
Abstract
After traumatic brain injury (TBI), lesions are anatomically heterogeneous, but the spatial heterogeneity of the post-traumatic brain's vulnerability to hypoxia-hypotension (HH) has been poorly studied. Our objective was to compare the effect of HH after TBI on brain energy metabolism into two regions: the frontal lobe and the thalamus. Twenty-eight Sprague-Dawley rats were randomized into four groups: sham, TBI (brain trauma alone, impact acceleration, 450-g weight drop from 1.8 m), HH (blood depletion to mean arterial pressure 40 mm Hg, FiO2 10%, 15 min), and TBI-HH (TBI followed by HH, 45-min delay). Cerebral perfusion pressure (CPP) was continuously measured. Brain microdialysis and brain tissue oxygen partial pressure (PtiO2) probes were both inserted stereotaxically into the right thalamus and frontal lobe. Except during the HH period, CPP was always above 60 mm Hg. During the hour following the HH period, significant increases in cerebral lactate-pyruvate ratio, glycerol, and glutamate were observed, and were always higher in the frontal lobe than in the thalamus (p<0.001). In the TBI-HH group and in the frontal lobe, increases in glutamate and glycerol were significantly higher than in the HH group (p<0.001). During the 30 min following the HH phase (reperfusion), an increase in PtiO2 was observed. In the TBI-HH group, this increase was significantly lower in the frontal lobe than in the thalamus. These findings demonstrate that in the early post-traumatic period, the metabolic cerebral response to HH is higher in the frontal lobe than in the thalamus, and is worsened by TBI, suggesting a higher vulnerability for the frontal lobes.
Introduction
F
Post-traumatic brain lesions are anatomically heterogeneous. They can affect grey matter (cortex and deep grey matter) and white matter. In TBI survivors presenting with neurocognitive sequelae such as learning difficulties and attention deficits, structural magnetic resonance imaging revealed reduced grey matter density in the basal forebrain, hippocampus, insula, and thalamus. 5 Even if primary lesions occur principally in cortical areas, secondary insults can affect basal ganglia, and particularly the thalamus. 5,6 Thalamic injury appears to have clinically relevant deleterious effects, as the amount of neuronal loss in the thalamus correlates well with the degree of incapacity after TBI. 6,7 Some brain regions could therefore be more vulnerable than others to secondary insults.
Studies have described the heterogeneity of the response to hypoxia and/or hypotension between brain regions in the non-traumatized brain. In humans and rats after cardiac arrest or models of global brain ischemia, selective subpopulations of vulnerable neurons have been described in the hippocampus, cortex, cerebellum, corpus striatum, and thalamus. 8 –10 However, the relative contribution of each cell death pathway remains controversial. 11 Differential vulnerability of the diencephalon and cortical regions has been suggested to be involved in the neuropathological status caused by global brain ischemia. 11 To our knowledge, the heterogeneity of vulnerability to a hypoxic insult has been poorly described in the traumatized brain. A better description of the changes that occur in brain metabolism and oxygen diffusion in different regions after injuries is of interest, as monitoring devices focusing on metabolism and oxygen (microdialysis and tissue oxygen partial pressure) are routinely used in humans. The location of the probes within normal-appearing brain is rarely taken into consideration for the interpretation of the values. Moreover, early brain metabolic disturbances could have an important impact on long-term neuronal and/or astrocyte cell loss.
Animal models of TBI combined with secondary insults such as HH can be used to study the cerebral consequences of associated injuries in standardized conditions. For several years our laboratory has used the adapted Marmarou model, which produces a diffuse TBI by impact acceleration, followed by a severe secondary injury consisting of hypoxia and hypotension. 12 –17 Using these associated injuries, we recently observed a post-traumatic impairment in brain energy metabolism (increased lactate-pyruvate ratio) in the thalamus. These perturbations observed in deep grey matter were not related to modifications in cerebral perfusion pressure (CPP) or in cerebral blood flow (CBF). 16,17 However, we did not measure brain energy metabolism in the frontal lobe. This study was performed to test the hypothesis that after TBI, the effects of HH can differ between the frontal lobe and the thalamus. We therefore aimed to compare the effect of HH after TBI on brain energy metabolism and oxygenation in two regions: the frontal lobe and the thalamus.
Methods
Animal preparation
This study was performed in accord with regulations of the French Ministry of Agriculture and the Helsinki Declaration on laboratory animal care and use. Adult male Sprague-Dawley rats (380–480 g) were anesthetized with 80 mg/kg intraperitoneal sodium pentobarbital (Sanofi, Libourne, France), followed by continuous intravenous infusion at 1 mL/h, corresponding to 14 mg/kg per hour, after catheterizing the femoral vein. The right femoral artery was catheterized for continuous arterial pressure monitoring and blood withdrawal. The catheter was flushed intermittently with heparinized solution (2 U/mL heparin sodium in isotonic saline solution). The right femoral vein was catheterized for infusion of anesthetic agents. Rectal temperature was continuously monitored by rectal probe and maintained between 37° and 38°C by a warming blanket. A tracheostomy was performed, and the animals were placed in the prone position and mechanically ventilated with room air (tidal volume 2.5 mL, respiratory rate 74 per min).
Adapted impact acceleration brain injury
Head trauma was performed according to the adapted method described by Marmarou and associates, producing a diffuse brain trauma by impact acceleration. 12 –17 A midline scalp incision was performed, followed by periosteal dissection, exposing a central area 1.5 cm in diameter between the coronal and the lambdoid sutures. A round steel disk (1 cm diameter, 3 mm thick) was fixed by surgical cement (Palacos R-40; Schering-Plough Europe, Brussels, Belgium) onto the central area of the skull. The animals were placed and secured in the prone position on a piece of foam under a hollow acrylic glass tube. A 450-g weight was allowed to drop freely from a height of a 1.80 m onto the center of the steel disk. Rebound impacts were prevented by moving the animal (and foam) immediately after the initial impact. The rats were rapidly transferred back to the warming blanket and mechanically ventilated with a room air and oxygen mixture (1 L/min). Rats with macroscopic skull fractures or rebound impacts and those who died within 5 h after trauma were excluded from the final analysis.
Hypoxia-hypotension phase
HH was induced by ventilating the rats with an O2-N2 mixture of 10%-90%, which decreased partial arterial oxygen pressure (Pa
Multimodal cerebral monitoring
Cerebral monitoring was performed immediately after head trauma. A stereotaxic device (1900 Stereotaxic Alignment System; David Kopf Instruments, Tujunga, CA) and the atlas of the rat brain were used for precise location of the intracerebral probes (see Supplementary Fig. S1; see online supplementary material at
Brain tissue oxygen partial pressure (PtiO2) was continuously measured in the right thalamus and the right frontal lobe using PtiO2 probes (Licox® CC1.R, 1 mm2 sensitive area, 0.5 mm diameter; Integra Neuroscience, Sophia Antipolis, France) at depths of 7 and 6 mm, respectively, and connected to the Licox CMP instrument.
Intracranial pressure (ICP) was analyzed using an intracranial microsensor (NeuroMonitor; Codman and Shurtleff, Randolph, MA) inserted 4 mm behind the coronal suture, 3 mm left of the sagittal suture, at a depth of 5 mm. The CPP was calculated as CPP=MAP – ICP, in mm Hg.
At the end of cerebral monitoring preparation (12 min maximum after trauma), surgical cement was added around the probes to guarantee skull watertightness and to avoid probe displacement.
Experimental procedure and measurements
The rats were randomized using a black box containing a total of 28 papers with 7 individuals for each of the four groups. If rats were excluded due to early death, the paper was replaced in the box for future randomization. Seven rats per group were selected as follows (Fig. 1): sham animals, in which the entire procedure was performed except head trauma and HH; TBI animals, in which head trauma (450 g, 1.8 m) alone was induced; HH animals, in which HH alone was induced at 45 min; and TBI-HH animals, in which head trauma (450 g, 1.8 m) was followed by HH with a 45-min delay.
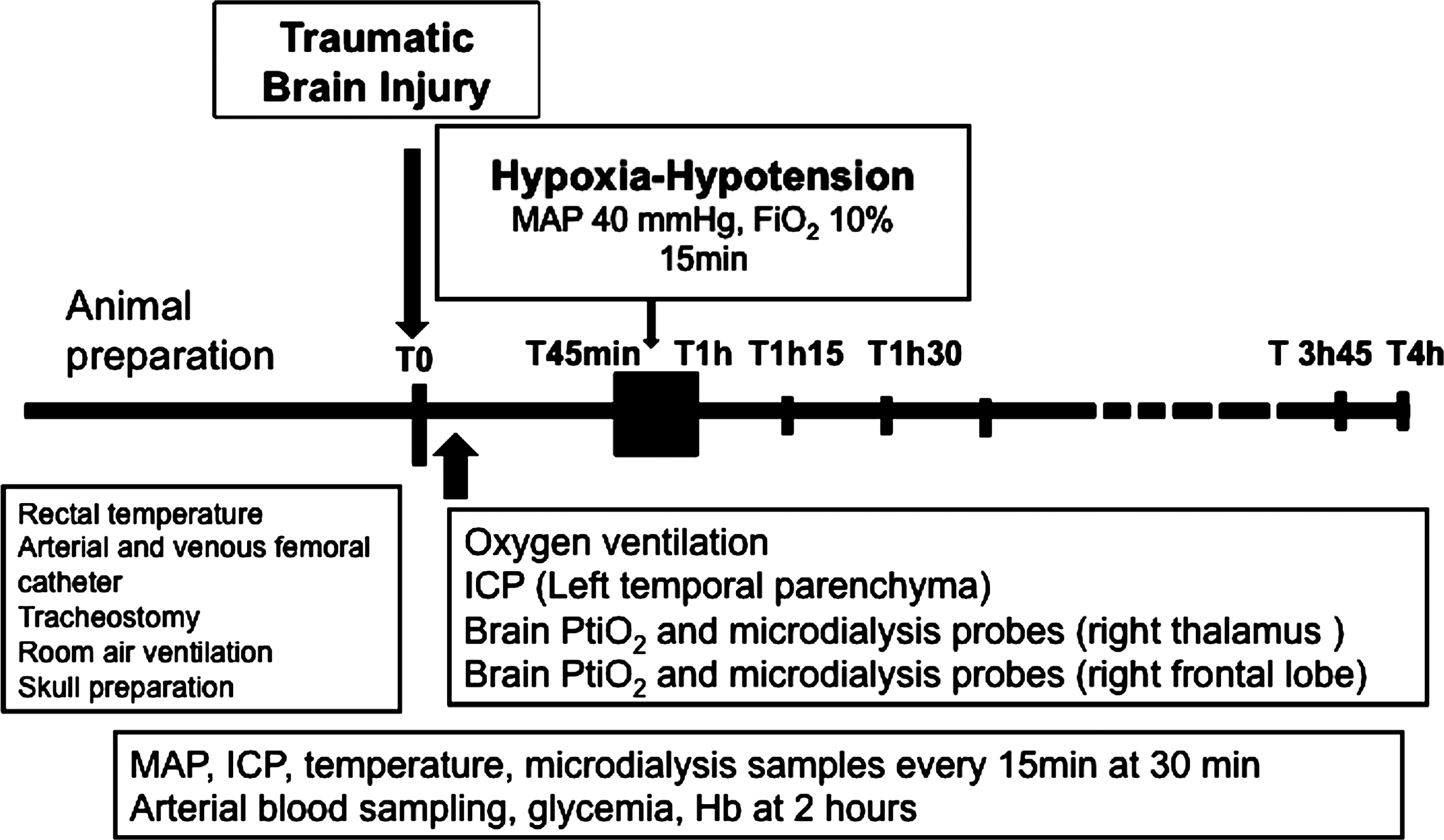
Experimental procedure and measurements (TBI, traumatic brain injury; HH, hypoxia-hypotension; Fi
MAP, ICP, and PtiO2 were recorded every 15 min for 4 h after TBI, with more frequent monitoring during the first hour after HH (every minute for 30 min). Microdialysis samples were taken every 15 min starting 45 min after TBI for 4 h. Arterial blood was sampled 2 h after TBI to determine partial arterial pressure of carbon dioxide (Pa
Statistical analysis
Comparisons between groups for general and basal hemodynamic data were performed using the Kruskal-Wallis test. The intergroup and time course comparisons for hemodynamic data, oxygenation, and metabolism, were performed using analysis of variance (ANOVA) using the overall dataset except HH periods. Post-hoc analysis was performed as necessary using Fisher's probabilistic least significant difference test. Areas under the curve were compared using the Kruskal-Wallis test. Tests were performed using StatView 5.0 software (SAS Institute Inc., Cary, NC). A p value<0.05 was considered statistically significant. Values are expressed as mean±standard error of the mean (SEM).
Results
A total of 50 rats were studied, but 22 were excluded from the final analysis because of early death or technical problems such as monitoring malfunctions (Table 1). The 300-min mortality rate was 22% in the TBI-HH group, and the skull fracture incidence was 25%, which was similar to those previously described in equivalent injury models. 12,16,17 Therefore, 28 rats were finally studied, with 7 allocated randomly into one of the following four groups: sham, HH, TBI, or TBI-HH. There was no significant difference between groups for rat weight (p=0.4501), or temperature. Due to technical problems during microvial analyses, 4/840 glycerol tubes, 2/840 glucose tubes, 4/840 lactate tubes, 8/840 pyruvate tubes, and 4/840 glutamate tubes were not studied.
TBI, traumatic brain injury; HH, hypoxia-hypotension.a
Cerebral hemodynamic
Basal MAP values were no different between groups (p=0.1037). After TBI but before HH, the traumatized groups (TBI and TBI-HH) had lower MAP and CPP levels than the non-traumatized groups (sham and HH; p=0.0093 for the MAP and p=0.0004 for the CPP; Fig. 2). During the HH periods, MAP and CPP were similar between the HH and TBI-HH groups (35.4±0.8 and 33.9±1.1 mm Hg, p=0.3625 for the MAP and 25.1±1.5 and 30±1.6 mm Hg, p=0.1102 for the CPP, for the HH and TBI-HH groups, respectively). After the HH phase, the MAP and CPP were no different between the HH and sham groups (p=0.5220 for the MAP and p=0.9325 for the CPP), but the MAP and CPP were lower with higher ICP in the traumatized groups (TBI and TBI-HH) compared to the non-traumatized groups (p<0.0001). In the study period (except during the HH period), the CPP was always higher than 60 mm Hg.
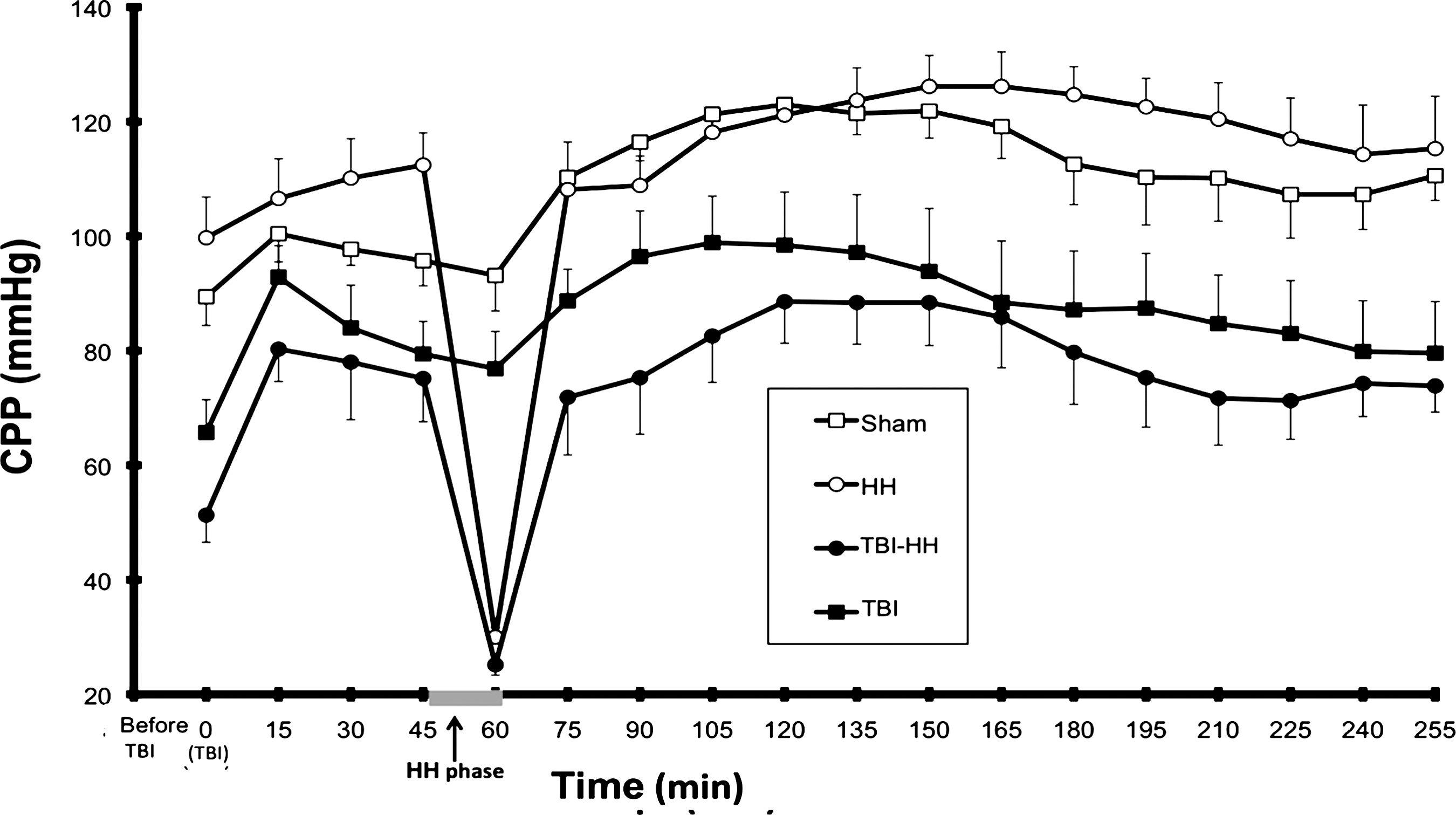
Cerebral pressure perfusion (CPP) during the procedure in the study groups. Values are mean±standard error of the mean (TBI, traumatic brain injury; HH, hypoxia-hypotension).
Brain energy metabolism
Glucose
During the hour following the HH period, brain glucose was higher in the thalamus than in the frontal lobe in the HH group (p<0.0001), and higher in the frontal lobe than in the thalamus in the TBI group (p<0.0001). Afterwards, cerebral glucose concentrations did not differ significantly between groups or between brain areas.
Pyruvate
For the entire study period, brain pyruvate levels did not differ significantly between the frontal lobe and the thalamus, nor did they differ between groups.
Lactate
During the hour following the HH period, a significantly higher increase in brain lactate in the thalamus and frontal lobe was observed in the groups undergoing HH (HH and TBI-HH), compared to the sham and TBI groups (p<0.0001 for all comparisons; Fig. 3). This lactate increase did not differ between the frontal lobe and thalamus (p=0.9741 in HH and p=0.9039 in TBI-HH). Moreover, the lactate production during these 60 min after reoxygenation, estimated by the AUC, did not significantly differ between the frontal lobe and thalamus in the groups undergoing HH.
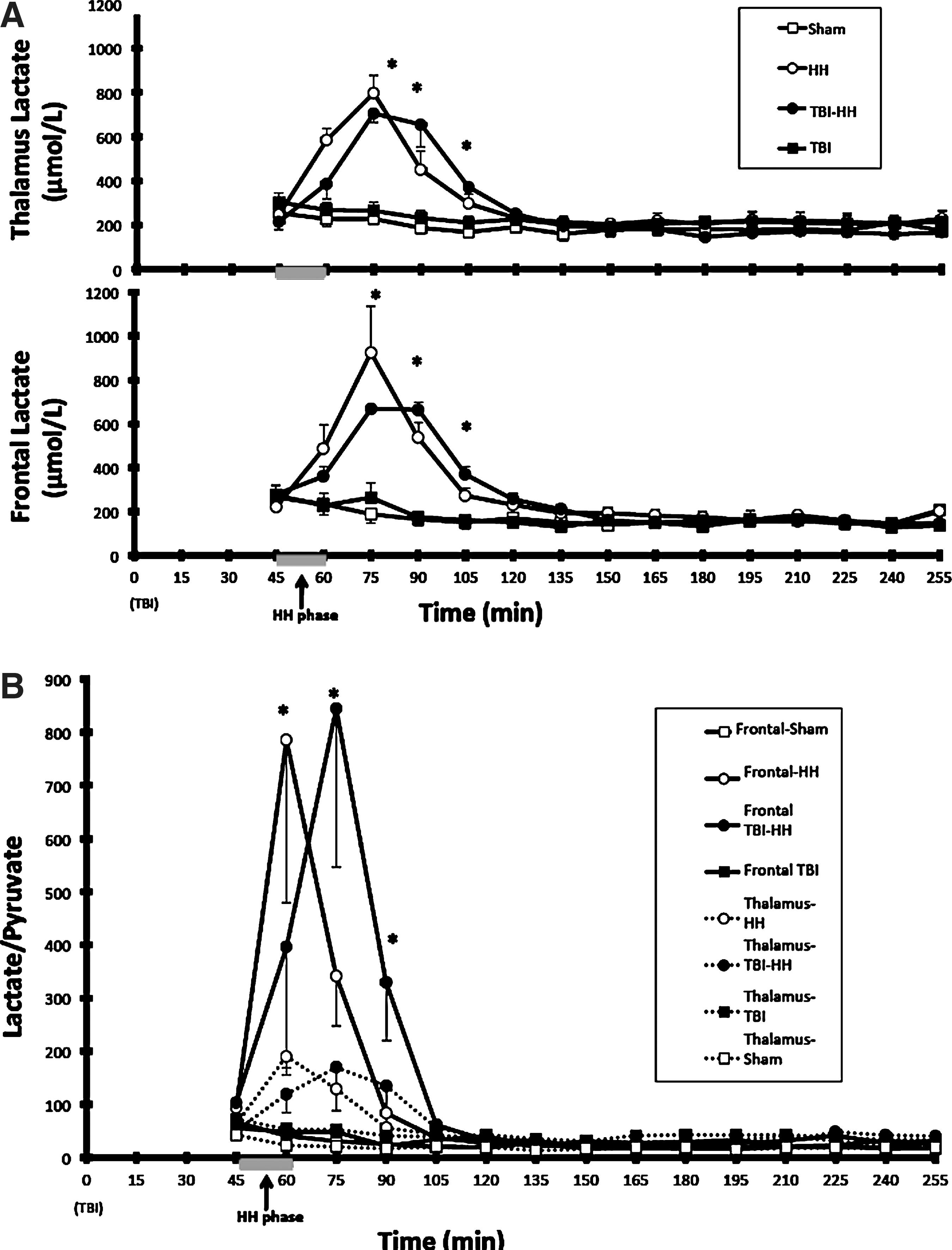
(
After these 60 min, brain lactate concentrations and brain lactate production did not differ significantly between the thalamus and frontal lobe and between groups (p=0.2335; AUC HH: 36,687±5372 and TBI-HH: 34,943±1173 μmol/L/min in the frontal lobe, and AUC HH: 36,776±2671 and TBI-HH: 35,281±1974 μmol/L/min in the thalamus, for brain lactate concentrations and brain lactate production, respectively).
Lactate-pyruvate ratio (LPR)
During the hour following HH, a different profile of LPR increases in the frontal lobe and thalamus was observed in the groups undergoing HH (HH and TBI-HH; Fig. 3). The LPR increase was significantly higher in the frontal lobe than the thalamus (p<0.0001). Moreover, the AUC of LPR was higher in the frontal lobe than the thalamus in the TBI-HH group (p=0.0181), but not in the HH group (p=0.0845).
After the first 60 min post-HH, brain LPR levels did not differ between the thalamus and the frontal lobe in all groups (p=0.3229).
Glycerol
During the hour following the HH period, brain glycerol concentrations and brain glycerol production were significantly higher in the frontal lobe than the thalamus in the groups undergoing HH (p<0.0001; AUC for HH: 2727±246, for TBI-HH: 3461±228 μmol/L/min in the frontal lobe, AUC for HH: 1345±211, for TBI-HH: 1666±220 μmol/L/min in the thalamus; p<0.0001, respectively, for brain glycerol concentrations and brain glycerol production; Fig. 4). In the frontal lobe, the glycerol increase was significantly higher in the TBI-HH group than the HH group (p<0.0001).
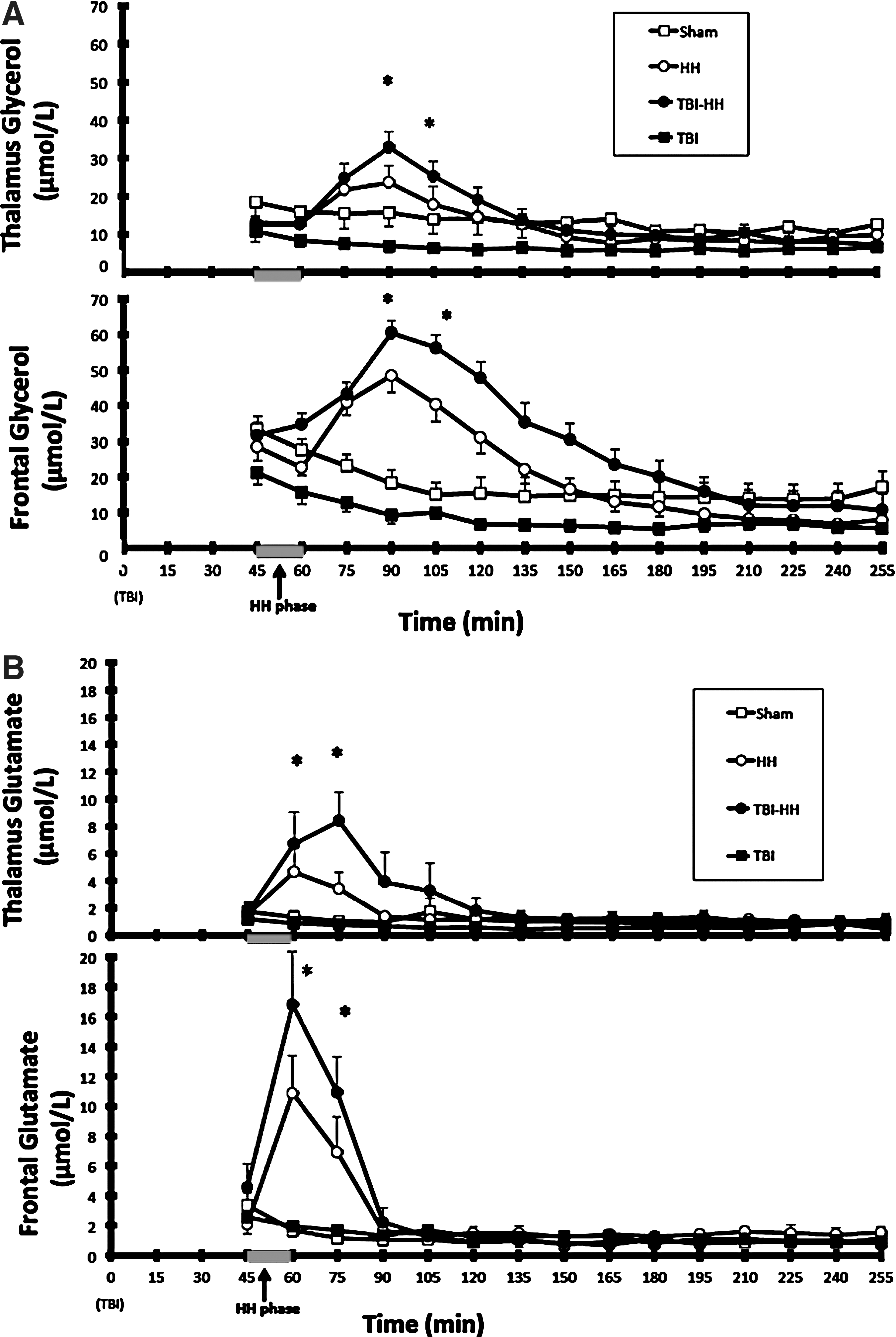
Brain glycerol (
After these 60 min, a higher glycerol concentration persisted in the frontal lobe than in the thalamus in the TBI-HH group (p<0.0001).
Glutamate
During the hour following HH, the increase in brain glutamate concentration was higher in the frontal lobe than in the thalamus in the groups undergoing HH (p=0.035 in the HH group and p=0.036 in the TBI-HH group; Fig. 4). In the frontal lobe but also in the thalamus, the brain glutamate increase during the hour following HH was higher in the TBI-HH group than in the HH group (p=0.019 in the frontal lobe and p=0.018 in the thalamus).
After these 60 min, brain glutamate concentrations did not differ between the frontal lobe and the thalamus, or between groups (p=0.4352).
Brain oxygenation
During the HH period, PtiO2 did not differ between the thalamus and the frontal lobe in the HH and TBI-HH groups (2.9±2.1 and 1.2±0.5 mmHg in the thalamus; and 3.8±2.2 and 1.5±1.3 mmHg in the cortex, p=0.5698) (Fig. 5).
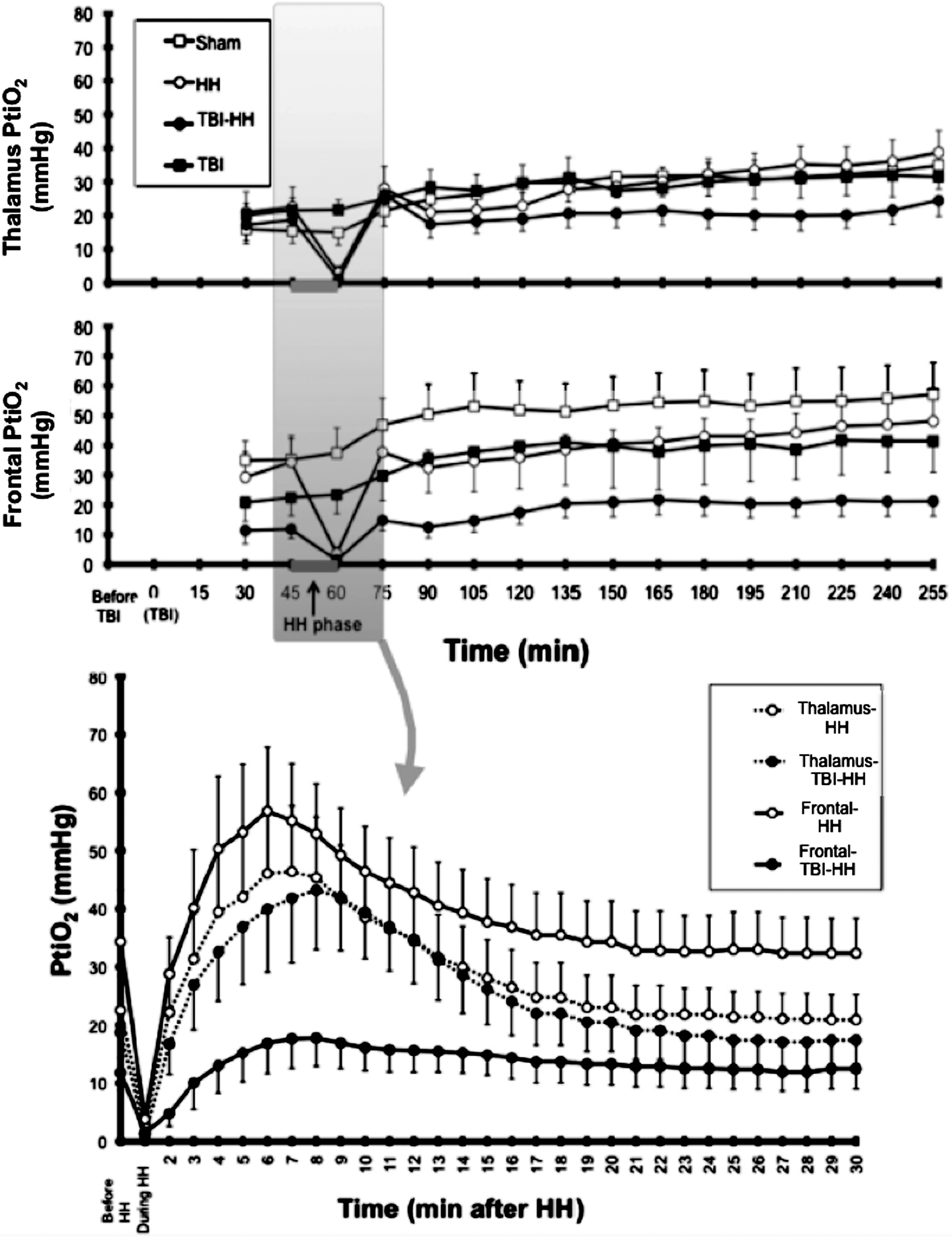
Brain tissue oxygen partial pressure (PtiO2) during the procedure in the study groups in the thalamus in the frontal lobe (top panels), with a zoomed view for the period before, during HH (15 min), and in the 30 min following HH (bottom panel). Values are mean±standard error of the mean (TBI, traumatic brain injury; HH, hypoxia-hypotension).
To study the early period of reperfusion, PtiO2 was measured every minute during the 30 min following blood restitution and reoxygenation in the HH and TBI-HH groups (Fig. 5). Following reperfusion, an increase in PtiO2 was observed in the frontal lobe and thalamus. This increase was significantly lower in the frontal lobe of the TBI-HH group (maximum PtiO2: 21.7±4.6 mm Hg) than in the HH group (maximum PtiO2: 59.8±11.7 mm Hg; p=0.04). Moreover, in the frontal lobe the AUC of PtiO2 for the 30 min after HH was significantly lower in the TBI-HH group than in the HH group (AUC: 396.5±93.4 and 1145.1±206.2 mm Hg/min, respectively; p=0.0088). In the thalamus, there was no significant difference in AUC of PtiO2 between the TBI-HH and HH groups (AUC: 768.5±169.4 and 856.7±181.1 mm Hg/min, respectively; p=0.7494).
In the period following the first 30 min after reperfusion, the PtiO2 was higher in the frontal lobe than in the thalamus in the sham, HH, and TBI groups (p<0.0001 for all comparisons). However, in the TBI-HH group, the PtiO2 was no different between the frontal lobe and thalamus (p=0.0769). Compared to the other groups, PtiO2 in the TBI-HH group was lower (p<0.0001). During the entire study period (except during the HH period) the PtiO2 was always higher than 10 mm Hg.
General data
Arterial sampling was done at a distance from the cerebral insults 2 h after TBI and 1 h after HH (Table 2). Hemoglobin, Pa
Values are mean±standard error of the mean. The p value represents the significant level for differences between groups.
TBI, traumatic brain injury; HH, hypoxia-hypotension; Pa
Discussion
This study shows that in rats after TBI, the consequences of secondary insults such as HH are different in the frontal lobe and in the thalamus, with a higher vulnerability of the frontal lobe. During the hour after HH, the LPR, glycerol, and glutamate increases were higher in the frontal lobe, and were worsened with associated TBI. This suggests that impairment of oxidative metabolism, cellular damage, and excitotoxicity are heterogeneous in the brain following such insults. After reperfusion, the increase in PtiO2 also differed from the frontal lobe to the thalamus, and was affected by the TBI. In the traumatized frontal lobe, the PtiO2 increase was low, while it was conserved in the thalamus and non-traumatized brain. This suggests that the hyperemia seen following brain reoxygenation may be affected by trauma, especially in the frontal lobe, with significant metabolic consequences.
Effect of hypoxia-hypotension
In the present study, the HH period induced a more significant increase in LPR in the frontal lobe (above 800) than in the thalamus (above 200). The extracellular LPR as estimated by microdialysis sampling reflects the intracellular redox state related to mitochondrial function. 19 Values higher than 30 are considered to be related to anaerobic conditions. 20 This significant increase indicates the severity of the HH phase, with a higher sensitivity to HH for the frontal lobe. Moreover, the HH period induced a significant and prolonged increase in glycerol and glutamate in the cortex. Glycerol is a marker of cell membrane degradation. Animal studies using brain ischemia models have shown that the increase in interstitial glycerol reflects the degree of ischemic brain damage and cell lysis. 21 Differences in glycerol interstitial concentrations in the present study may reflect differences in the degree of cell damage. Glutamate is a physiological excitatory neurotransmitter that is rapidly and massively produced in the case of ischemia. 22 These results taken together suggest that the frontal lobe is more susceptible to HH than the thalamus.
PtiO2 reflects oxygen availability for cerebral oxidative metabolism and for mitochondrial ATP synthesis. In rats, the ischemic threshold appears to be 10 mm Hg. 22 In the present study, PtiO2 decreased to below the ischemic threshold during the HH phase, verifying the achievement of HH. During the HH phase, there was no difference in PtiO2 between groups, and no difference in PtiO2 between the frontal lobe and the thalamus. Thus an increase in PtiO2, probably corresponding to a hyperemic post-ischemic phase, was observed in the groups subjected to HH, with similar profiles seen in the thalamus and the frontal lobe (as previously described by Julien-Dolbec and associates 23 ). Therefore, the observed differences in the metabolic response to HH between the frontal lobe and the thalamus could not be attributed to oxygenation differences during the HH phase, or to differences in the hyperemic response after HH.
In humans and rats after cardiac arrest or models of global brain ischemia, selected subpopulations of vulnerable neurons have been described in the hippocampus, cortex, cerebellum, corpus striatum, and thalamus. 8 –10,24 Important abnormalities in cortical areas including the occipital, parietal, and frontal lobes, have been detailed using magnetic resonance imaging in humans after cardiac arrest. 25,30 However, in comatose survivors of cardiac arrest, diffuse abnormalities in the thalamus and frontal and parietal cortices have also been described. 26 Higher vulnerability of cortical areas may lead to seizures and status epilepticus, whereas insults to the diencephalon that occur with more prolonged ischemic events may lead to a vegetative state. The present study adds important and new information on the relative vulnerabilities of cortical and thalamic regions to standardized hypoxic and hypotensive insult, clearly showing higher vulnerability in the cortex.
Effects of traumatic brain injury
The head trauma model used in this study was performed according to an adapted method described by Marmarou and associates consisting of a diffuse brain trauma produced by impact acceleration. 12,13 The height of the weight drop affects the severity of brain trauma, and corresponded in our study to severe head trauma. In cases of head trauma alone, severe behavioral consequences are observed, and are related to diffuse axonal injuries. When TBI was applied alone (without HH), the pattern of metabolic (LPR, glycerol, and glutamate), and oxygenation (PtiO2) changes were similar between the frontal lobe and the thalamus. Therefore, TBI alone does not appear to have different effects on the frontal lobe and thalamus in the early post-traumatic period.
Effects of combined injuries
In the present study, the brain response to HH appeared to be worsened with associated TBI. During the hour after HH, the AUC of the LPR was significantly higher in the frontal lobe than in the thalamus in the TBI-HH group, but not in the HH group. Moreover, the increase in glycerol and glutamate induced by HH was more significant when rats were previously subjected to TBI, in both the frontal lobe and the thalamus.
During the first 30 min following reperfusion, and when TBI was applied, different profiles of PtiO2 increases during the hyperemic phase were observed between the frontal lobe and thalamus. In the thalamus of the traumatized brain, the PtiO2 increase appeared to be preserved (similarly to the HH-alone group), while it appeared to be altered in the frontal lobe of the traumatized brain. The observed differences in oxidative metabolism, excitotoxicity, and cellular damage, were therefore probably not related to differences in oxygen supply, but were in part due to differences in the hyperemic phase just after brain reoxygenation. An alteration of the post-ischemic hyperemic phase in the traumatized cortex could have led to pronounced metabolic perturbations in the post-ischemic period. This could be related to the cumulative effects of TBI and HH on the microcirculation in the cortex, and not in the thalamus. 27 Microcirculatory (and possibly mitochondrial) failure could have been more important in the superficial frontal cortex than in the deep grey matter after TBI, and may have induced more deleterious effects of HH on brain energy metabolism.
Disturbances in oxidative metabolism and excitotoxicity, and increased cell damage could also be attributed to mitochondrial dysfunction. This mitochondrial dysfunction has been observed after TBI, in the absence of brain ischemia. 20,31 –33 The importance of the brain's vulnerability to HH after TBI in the frontal lobe suggests more mitochondrial dysfunction in this area. Finally, differences in cell types between the frontal lobe and thalamus could explain the heterogeneity of brain vulnerability.
This experimental study has some limitations. For obvious technical reasons, cerebral monitoring was not begun before TBI. The real-time metabolic consequences of TBI were therefore not studied, and it was impossible to assess metabolic basal status (before TBI) in the traumatized groups. Moreover, inserting invasive probes such as microdialysis or PtiO2 probes may induce cerebral lesions leading to metabolic disturbances. No relevant brain consequences due to the use of probes in rats (0.5 mm diameter for microdialysis, PtiO2, and ICP probes) have been described in the literature. 28,29 Moreover, sham-operated rats did not exhibit major metabolic disturbances. However, it can be assumed that lesions induced by the placement of probes are more important in the frontal lobe (the area surrounding the impact after TBI) than in the thalamus, a deeper area of the brain.
Conclusion
This animal study suggests that the consequences of HH are more important in the frontal lobe than in the thalamus, and are worsened by TBI. We have studied the spatial heterogeneity of brain vulnerability using microdialysis parameters such as LPR, glycerol, and glutamate, and with multimodal monitoring with ICP and PtiO2 probes inserted at the same locations as the microdialysis probes. The frontal lobe appears to be more susceptible to HH than the thalamus. During the hour after HH, the LPR, glycerol, and glutamate increases seen were higher in the frontal lobe, and worsened with associated TBI. Moreover, hyperemia after HH appears to be altered in the traumatized frontal lobe, but conserved in the thalamus and in the non-traumatized brain (frontal lobe and thalamus). Following brain reoxygenation, the hyperemic phase was affected by trauma, especially in the frontal lobe, with significant metabolic consequences.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.