × TBS with protease inhibitor. As previously described (Dawson et al., 2007), equal amounts of protein samples (50 μg) were electrophoresed on a denaturing 4–20% SDS-PAGE gel and transferred onto nitrocellulose membranes. The membranes were then blocked in 5% milk and incubated in AT8 anti-phosphorylated tau antibody (1:150) overnight at 4°C. After incubation in HRP-conjugated anti-mouse secondary antibody (1:2000) for 1 h at room temperature, the blot was washed in TBST and developed using the ECL plus Western blot detection kit (GE Healthcare, Amersham, U.K.). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 1:1,000,000 mouse anti-GAPDH; Advanced ImmunoChemical, Long Beach, CA) was used to ensure accurate gel loading.
Aβ ELISA
Total amounts (both soluble and insoluble pools) of Aβ1–40 and Aβ1–42 were measured by sandwich ELISA as described previously (Sadowski et al., 2006). Briefly, approximately 40 mg of brain tissue was added to 350 μL of 70% cold formic acid, sonicated on ice for 1 min and then spun at 100,000g for 1 h at 4°C with a Sorvall RC M120EX micro-ultracentrifuge. Supernatants were neutralized with a 20-fold excess of 1 M Tris Base, containing 0.5 M Na2HPO4 and 1 × protease inhibitor cocktail. Levels of total Aβ1–40 and Aβ1–42 were detected using sandwich ELISA kits (Covance, Princeton, NJ). The final values of brain Aβ were expressed as picograms per milligram protein (n = 4 per genotype per time point).
Tumor necrosis factor-α ELISA
Pulverized brain hemispheres were sonicated on ice in homogenization buffer (containing 20 mM Tris-HCL [pH 8.0], 137 mM NaCl, 2 mM EDTA, 10% glycerol, 1% Triton X-100, and Complete™ protease inhibitor mixture tablets; Roche Applied Science, Basel, Switzerland) for 20 sec. Brain homogenates were incubated on ice for 20 min and then spun at 10,000 rpm at 4°C for 10 min. The supernatant was removed and stored at −70°C until ELISA analysis. The TNF-α level in brain homogenate was determined using an ELISA kit for mouse TNF-α per the manufacturer's specifications (Invitrogen). Results were expressed as the amount of TNF-α per nanogram of protein (n = 4 per genotype).
Cytokine mRNA quantification
The level of TNF-α and IL-6 gene expression after TBI was measured by running quantitative real-time PCR. RNA was extracted from pulverized frozen hemispheres using the PerfectPure RNA Tissue Kit (5 PRIME, Gaithersburg, MD) according to the manufacturer's instructions. RNA was reverse-transcribed to cDNA using the High-Capacity cDNA Archive Kit (Applied Biosystems, Carlsbad, CA). The levels of mRNA expression were determined by quantitative real-time polymerase chain reaction performed on a ABI 7300 Sequence Detection Software system (Applied Biosystems), using 100 ng cDNA per reaction, adding TaqMan Universal PCR master mix and TaqMan Assays-on-Demand Gene Expression primer/probe sets (all from Applied Biosystems) for the murine TNF-α. Relative mRNA quantification was calculated using the 2(-delta delta c[t]) method (Livak and Schmittgen, 2001). The threshold cycle number of the target gene for each sample was normalized using a housekeeping gene (endogenous 18S) and calibrated to the control samples (procedurally naïve uninjured animals). Final results were expressed as fold changes over uninjured animals (median n = 4 animals per genotype per time point).
Quantification
For all histological quantification, a minimum of three animals were used per group. Stereological analysis was performed on microglia that were positive for F4/80 immunostaining (the whole hippocampus, median n = 5), neurons that were positive for NeuN staining (hippocampal pyramidal layer of CA1–CA3, median n = 3), and intracellular accumulations that were 4G8-immunopositive (hippocampal pyramidal layer of CA1–CA3, median n = 5). Stereological analysis was performed using the optical fractionator method on a Nikon 218912 light microscope interfaced with the StereoInvestigator software package (MicroBrightField, Williston, VT) as described previously (Dawson et al., 2007, 2010). The optical fractionator is an unbiased counting method, which is independent of the size, shape, and orientation of the cells to be counted. The parameters of the fractionator-sampling scheme were established in a pilot experiment and were uniformly applied to all animals. Before counting, all slides were coded to avoid experimenter bias. Objects were counted on the sections that contained both the pyramidal layer and the dentate gyrus of the hippocampus. As we did not quantify through the entire hippocampus, results are presented as objects per unit volume to allow comparability between animals. The quantification of synaptotagmin immunoreactivity in the stratum radiatum of the CA1 region (median n = 4) was performed using the NIH Image-Pro Plus 6.1 software (Wilcock et al., 2006). Western blot AT8 immunoreactivity (n = 3) was quantified using densitometry analysis with Kodak Molecular Imaging software (Carestream Health, Rochester, NY). Immunoreactivity of AT8 was expressed as the ratio of AT8 band to GAPDH band intensity.
Statistical analysis
All immunohistochemical, stereological, ELISA, and Western blot data were analyzed by one-way analysis of variance (ANOVA) using GraphPad Prism 4 software (GraphPad, San Diego, CA). When intergroup comparisons were statistically significant as determined by the F-statistic, pairwise testing was performed with correction for multiple comparisons using the post-hoc Tukey test. Rotarod performance was analyzed with two-way repeated-measures ANOVA with day as the repeated variable. Data are presented as mean ± SEM; significance was set at p < 0.05.
Results
Uninjured 12-week-old male PDAPP/APOE33TR and PDAPP/APOE44TR mice, which overexpress human amyloid precursor protein (APPV7l7F) and human apoE at physiological levels similar to primates and humans in both temporal and spatial patterns (Sullivan et al., 1997, 2004), had comparable whole-brain levels of total Aβ as assessed by ELISA (Fig. 1). There was no significant increase in total Aβ level in either of the double-transgenic lines at 16 weeks. However, compared to procedurally-naive uninjured animals, both 16-week-old PDAPP/APOE33TR and PDAPP/APOE44TR mice that had undergone TBI at 12 weeks of age had evidence of increased total Aβ and Aβ (1–40) levels (Fig. 1).

Total Aβ levels increase post-TBI.
Accumulation of Aβ in cells and parenchyma is a characteristic of TBI, and has been suggested to be an early event in AD pathology, preceding the development of mature amyloid plaques (Gomez-Ramos and Asuncion Moran, 2007; LaFerla et al., 2007; Wirths et al., 2008). To assess whether APOE polymorphisms modified intracellular Aβ accumulation after TBI, animals were sacrificed at 1, 2, 4, and 12 weeks after TBI and brain slices were immunostained with 4G8 (antibody against Aβ). Punctate staining was clearly visible in both PDAPP/APOE33TR and PDAPP/APOE44TR mice at all time points post-TBI (Fig. 2A), whereas only minimal staining was seen in the procedurally-naïve uninjured controls (data not shown). The staining was evident throughout the whole brain, but was most intense in the hippocampus (Fig. 2A) followed by layer 5 of the cortex. Co-localization of 4G8 and GRP78 BiP (an endoplasmic reticulum marker) immunoreactivity demonstrated that the Aβ accumulation was primary intraneuronal, with the majority located in the endoplasmic reticulum (Fig. 2B). Since the epitope of 4G8 is present in the Aβ peptide and APP, brain slices were immunostained with anti-APP antibodies specific to epitopes of the N-terminus of APP, and at the C-terminus of APP. There was no immunostaining with the N-terminal-APP antibody (data not shown), and the immunostaining pattern of C-terminal-APP was not punctate (Fig. 2C). Taken together, these results suggest that the punctate accumulation visible with 4G8 immunostaining was most consistent with Aβ accumulation, rather than with the APP holoprotein or C-terminal fragments. We used unbiased stereology to quantify the number of punctate 4G8-immunopositive material in the pyramidal layer of the hippocampus (Fig. 2D). At every time point, PDAPP/APOE44TR mice had more 4G8 staining than PDAPP/APOE33TR mice, suggesting a primary genetic effect of APOE polymorphism on intracellular Aβ accumulation. Moreover, the 4G8-immunopositive concentration continued to increase with time in the APOE44TR but not APOE33TR mice for up to 12 weeks following TBI (Fig. 2D). Thioflavin-S staining of animals sacrificed at 12 weeks post-TBI failed to reveal any extracellular amyloid plaques in either line (data not shown).
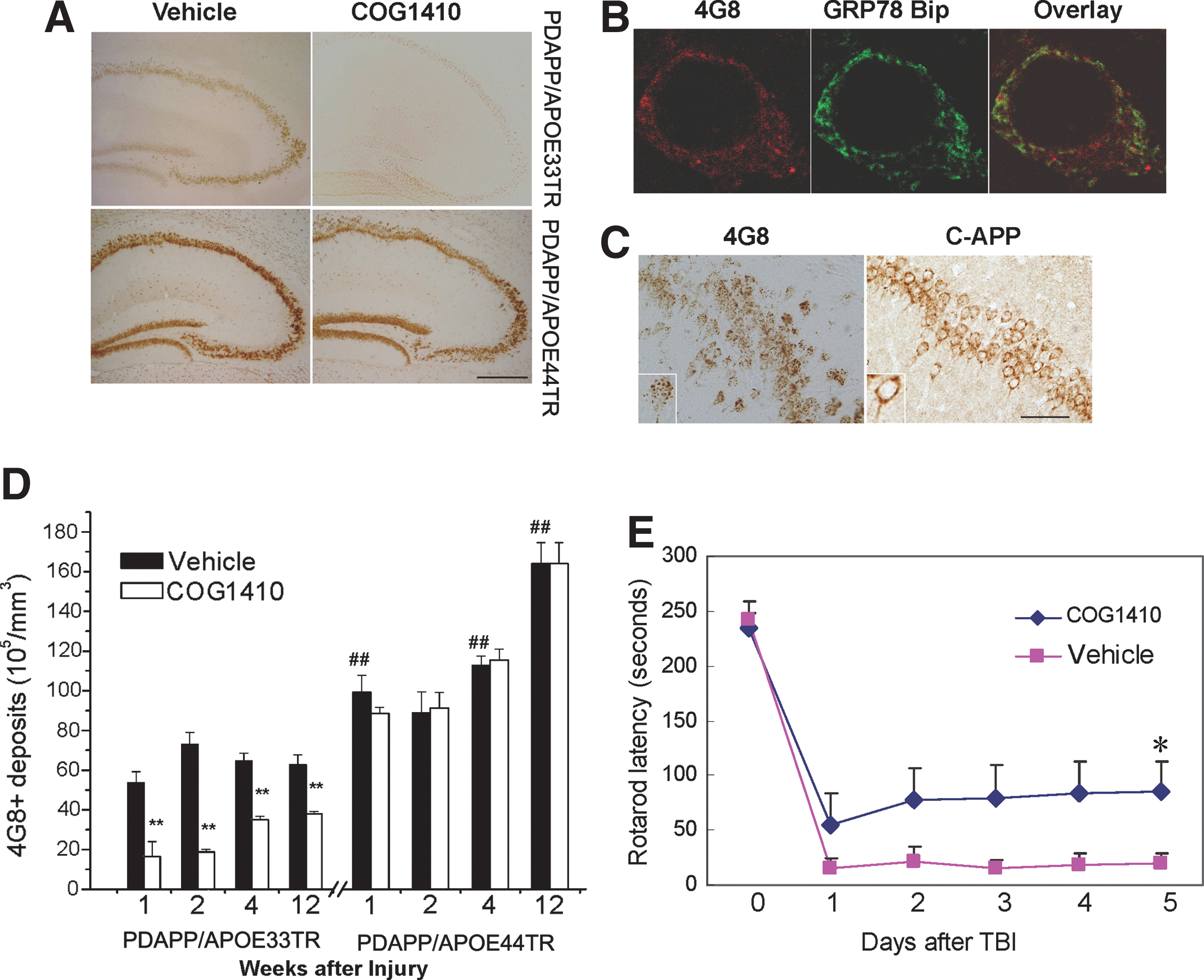
Intracellular Aβ accumulates in PDAPP/APOETR mice after TBI.
A more complete understanding of the role that endogenous apolipoprotein E plays in modifying CNS responses to acute brain injury may also have therapeutic significance, and a series of apoE-mimetic peptides have recently been developed that suppress glial activation (Laskowitz et al., 2001, 2006; Lynch et al., 2003), and improve functional recovery in murine models of TBI (Laskowitz et al., 2001, 2006; Lynch et al., 2003, 2005). More recently, an apoE-mimetic peptide (COG1410, derived from apoE residues 138–149; Wang et al., 2007) was demonstrated to reduce Aβ (1–42) accumulation and microgliosis in brain-injured mice (Wang et al., 2007). To assess whether the administration of COG1410 modified hippocampal Aβ accumulation after TBI in the double-transgenic mice, animals were treated intravenously with 0.6 mg/kg COG1410 or vehicle immediately following head injury, and then daily for 7 days. COG1410 administration was associated with improved vestibulomotor function in PDAPP/APOE33TR mice as assessed by rotarod performance, an effect that was sustained throughout the 5-day testing period (Fig. 2E). Administration of COG1410 also dramatically reduced intracellular Aβ accumulation in PDAPP/APOE33TR mice (Fig. 2A and D). This effect was durable, persisting for 12 weeks after TBI. COG1410 had no effect on functional recovery or Aβ deposition in the APOE44TR mice during this time period, suggesting a pharmacogenomic interaction between APOE polymorphism and COG1410.
Microgliosis is the cornerstone of the endogenous CNS inflammatory response, and is a feature of both acute brain injury and neurodegenerative pathology. To characterize this aspect of the CNS inflammatory response, brain slices were immunostained with F4/80, which recognizes microglia and cells of monocyte lineage (Fig. 3A). Stereological quantification demonstrated that head injury resulted in a robust increase in F4/80-positive cells compared to age-matched procedurally-naïve uninjured controls in both double-transgenic lines (Fig. 3B). This response peaked at 2 weeks after injury. Administration of COG1410 reduced microgliosis in the APOE33TR, but not APOE44TR, mice (Fig. 3A and B). To further define the effect of treatment on neuroinflammatory responses, real-time PCR was performed to quantify mRNA levels of the inflammatory cytokine TNF-α at 1 and 14 days post-injury. Treatment with COG1410 reduced levels of TNF-α mRNA at 1 day in both lines (Fig. 3C). The reduction of IL-6 was only significant at day 1 in the PDAPP/APOE44TR animals (Fig. 3D). Interestingly, this decrease in inflammatory cytokine RNA was not associated with a corresponding decrease in microgliosis in treated PDAPP/APOE44TR animals. This suggests a dissociation between early cytokine production and the later evidence of microgliosis, as has been described previously (Wang et al., 2007).
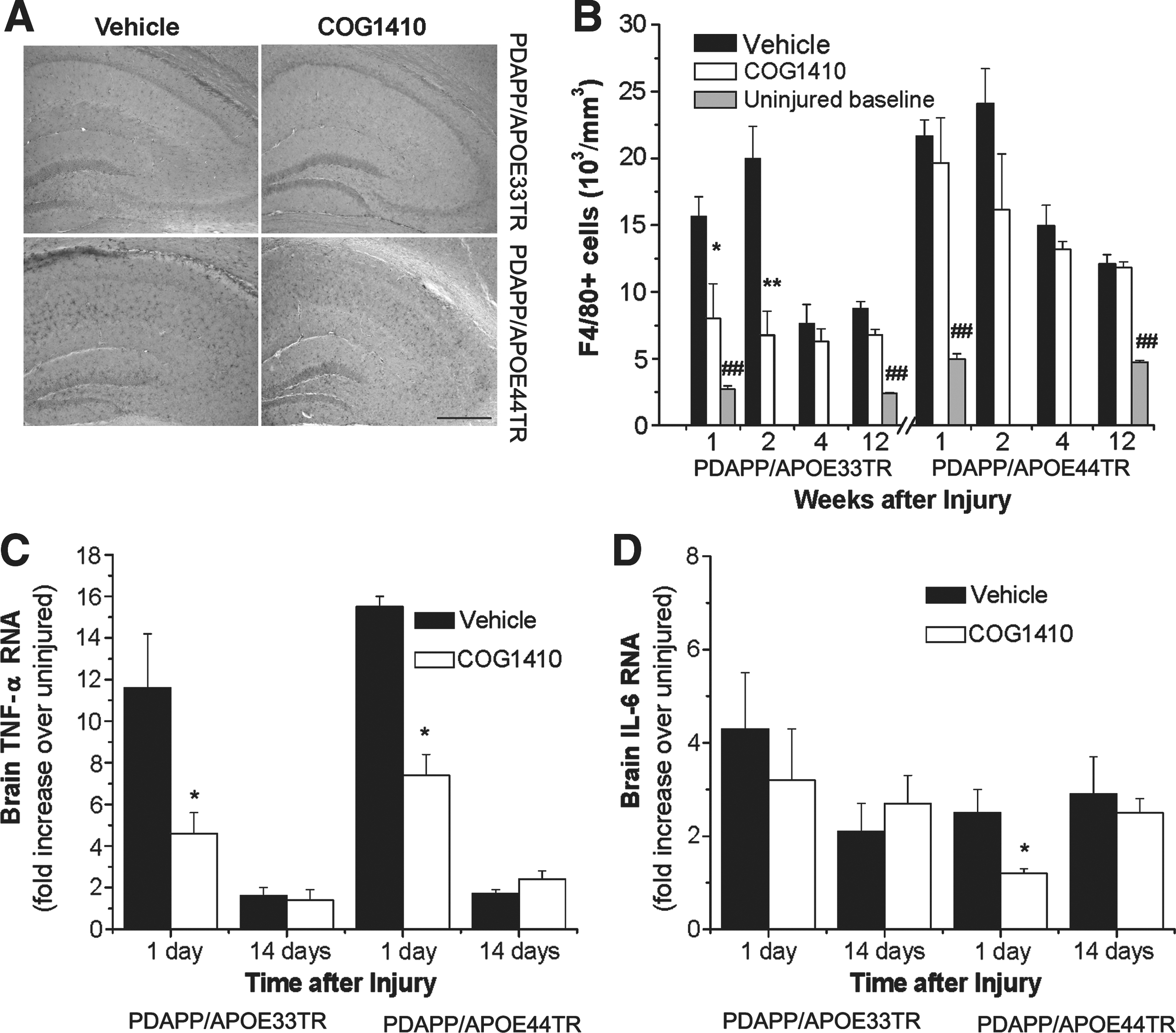
TBI induces microgliosis in PDAPP/APOETR mice.
To assess the effect of COG1410 and APOE polymorphism on neuronal degeneration following TBI, a stain specific for early degeneration of neurons, Fluoro-Jade B (FJB), was performed 24 h following injury. FJB staining revealed a comparable degree of neuronal injury in PDAPP/APOE33TR and PDAPP/APOE44TR animals following TBI. However, a single administration of COG1410 at 30 min after injury reduced the number of degenerating hippocampal neurons in PDAPP/APOE33TR, but not PDAPP/APOE44TR, animals at 24 h post-TBI (Fig. 4A and B). Although the number of FJB-positive neurons decreased significantly by 1 week post-TBI as expected, there was still a persistent effect of COG1410 treatment on reducing FJB-positive degenerating neurons in PDAPP/APOE33TR mice (82 ± 16 versus 39 ± 8 FJB-positive neurons in the hippocampal area for untreated versus treated mice; p = 0.041). Because FJB staining is transient, we evaluated long-term neurodegenerative changes following TBI by quantifying neuronal and synaptic density. Neuronal density was assessed by performing unbiased stereological quantification of NeuN-immunopositive neurons (an antibody specific for neurons) in the entire pyramidal layer of the hippocampus, which revealed decreases in the hippocampal neuron numbers in both the PDAPP/APOE33TR and PDAPP/APOE44TR animals at 3 months following injury (Fig. 4C). Treatment with COG1410 restored neuronal density in PDAPP/APOE33TR animals to near pre-injury levels, but once again, it had no effect on the PDAPP/APOE44TR animals (Fig. 4C). Representative photomicrographs of NeuN staining are shown in (Fig. 5). As another measure of long-term changes in plasticity, synaptic density was quantified in the cortex and stratum radiatum of the CA1 hippocampal region, because a large percentage of the CA3 pyramidal neurons project to the stratum radiatum of the CA1 region, and most of the positive Aβ immunoreactivity is located in the cortex and in the pyramidal layer of the hippocampus. As assessed by staining with synaptotagmin, a synaptic protein, the synaptic density in the frontal cortex and the stratum radiatum of the CA1 region of the hippocampus was significantly reduced at 3 months post-injury in PDAPP/APOETR animals. This deficit was restored by post-injury treatment with COG1410 in PDAPP/APOE33TR animals (Fig. 4D), but not in the PDAPP/APOE44TR animals (data not shown).
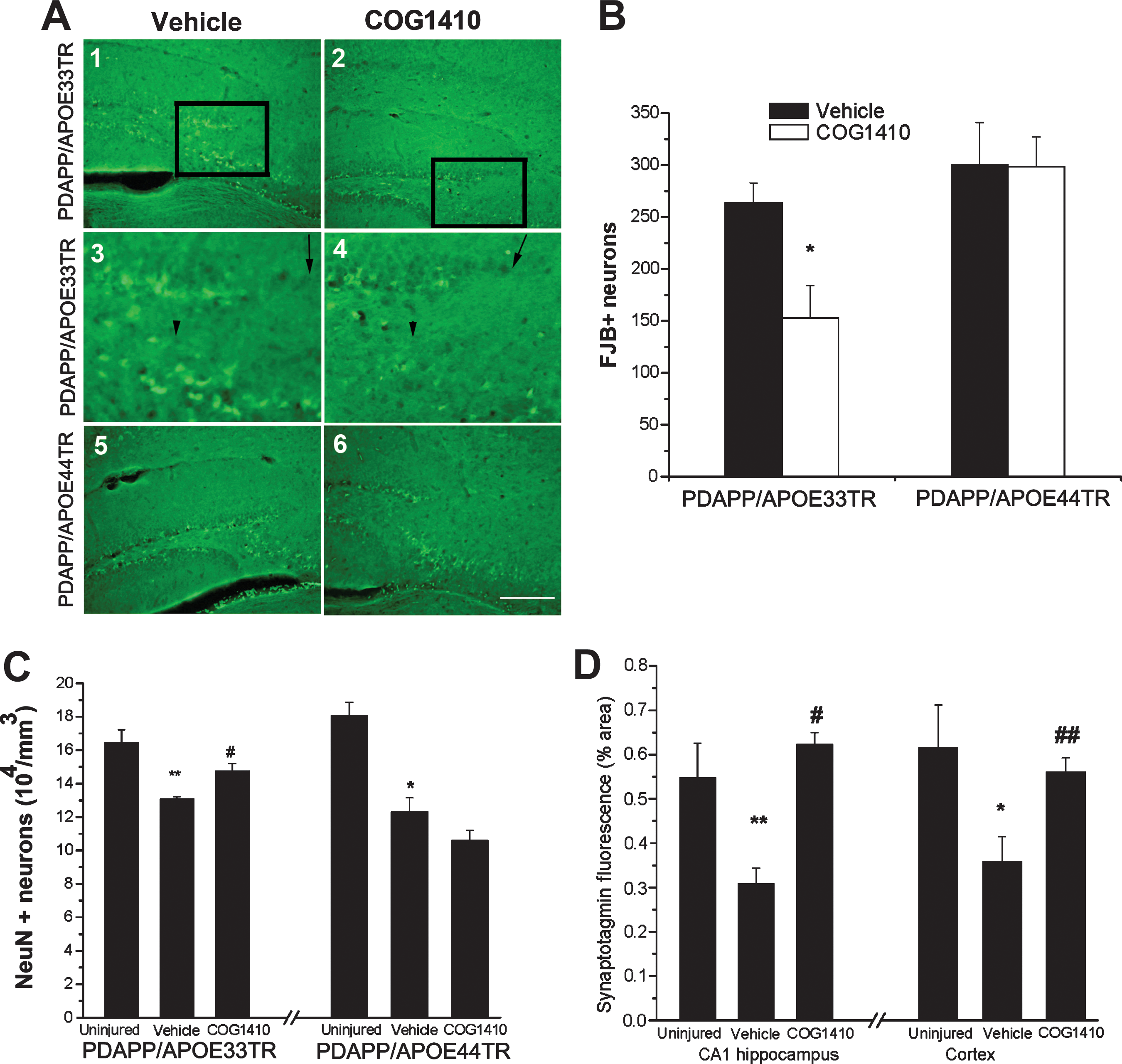
TBI elicits neurodegeneration in PDAPP/APOETR mice.
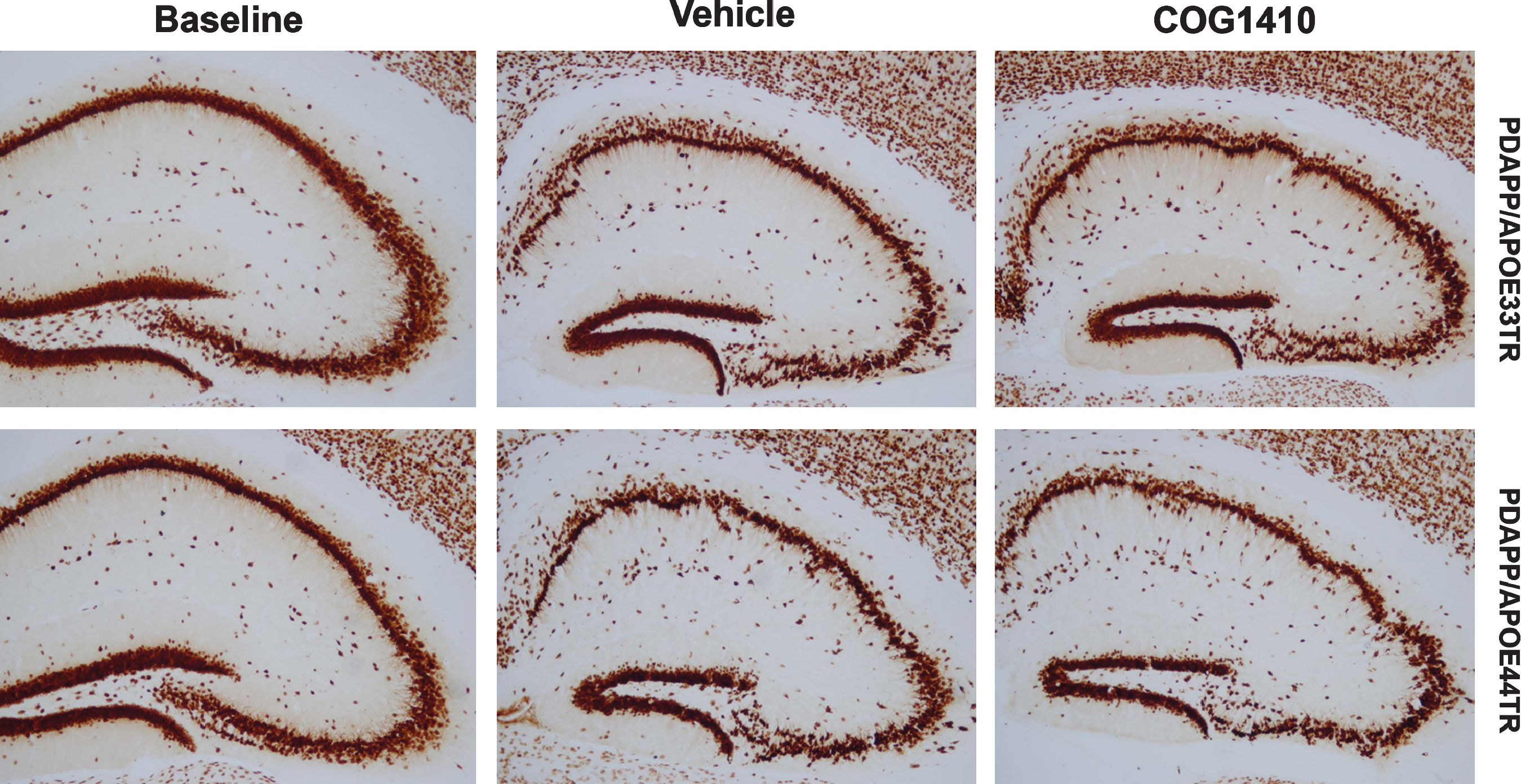
Decreased NeuN immunostaining in PDAPP/APOETR mice at 3 months post-TBI. Images of hippocampi immunostained with NeuN antibody from PDAPP/APOE33TR and PDAPP/APOE44TR uninjured baseline, vehicle-treated, and COG1410-treated animals at 3 months post-TBI (scale bar = 100 μm; TBI, traumatic brain injury; PDAPP/APOE33TR, platelet-derived growth factor amyloid precursor protein/APOE33 targeted replacement; PDAPP/APOE44TR, platelet-derived growth factor amyloid precursor protein/APOE44 targeted replacement; NeuN, neuronal-specific nuclear protein).
Another pathological feature common to acute brain injury and neurodegenerative disease is hyperphosphorylation of tau protein, which is believed to represent early tau pathology. AT8 (an antibody specific for phosphorylated tau) immunostaining was apparent in brain slices of both double-transgenic lines at 3 months post-injury (Fig. 6A). We next quantified the effect of APOE genetic background and apoE-mimetic treatment on tau phosphorylation by Western blotting (Fig. 6B and C). Low levels of AT8 immunostaining were comparable in 6-month-old procedurally-naïve uninjured PDAPP/APOE33TR and PDAPP/APOE44TR animals. As predicted by immunostaining, tau phosphorylation was increased in both transgenic lines at 3 months post-injury. However, following treatment with COG1410, PDAPP/APOE33TR animals demonstrated a significant reduction in tau phosphorylation, although there was no observable effect of treatment on the PDAPP/APOE44TR animals (Fig. 6B and C).
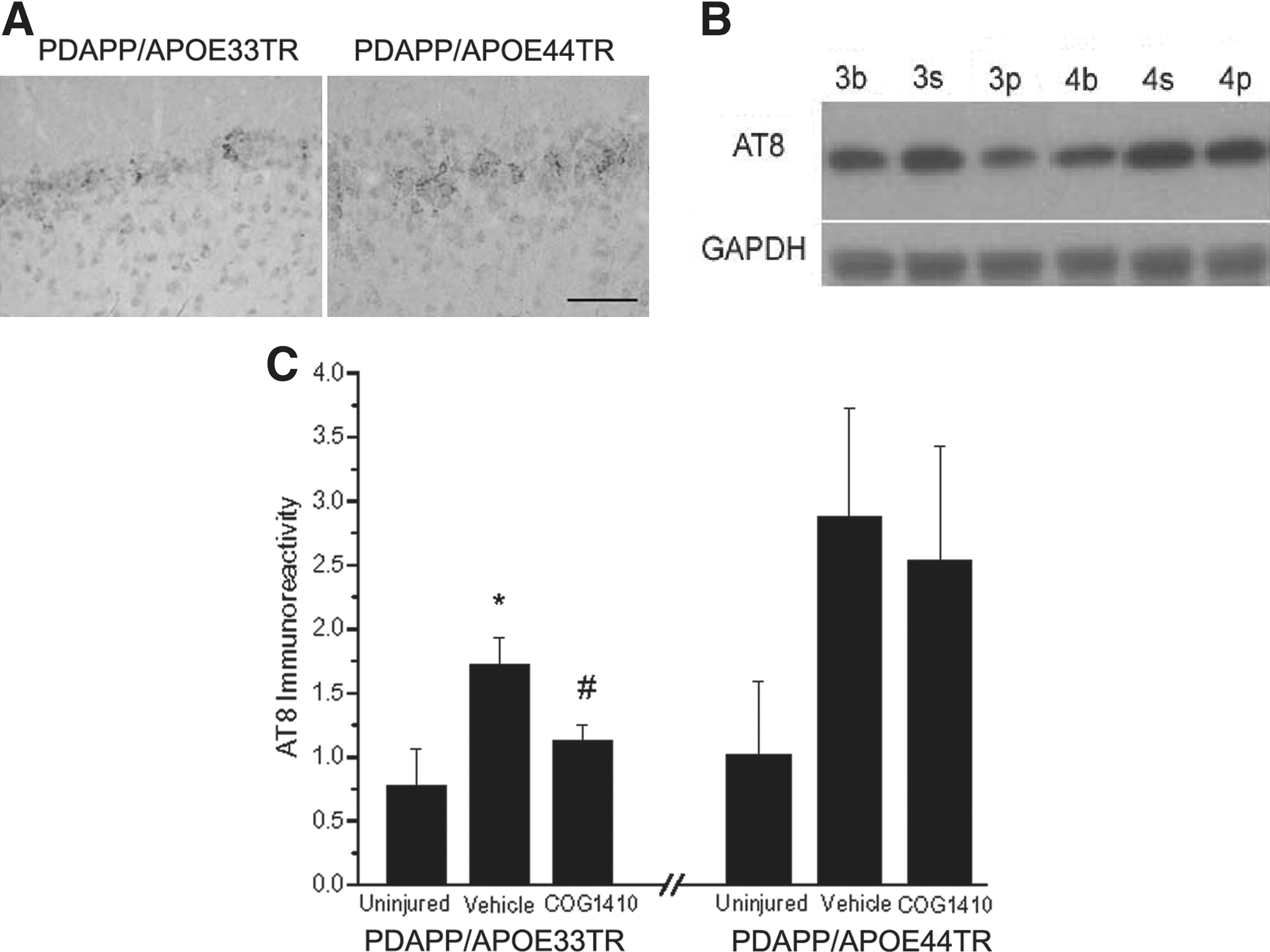
Increased tau hyperphosphorylation in PDAPP/APOETR mice 3 months post-TBI.
Although our results demonstrate that the apoE-based peptide COG1410 resulted in a reduction of intracellular Aβ accumulation, microgliosis, TNF-α, tau phosphorylation, and neuronal and synaptic loss in PDAPP/APOE33TR animals, there was only a minimal effect seen in PDAPP/APOE44TR mice (homozygous for the APOE4 transgene). Since the majority of human carriers only have one APOE4 allele, establishing the efficacy of COG1410 in heterozygous animals would be critical for potential clinical translation. Thus, 12-week-old triple-transgenic animals heterozygous for the APOE polymorphisms (PDAPP/APOE3TR/APOE4TR) were tested using the same experimental paradigm. Administration of COG1410 significantly reduced both Aβ accumulation and microgliosis at 2 weeks post-TBI in these animals (Fig. 7A and B). To assess the durability of the anti-inflammatory responses, TNF-α protein was quantified from brain homogenates of PDAPP/APOE3TR/APOE4TR animals at 14 days post-injury. Brain TNF-α was robustly upregulated after injury, an effect that was blunted by COG1410 administration (Fig. 7C).
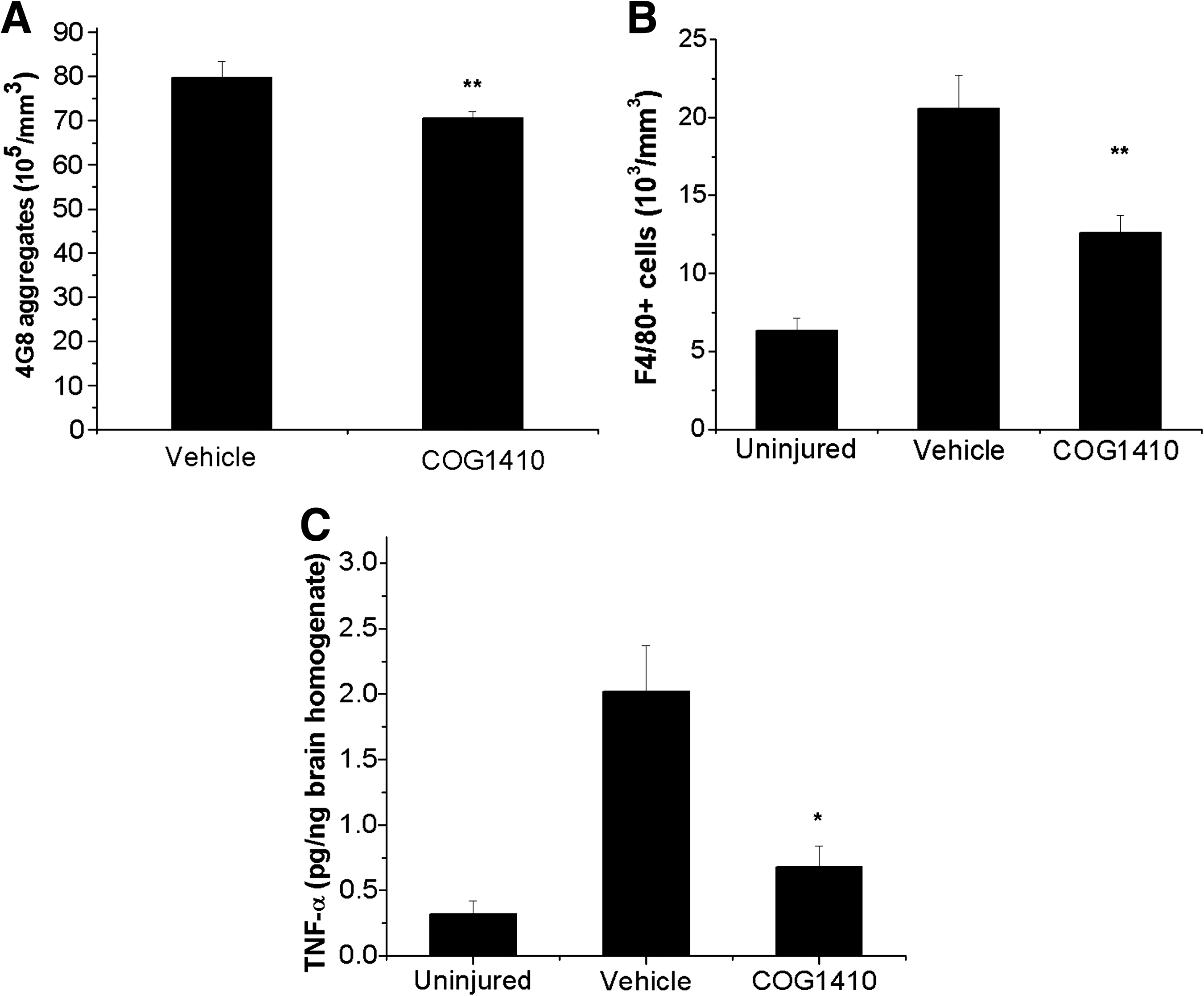
Stereological quantification of Aβ accumulation and microgliosis in PDAPP/APOE3TR/APOE4TR animals at 2 weeks post-TBI.
Discussion
Neurocognitive impairment remains an important sequela of traumatic brain injury that may significantly impact quality of life, even in the absence of objective motor impairment. Although an acute neurocognitive deficit may be a static phenomenon due to injury of preferentially susceptible hippocampal and cortical neurons, there is evidence that brain trauma may be associated with later development of neurodegenerative disease in a subset of patients (Guo et al., 2000; Plassman et al., 2000). One postulated mechanism is that a prolonged CNS inflammatory response may result in the increased deposition of inflammatory mediators, oxidative stress, excitotoxins, and acute phase reactants such as Aβ. Accumulation of Aβ may serve to maintain microglia in an activated state and promote later neurodegeneration. A better understanding of the role that post-traumatic neuroinflammation plays in promoting neurodegenerative pathology may allow us to identify genetic influences that predispose a subset of head-injured patients to the later development of neurodegenerative disease. Thus it is of interest that APOE polymorphisms, genetic modifiers of outcomes in both acute brain injury and neurodegenerative disease, play a central role in modifying CNS inflammatory responses (Laskowitz et al., 1997; Lynch et al., 2003).
A more complete understanding of the relationship between acute brain injury and the development of neurodegenerative pathology is important both from a mechanistic standpoint and for the rational development of novel therapeutic strategies and clinical trial designs. For example, the possibility of a pharmacogenomic interaction between genetic factors that modulate CNS inflammatory responses and anti-inflammatory interventions is consistent with recent data from the Cardiovascular Health Study, which found that nonsteroidal anti-inflammatory agents only reduced the risk of Alzheimer's disease in patients possessing the APOE4 allele (Szekely et al., 2008). Conversely, the possibility that APOE4 is associated with increased neuroinflammation is consistent with recent observations demonstrating that APOE4 was associated with an increase in vasogenic edema in a clinical trial of bapineuzumab, a monoclonal antibody designed to promote clearance of Aβ (Salloway et al., 2009). Moreover, long-term clinical end points may be appropriate when anti-inflammatory strategies are used in acute brain injury trials, as it is possible that intervention will have a disease-modifying effect on the later development of neurodegenerative disease.
Although the mechanism by which APOE polymorphisms influence susceptibility to neurodegenerative pathology and poor outcome after TBI has not been fully defined, one plausible hypothesis is that apoE modifies glial activation and subsequent CNS inflammatory responses in an isoform-specific manner (Barger and Harmon, 1997; Laskowitz and Vitek 2007; Laskowitz et al., 2001, 1997; Lynch et al., 2003; Vitek et al., 2009). Glial activation plays an important role in mediating secondary neuronal injury in both acute brain injury (Kelley et al., 2007), as well as in more chronic neurodegenerative processes. Thus an increase in APP expression and neuroinflammation may contribute to a cycle of Aβ deposition and resultant microglial activation that contributes to chronic neuropathology (Brugg et al., 1995). Our current results demonstrate that apoE modifies inflammatory responses in an isoform-specific fashion, as the presence of the APOE4 allele was associated with an increased level of microgliosis and intracellular Aβ accumulation following brain injury. This is consistent with independent observations demonstrating isoform-specific effects of apoE on modulating amyloid-induced glial activation in vitro (Barger and Harmon, 1997; Laskowitz et al., 2001), and neuroinflammatory effects after acute brain injury in vivo (Gao et al., 2006; Lynch et al., 2003). Alternatively, increased levels of Aβ associated with APOE4 may also have contributed to microglial activation (Nicoll et al., 1995; Roberts et al., 1994). Importantly, our current study suggests that although TBI increases Aβ accumulation acutely independent of APOE background, only the APOE4 allele is associated with a progressive subacute increase after injury, which may correlate with clinical studies suggesting a synergistic relationship between APOE4 and TBI in modifying the later development of AD (Mayeux et al., 1995; Tang et al., 1996).
Over the past decade, numerous transgenic models have been developed that recapitulate key features of neurodegenerative pathology, such as increased Aβ deposition. One limitation to these models is the fact that the development of pathology is often inconsistent and age-dependent, making it difficult to evaluate the effect of pharmacological interventions. In the current study, we demonstrate that TBI accelerates the pathological features of PDAPP/APOETR animals at a young age that would otherwise not be associated with neuropathology. By compressing the time course of the development of this characteristic pathology in a reproducible fashion, our model is more suitable for the evaluation of therapeutic interventions that suppress the development of neurodegenerative pathology. The use of double-transgenic targeted replacement (TR) animals expressing the human APOE3 and APOE4 alleles also provides a powerful tool for examining pharmacogenomic interactions (Laskowitz and Vitek, 2007). These TR animals express the human apoE3 or apoE4 protein isoforms at physiological levels in a temporal and spatial pattern similar to that observed in primates and humans (Sullivan et al., 1994, 1997). Although in the current paradigm we utilized a single pneumatic impact that replicates mild to moderate TBI, our findings are consistent with prior pre-clinical studies demonstrating that animals with genetically modified APP have accelerated Aβ accumulation following repetitive head trauma (Uryu et al., 2002). For example, although single-transgenic PDAPP animals do not spontaneously develop Aβ deposition prior to 6 months of age (Games et al., 1995), TBI markedly accelerates Aβ deposition and hippocampal cell death in amyloid-overproducing animals (Uryu et al., 2002). Interestingly, the presence of the human apoE protein retards extracellular amyloid deposition in non-injured PDAPP animals (Fagan et al., 2002; Holtzman et al., 2000).
A more complete understanding of the isoform-specific role that brain apolipoprotein E plays in mediating neuroinflammatory responses may also aid in the development of novel therapeutic strategies. Given its size, the apoE holoprotein does not readily cross the blood–brain barrier (Linton et al., 1991). However, prior observations suggest that the immunomodulatory and neuroprotective effects of apoE are mediated by specific receptor interactions (Hoe et al., 2005; Misra et al., 2001), and a small peptide derived from the receptor binding region mimics the adaptive effects of the intact protein. It is worth noting that although COG1410 is derived from the receptor binding region of apoE, it does not contain the polymorphic regions that define the common human polymorphisms, and would likely have different receptor-binding characteristics than the holoprotein. The approach of using a therapeutic apoE-mimetic peptide has previously proven successful in improving outcomes in murine models of TBI (Hoane et al., 2007; Kaufman et al., 2010; Laskowitz et al., 2007; Lynch et al., 2005), subarachnoid hemorrhage (Gao et al., 2006; Mesis et al., 2006), and experimental allergic encephalomyelitis (Li et al., 2006). The apoE-mimetic peptide used in the current experiment, COG1410, is a second-generation therapeutic that was rationally developed to result in enhanced potency and CNS penetration (Laskowitz et al., 2006).
In the current study, we demonstrated that intravenous administration of COG1410 for the first 7 days after TBI dramatically reduced intracellular hippocampal Aβ accumulation in mice expressing the APOE3 allele. These effects were durable, and persisted throughout the 3-month period during which testing was performed. One possible explanation for the reduction of Aβ was that COG1410 blunted the neuroinflammatory cascade, resulting in decreased Aβ accumulation. This is suggested by the reduction in microgliosis and TNF-α mRNA levels associated with treatment. The therapeutic effects of the peptide also extended to other features of neurodegenerative pathology in PDAPP/APOE33TR animals. Tau phosphorylation, a feature of AD pathology, was increased in our double-transgenic mice following injury, and treatment with COG1410 reduced tau phosphorylation in PDAPP/APOE33TR animals. Immunohistochemistry performed 3 months after injury confirmed that peptide treatment prevented the long-term reduction in neuronal and synaptic density associated with TBI. However, a pharmacogenomic interaction was observed such that the apoE-derived peptide had maximal efficacy in the APOE3-homozygous mice, and minimal activity in the APOE4-homozygous mice. Since a large majority of human individuals possessing the APOE4 allele is heterozygous, and this population is overrepresented in AD, it is significant that this therapeutic approach also reduced intracellular Aβ, microgliosis, and TNF-α in PDAPP mice possessing both the APOE3 and APOE4 alleles.
There are several limitations to this study that should be addressed. First, as noted previously, the effect of COG1410 was greatest in the APOE3 homozygotes, intermediate in the APOE3/APOE4 heterozygotes, and minimal in the APOE4 homozygotes. Thus it is possible that similar differential efficacy might be expected were this approach to be translated to the clinical setting. The nature of our study was also relatively short-term, and prior to clinical translation it would be important to demonstrate long-term improvements in learning and memory function. The intracellular punctate accumulations that we observed were immunoreactive with 4G8 and most likely represented Aβ, given the absence of immunoreactivity with antibodies directed at either the C- or N-terminus of intact APP; however, we cannot absolutely exclude the possibility that it represents an unrelated cleavage product of APP. It has been postulated that the intraneuronal accumulation of β amyloid is the precursor to mature plaques (Gomez-Ramos and Asuncion Moran, 2007; LaFerla et al., 2007; Wirths et al., 2004), although we did not observe significant extraneuronal Aβ or plaque burden in any of the animals tested by 6 months of age, when the study was terminated. Similarly, although increased tau phosphorylation is generally accepted as the initiating step of tau pathology, we did not observe the formation of neurofibrillary pathology prior to 6 months of age, when the oldest animals were sacrificed.
Another potential limitation in the study design was that the control group received vehicle injection rather than scrambled peptide, and thus we cannot exclude non-specific peptide effects. However, previous data strongly suggest the specificity of peptide sequence for therapeutic effect. For example in prior studies, an apoE-receptor-based protein, but not scrambled control peptide, suppressed microglial secretion of NO and TNF-α in a cell-culture-based system (Laskowitz et al., 2001). Similarly, in a pre-clinical model of TBI, COG 1410, but not a control peptide of similar amino acid content and number of Aib substitutions (acetyl-AS-Aib-LRKL-Aib-KR-amide), reduced neuronal injury in the hippocampus following injury (Laskowitz et al., 2007).
In summary, here we demonstrate that a single pneumatic closed-head injury accelerates neurodegenerative pathology in animals genetically predisposed to overexpress human APP. This model may serve as a paradigm to test therapeutic interventions designed to delay or stop the progression of neurodegenerative pathology. In our model, following acute brain injury, PDAPP/APOE44TR mice had a greater degree of intracellular Aβ accumulation, tau phosphorylation, microgliosis, and neuronal injury than their PDAPP/APOE33TR counterparts. Moreover, a therapeutic peptide derived from the receptor-binding region of the apoE holoprotein reduced many of the cardinal features of neurodegenerative pathology in both PDAPP/APOE33TR and PDAPP/APOE3TR/APOE4TR animals, suggesting that this may be a promising new strategy for future translation into clinical use.
Footnotes
Acknowledgments
This work was supported by a grant from the Institute for the Study of Aging (to D.T.L.) and the Alzheimer's Association (to M.P.V.). The mice were generously provided by Eli Lilly & Company. COG1410 was provided by Cognosci, Inc.
Author Disclosure Statement
Dr. Vitek is an officer of Cognosci, Inc., and Dr. Laskowitz has served as a consultant for Cognosci, Inc.