
Abstract
The complement system plays an important role in the inflammatory response activated by many central nervous system disorders. However, its significance in traumatic diffuse traumatic axonal injury (TAI) is not fully known. Here we analyze the complement activity in two rat models of traumatic brain injury (TBI); a focal penetration injury (pen-TBI) and a rotational acceleration injury (rot-TBI) that leads to a mild TAI. We used in situ hybridization to examine the distribution of mRNA for C1q and C3 and immunohistochemistry to examine the presence of the C3 protein and C5b-9 complex at 1–5 days after injury. We found a time-dependent complement activity in both models. However, the responses caused by the two models were different. We detected C5b-9 surrounding the cavity in pen-TBI, but C5b-9 was not found in the rot-TBI. Our findings suggest that the terminal complement pathway is progressed to the formation of the C5b-9 membrane attack complex only in the penetrating TBI but not in isolated TAI model. This indicates that the complement activation does not lead to membrane-damaging effects and a subsequent secondary axotomy in TAI by the terminal complex C5b-9. The role of complement activation in TAI is unclear, but might indicate an alternative function following rot-TBI, such as opsonizing the synapses for elimination.
Introduction
A
The extensive investigations of the underlying cause of traumatic brain injury (TBI) pathobiology during the past decades have shown neuroinflammation to be a hallmark of secondary events in TBI. 3 The complement system, which is a powerful pillar of the innate immune system, has been shown to play a crucial role in many nervous system pathologies such as Alzheimer's disease, 4 –6 spinal cord injury, 7,8 peripheral nerve axotomy, 9 and cerebral contusions. 10 In a previous study using a weight drop model to induce DAI, an increased expression of neuronal C5a receptors was shown, indicating that complement-mediated neurotoxic effects could result in secondary neuronal cell death. 11
The complement system is activated through different pathways. The classical-, the alternative-, and the mannose-binding lectin pathway are the three most well known, but activation through coagulation enzymes of the extrinsic protease pathway and the C3-indedendent thrombin pathway has recently been suggested (Fig. 1). 12 Neurons and glial cells are able to synthesize the complete set of complement proteins. 13 –15 The initial component of the classical pathway is C1q, which mediates the phagocytic or lytic pathway by cellular tagging. 16,17 The first three well-known complement pathways converge at C3, which generates the cascade toward the cleavage of C5, initiating the terminal or cytolytic pathway and formation of C5b-9, the membrane attack complex (MAC), which creates pores in the target cells leading to a subsequent cytolysis. Complement is activated in the border zones of cerebral contusions in experimental animal models 18 and in humans with TBI. 10 Furthermore, in a recent study, elevated levels of local brain C5b-9 and systemic C5b-9 were shown in experimental blast TBI. 19 Disturbed blood–brain barrier (BBB) integrity has been shown to correlate with intrathecal levels of the soluble membrane attack complex (sC5b-9) in patients with TBI. 20 Secondary insults in patients following TBI correlate to increased levels of complement and the biomarker S100B in cerebrospinal fluid (CSF). 21 In an experimental focal closed head injury animal model, it has been shown that mice lacking the C5b-9/MAC inhibitory molecule CD59 showed a significantly impaired neurological outcome and neuronal loss following injury. 22 The balance between complement activation and complement inhibitors has been suggested to play an important role in the development of secondary brain damage. 18,23 In addition to the complement proteins cytolytic effect, recent studies have shown that complement proteins also play a role in neuronal plasticity, neuroprotection, and neurogenesis. 24 –26 Furthermore, a recent study has suggested that complement activation is involved in axotomy-induced adult synapse removal promoting functional recovery. 27 C-reactive protein (CRP) is an activator of the complement systems classical pathway, 28 and is a well-known marker of systemic inflammation in humans as well as an excellent marker of experimental inflammation in rats. 29 CRP was used in the present study to detect inflammatory response to the surgical procedure or the TBI.
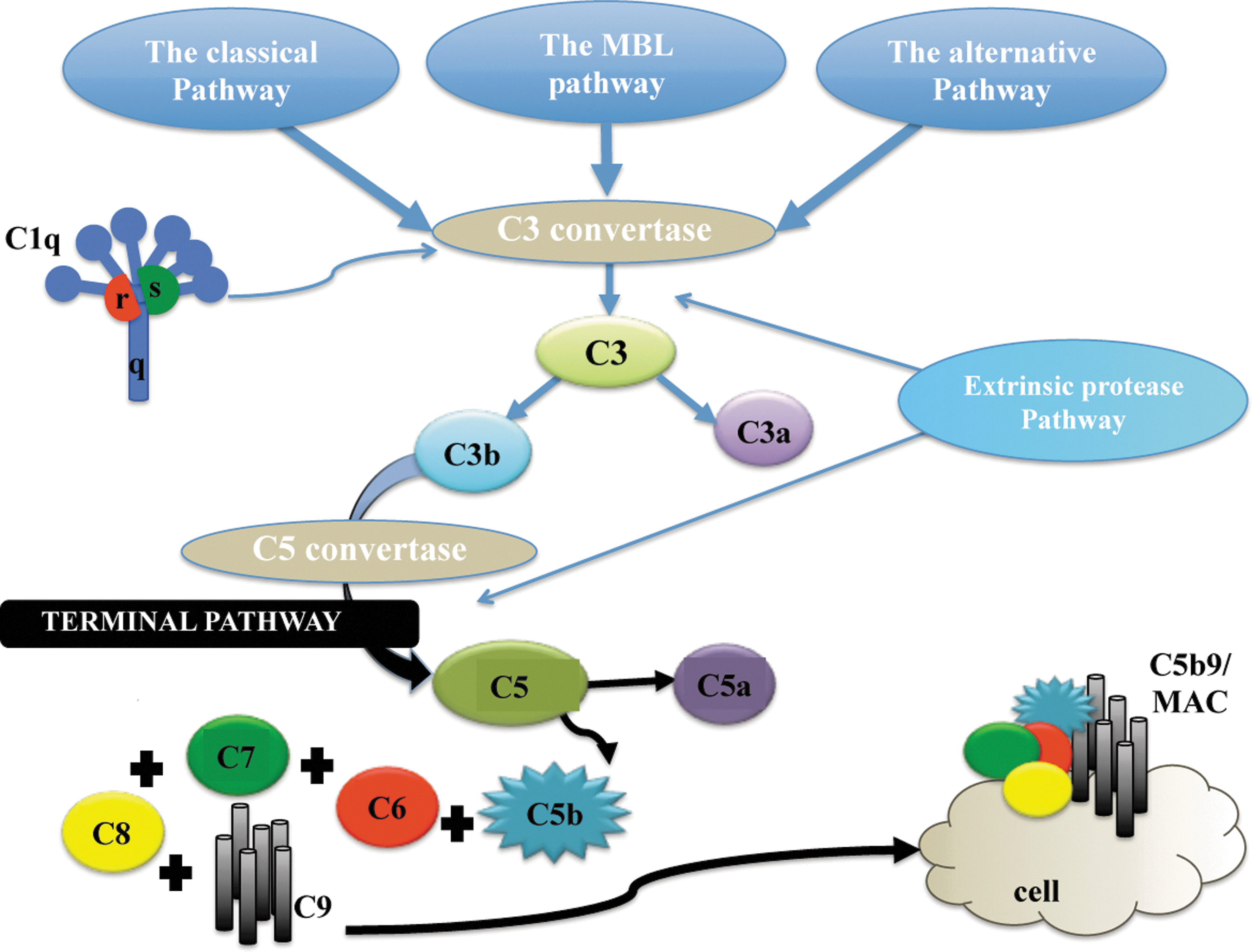
Three main pathways initiate the complement cascade, when, upon activation, all generate the protease C3-convertase. The classical pathway is activated through the C1-complex that consists of C1q, C1r, and C1s. The lectin pathway is activated by mannose-binding lectin (MBL) instead of C1q. The alternative pathway is continuously active at a low level, but the C3b that is generated from C3 by the C3 convertase is rapidly inactivated. The generation of C3-convertase by all pathways leads to cleavage of C3 into C3a and C3b. All complement proteins with the initial (a) are anaphylotoxins, whereas (b) stands for opsonins. Upon further activation of C5 convertase, it cleaves C5 into C5a and C5b and the terminal pathway is activated. The C5b assembles additional complement proteins and C9 and forms the membrane attack complex (MAC) or C5b9. This creates a hole in the cell membrane that eventually leads to cell death.
Here, we examine the role of the complement system in two distinct TBI models that mimic two important types of TBI; a focal penetrating and a diffuse rotation acceleration TBI model. 30 –33 Both models can create a graded injury. The rotational TBI model in this study is considered to lead to a mild type of brain injury without any contusions or disturbed integrity of the BBB. Based on previous studies on TBI, we expected to detect complement activation down to the terminal pathway in the penetrating TBI. 10,15
Methods
Experimental TBI
Adult Sprague–Dawley male rats (352–518 g, n=132) were used (Table 1). The animals were deeply anaesthetized by an intra-abdominal injection of a 2.4 mL/kg mixture of 1 ml Dormicum® (5 mg/ml Midazolam, Roche), 1 mL Hypnorm® (Janssen), and 2 mL distilled water. Thereafter, the animals were given 0.2 mL/kg intramuscular injections of the above-mentioned mixture every 0.5 h until trauma and surgery were completed. After euthanizing the animals, the brains were used for analysis with immunohistochemistry (IHC), in situ hybridization, and Western blot were dissected and fresh-frozen on dry ice. The work was performed in accordance with the Swedish National Guidelines for animal experiments, and was approved by the Animal Care and Use Ethics Committee in Stockholm. The methods have been described in detail previously. 30 –32, 34
Note that same animals were used for reverse phase protein microarray (RPPA) analysis and Aushon BioSystems multiplex immunoassay (ABMI); therefore, these two analyses include 42 animals in total.
APP, amyloid precursor protein.
Penetration injury model (pen-TBI)
A midline incision was made through the skin, and a 2.8 mm hole was drilled with a center ∼3 mm posterior and 3 mm lateral to bregma. The animals were placed in a frame in order to avoid acceleration injury. A lead bullet was accelerated by air pressure in a specially designed rifle and impacted a secondary projectile. The second projectile consisted of a metal cylinder with an attached carbon fiber pin (length 24 mm, diameter 2 mm) with a tip shaped like a pencil. The tip angle was 30 degrees. The pin of the secondary projectile, guided by a narrow tube, penetrated the brain of the rats with an initial speed of 90 m/sec. The design of the narrow tube provided good control of the depth of brain penetration, which was 5.4±0.4 mm (mean±SD) mm from the dura. This model has previously been presented in detail. 33
Rotational TBI model (rot-TBI)
A midline incision was made through the skin and periosteum on the skull vault, and parts of the frontal, nose, and parietal bones were freed from adherent tissue. The exposed bone was then treated with a weak phosphoric acid, dried, and gently sanded prior to the fixation of a curved aluminum plate, which was shaped to match the contour of the exposed skull. The plate was attached to the bone using dental glue (Super-Bond C & B; Sun Medical Co., Shiga, Japan). Subsequently, the attachment plate was inserted and secured to a rotating bar that enabled rotation of the head in the sagittal plane. During the trauma, a solid brass weight impacted the rotating bar, and the impulse produced subjected the animal heads to a short-lasting sagittal plane rearward rotational acceleration. The peak value of the rotational acceleration was 1.51±0.13 mrad/sec2 (mean±SD) and lasted for ∼0.4 ms. This model has previously been presented in detail. 30,34
In situ hybridization
A total of 30 animals were used for in situ hybridization, 3 injured and 2 sham- operated animals for each time point (1, 3, and 5 days) and each type of injury (rot-TBI and pen-TBI). The frozen brain tissue was cut by Cryo-Star HM 560 M (MICROM International GmbH) in coronal sections with a thickness of 14 μm, and placed on Superfrost Plus slides. Synthetic oligonucleotides were synthesized (CyberGeneAB, Huddinge, Sweden). The sequence of the probes was checked in a GeneBank database search to exclude significant homology with unrelated genes. The synthesized oligonucleotides were 11 CCA TCC CTG AGA GGT CTC CAT GAT GTT CCT GCC TCC GTG GTC CGC TCG, complementary to nucleotides of the mRNA encoding Rattus norvegicus complement component C1q (GeneBank accession no. NM_001008515.1), oligonucleotides 2028 CCT CCT TTC CAT CAA CTG CAC TGA GCG ACG GCG GCG GGC AGC TGG CTT, complementary to nucleotides of mRNA encoding Rattus norvegicus complement component C3 (GeneBank accession no. NM_016994.2). The probes were labeled at the 3′-end with deoxyadenosine- α-(thio)triphosphate[33P] (NEN, Boston, MA) using terminal deoxynucleotidyl transferase (Amersham-Pharmacia, Uppsala, Sweden) to a specific activity of 1.5–6×105 cpm/μL and hybridized to the sections, without pretreatment, for 16–18 h at 42°C. The hybridization mixture contained: 50% formamide (Fluka / Sigma-Aldrich, Sweden), saline sodium citrate buffer (0.15 m NaCl and 0.015 m sodium citrate), Denhardt's solution (0.02% each of polyvinyl-pyrrolidone, bovine serum albumin and Ficoll), 1% sarcosyl (N-lauroylsarcosine; Sigma-Aldrich), 0.02 m phosphate buffer (pH 7.0), 10% dextran sulphate (Amersham-Pharmacia), 500 μg/mL sheared and heat-denatured salmon sperm DNA (Sigma-Aldrich) and 200 mM dithiothreitol (Sigma-Aldrich). Following hybridization, the sections were washed several times in saline sodium citrate buffer for 15 min at 60°C, rinsed in distilled water, and dehydrated in ascending concentrations of ethanol. The sections were then coated with NTB2 nuclear track emulsion (Kodak, Rochester, NY). After 2–4 weeks, the sections were developed in D-19 developer (Kodak) for 4 min at room temperature, fixed in AL-4 fixative (Kodak) for 5 min and cover-slipped. Some of the slides were counterstained with cresyl violet (C5042, Sigma, USA), dehydrated in ascending concentrations of ethanol, and mounted in Entellan (Histolab Products AB, Gothenburg, Sweden). Sense probes for C1q and C3 were also used as negative controls. Photomicrographs were captured in a microscope in bright field or cold light dark field illumination (Nikon), digitized by using a digital camera (Digital Sight, U1, Nikon) and analyzed U1 with Eclipse.net software.
IHC
A total of 48 animals were used for IHC. A total of four injured and four sham-operated animals for each time point (1, 3, and 5 days) and each type of injury (rot-TBI and pen-TBI) were used. The frozen brain tissue was cut by Cryo-Star HM 560 M (MICROM International GmbH) in coronal sections with a thickness of 14 μm, and placed on Superfrost Plus slides. The tissue was air dried and thereafter soaked in 0.01 M phosphate-buffered saline (PBS) for 20 min. The sections were fixated in 4% formaldehyde for 1 min and then rinsed in 0.01 M PBS. They were subsequently incubated in a humid chamber at 4°C for 24 h with either a rat monoclonal antibody against mouse C3b/iC3b/C3c (Hycult biotechnology, dilution 1:50), mouse monoclonal antibody against rat C5b-9 (Hycult biotechnology, dilution 1:50), or rabbit polyclonal antibody against β-amyloid precursor protein (APP) (Zymed, dilution 1:100), monoclonal mouse ED1 (AbD Serotec, dilution 1:1000), and monoclonal mouse CD11b (AbD Serotec, dilution 1:100).
All primary antibodies were diluted in a solution of 0.3% Triton, 5% bovine serum albumin, and 0.1% sodium azide in 0.01 M PBS. Donkey serum (5%) was added to minimize background staining. The sections were then rinsed in 0.01 M PBS and incubated for 30 min at 20°C with 0.01% PBS, 0.1% sodium azide, and 0.3% Triton containing Cy3 donkey anti-rabbit/mouse/rat-conjugated IgG (Jackson Immuno-Research, Inc., West Grove, PA, dilution 1:400). For APP, a Cy2-conjugated donkey anti-rabbit IgG (Jackson Immuno-Research, Inc., dilution 1:100) was used. The secondary antibodies were pre-absorbed against the tissue specified by the manufacturer. After the sections were rinsed in PBS, they were mounted in a mixture of glycerol and PBS (1:2) and cover-slipped.
The specimens were examined in a microscope equipped with epifluorescence, using filter combinations appropriate for the fluorophores used (Eclipse E600, Nikon).
Western blot
After protein content determination of brain tissue samples, 10 μg of total protein from each sample were electrophoresed in loading buffer (LDS sample buffer, NuPAGE®, β-mercaptoethanol, deionized water) across a Bis-Tris gel NuPAGE® for 1 h at 200 V and 150 mA. Proteins were transferred to a polyvinylidene difluoride (PVDF) membrane according to the manufacturer's instructions (Invitrogen). Membranes were then blocked in Odyssey blocking buffer for 1 h before incubation with a primary antibody C5b-9 (Mouse,1:500, Santa Cruz sc-66190, ∼65kDa) overnight. Membranes were washed and then incubated with secondary antibodies (donkey anti-mouse IRDye 926-32212, 1:5000, 1 h; LiCor Biosciences) and visualized using Odyssey Infrared imaging system (LiCor Biosciences). Loading control was performed by overnight incubation with primary antibody GADPH (Mouse, 1:1000, Novus Biologicals NB300-221, ∼37kDa). Liver tissue known to contain high levels of C5b-9 was used as control tissue.
CRP
To measure the levels of CRP in serum, we used Aushon BioSystems multiplex immunoassay testing service that uses Aushon protein arrays (Aushon BioSystems, Billerica, MA). Briefly, samples were incubated for 3 h on the array plates that were pre-spotted with capture antibodies specific for CRP. Plates were decanted and washed four times before adding a cocktail of biotinylated detection antibodies to each well. After incubating with detection antibodies for 30 min, plates were washed four times and incubated for 30 min with streptavidin-horseradish peroxidase conjugate. All incubations were performed at room temperature with shaking at 200 rpm. Plates were again washed four times before adding a chemiluminescent substrate. The plates were immediately imaged using the Aushon CCD imaging system, and data were analyzed using Aushon Array Analyst software. The amount of luminescent signal produced is proportional to the amount of protein present in the original standard or sample. Concentrations are interpolated from a standard curve.
Reverse Phase Protein Microarray (RPPA)
A total of 42 animals were used for RPPA analysis, 18 animals with different surviving time in rot-TBI and pen-TBI, respectively, and 6 normal controls. Injury groups were: 1 day (n=3), 3 days (n=3), 14 days (n=3) and sham groups; 1 day (n=3), 3 days (n=3), and 14 days (n=3).
The animals were deeply sedated by a 2.4 mL/kg intra-abdominal injection of a mixture of 1 mL Dormicum® (5 mg/mL Midazolan, Roche), 1 mL Hypnorm® (Janssen), and 2 mL of distilled water, and euthanized through vast drainage of peripheral blood. The blood was drained and centrifuged and serum was removed, aliquoted, and frozen on dry ice. The detailed method of proteomics has been described previously. 35 Samples were transferred into a JANUS Varispan Integrator and Expanded Platform Workstation (PerkinElmer, Waltham, MA) for serial dilution, and transferred into Genetix 384-well plates (X7022, Fisher Scientific, Pittsburg, PA). Plates were centrifuged at 4°C at 1500g for 5 min, and transferred into a Q-array Mini microarray printer (Genetix, Boston, MA). Nitrocellulose-coated glass slides (Whatman FAST, Fischer) were stained with primary antibodies for C5b-9 (C5b-9, 1:20, Santa Cruz sc-66190) and C3 (C3, 1:20, Santa Cruz sc-14612). The slides were incubated with primary antibodies in a humidity chamber at 4°C overnight, while gently rotated. The slides were finally incubated with secondary antibodies gently rotated for 1 h at room temperature. The fluorescent signals were measured by scanning the slides with a 633-wavelength laser using a 647 nm filter in a Scan Array Express microarray scanner (Perkin Elmer, Waltham, MA).
Data were imported into a Microsoft Excel-based bioinformatics program developed in-house for analysis. The tool imported intensity data from the scanner output and calculated the total net intensity after local background subtraction for each spot. The background subtracted intensity data from the dilution series of each sample were then plotted against dilution on a log10 graph. Linear regression of the log10 data was performed after removal of flagged data. Flagged data included spot intensities in the saturation range or noise range, signal-to-noise ratio <2, or high variability between duplicate spots (>10–15%). The total amount of the antigen was determined by the Y-axis intercept, that is, extrapolating the regression to zero (the undiluted sample). The Y-intercept values were given in log10, hence the power of each fold change was vast.
Statistical analysis
Statistical analysis was conducted using SPSS 19.0 and α was set to p<0.05 for all analyses. For each of the biomarkers, a one-way analysis of variance (ANOVA) was performed to compare values of sham and normal controls. After that, no significant differences were obtained between sham and controls, and the log10 values obtained for each biomarker in injured animals were normalized (z-transformation) in comparison to values obtained from normal controls. For each type of TBI and the biomarkers, a two-way ANOVA with time and group was performed. The ANOVA analysis was followed up by pairwise comparison based on estimated marginal means, and Bonferroni correction was included in all analyses.
Results
Brain tissue
In situ hybridization: Increased expression of C1q and C3 mRNA in rot-TBI and pen-TBI
Increased expression of the classical complement pathway was shown by in situ hybridization for C1q mRNA and the progression of the complement cascade by C3 mRNA. In the rot-TBI model an increased expression of C1q was found in the cingulate cortex, the hippocampus, the corpus callosum, the thalamus, and the amygdala. This expression decreased over time and by day 5 the C1q expression could mainly be seen in the centroaxial line structures and the cingulate cortex. In the hippocampus positive cells were mainly found in the neurons of the dentate gyrus (DG) (Fig. 2). The time course for C3 expression in the different areas varied. Hippocampus showed a clear expression of C3 in the polymorph and granular layer of DG at day 1 that was not observed at day 3 or 5. There was also a clear expression of C3 in the centroaxial line structures, including the different thalamic nuclei and the amygdala. Contrary to what occurred in the hippocampus, this expression increased over time.
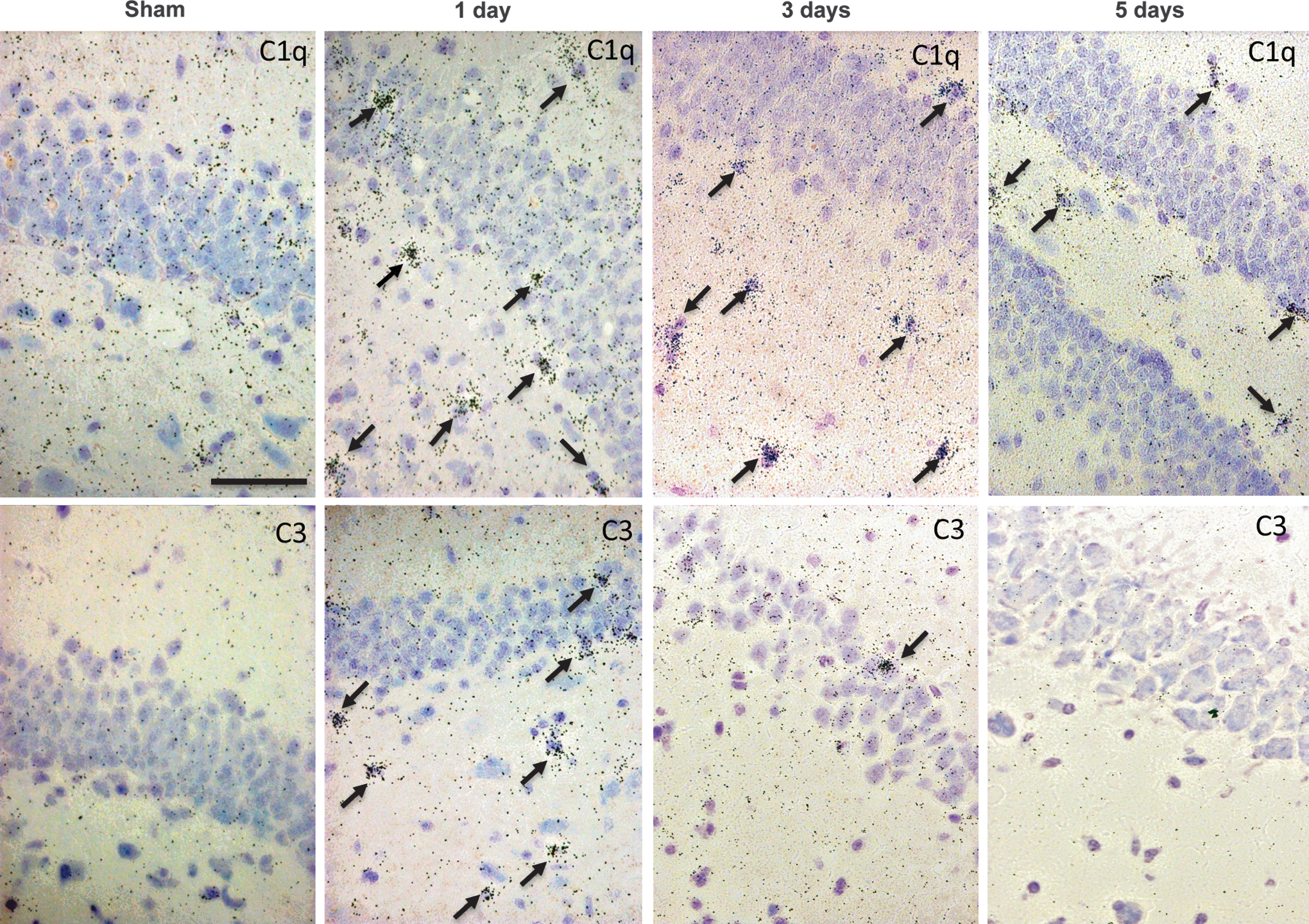
In situ hybridization of C1q and C3 in rotational acceleration traumatic brain injury (rot-TBI). The upper row presents C1q and the lower row C3 in situ hybridization (bright field microscopy). Picture illustrates the dentate gyrus of the hippocampus in the rot-TBI model sham and injured animals 1 day, 3 days, and 5 days following injury. Scale bar=50 μm.
In the pen-TBI model, an increased expression of C1q was detected in the corpus callosum and hippocampus ipsilateral and, to a lesser extent, contralateral to the injury. At day 1 and day 3 the C1q increased and peaked at day 5 in the area surrounding the cavity (Fig. 3). C3 showed an expression in the area surrounding the cavity and increased over time. Almost no expression was noted at day 1; however, C3 was clearly observed on day 3 and reached a maximum by day 5 (Fig. 3). C3 was found in the corpus callosum and the hippocampus contralateral to the injury site by day 1.
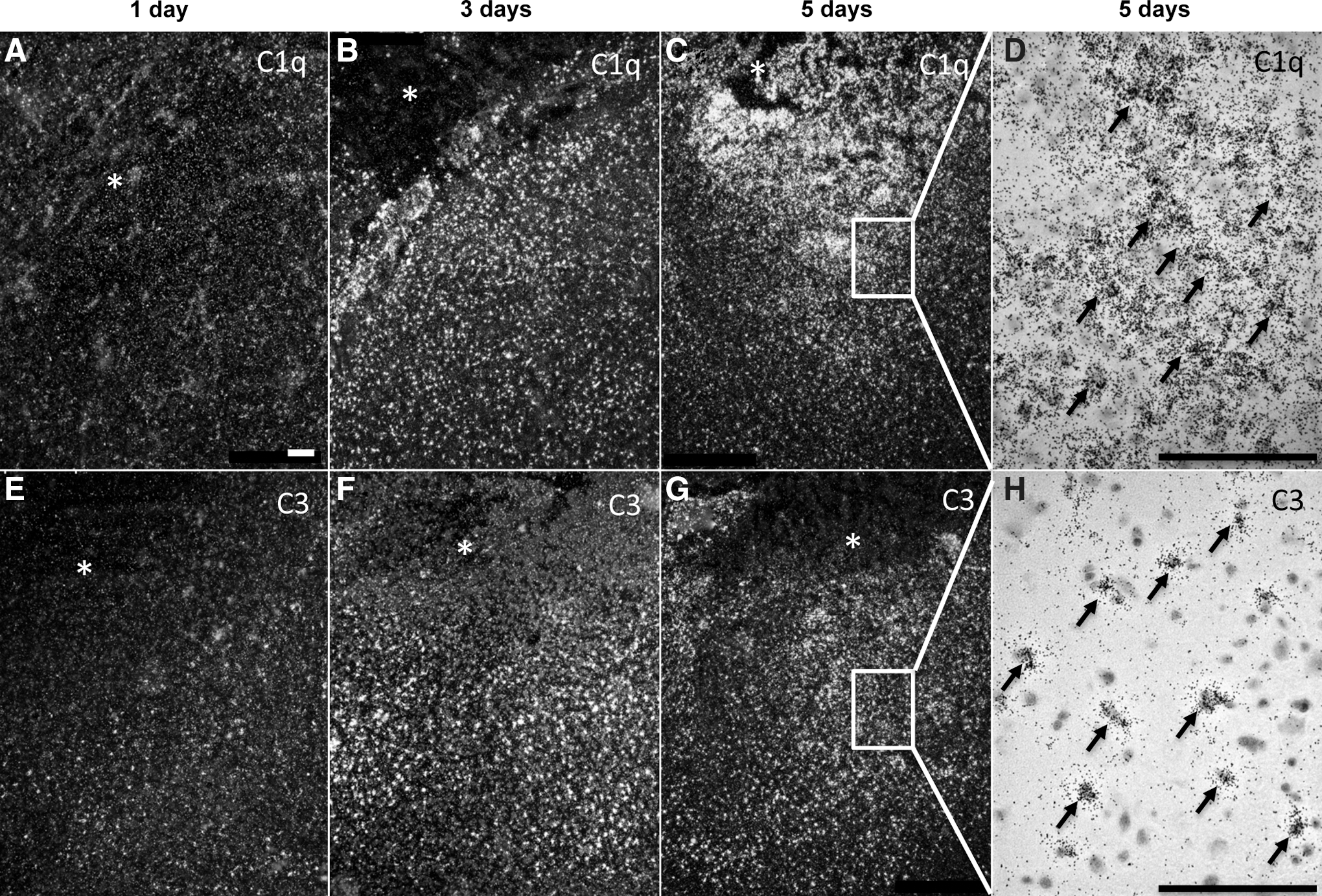
In situ hybridization of C1q and C3 in focal penetration traumatic brain injury (pen-TBI). In situ hybridization of C1q at day 1
IHC: β-APP immunostaining showed axonal injury
The occurrence of axonal injury in the brain tissue was analyzed using β-APP. The temporal and spatial distribution of β-APP in rot-TBI corresponded well with previous findings. 30 In the rot-TBI model, an increase of β-APP immunoreactivity was found in the corpus callosum and at the border zone of the gray-white matter of the corpus callosum and the hippocampus. This increase was time dependent, and showed a similar pattern as in the pen-TBI model, and had its peak at 1–3 days. By day 5, β-APP immunoreactivity appeared to have declined, and was more scattered. Positive IHC for β-APP in the pen-TBI was observed in the area surrounding the cavity, as well as in thalamus and hippocampus, ipsilateral to the injury. The β-APP immunoreactivity was substantial by day 3 and decreased by day 5 in the pen-TBI model (Fig. 4).
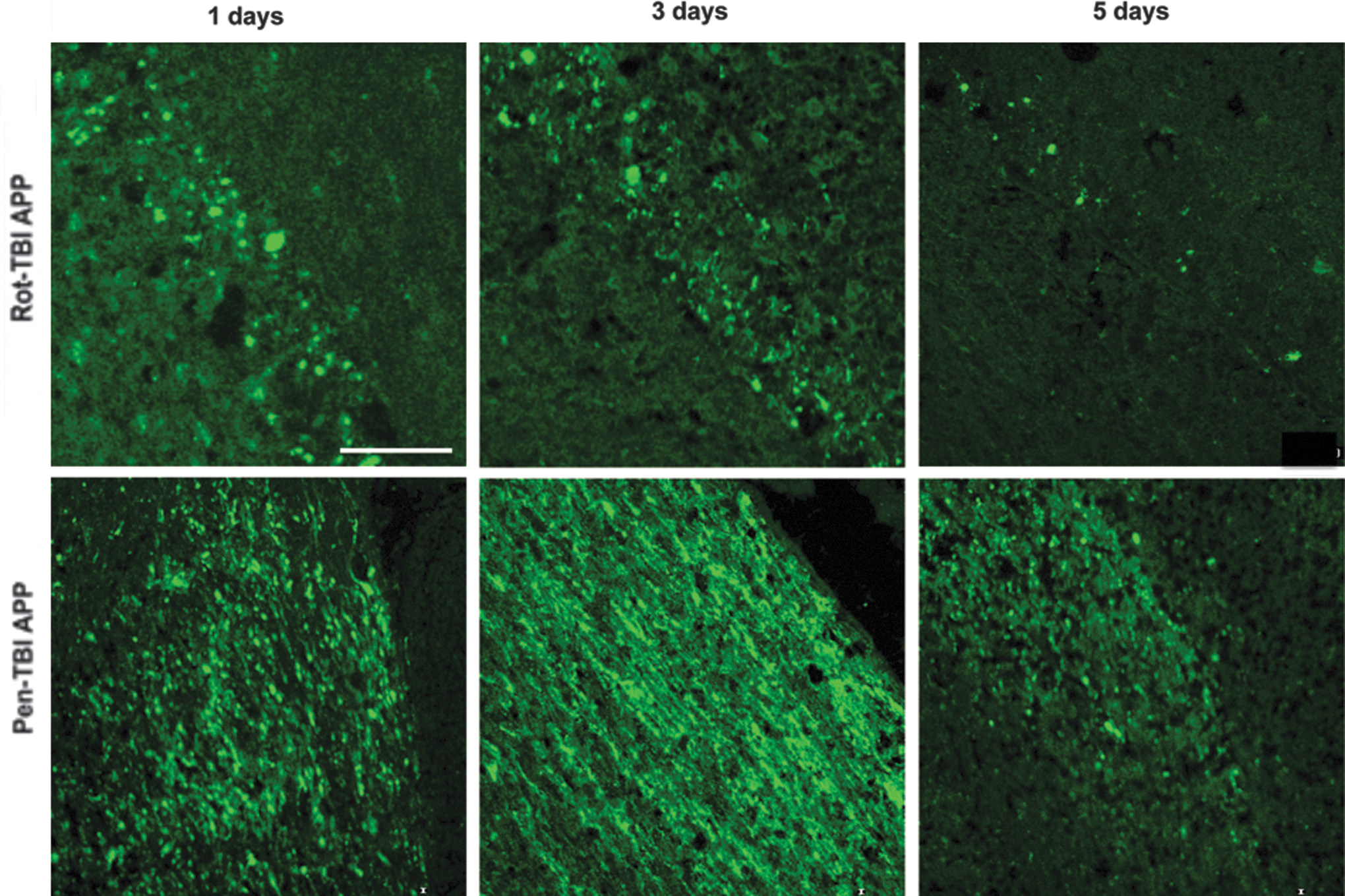
Detection of axonal injury by amyloid precursor protein (APP) immunohistochemistry. In the rotational acceleration traumatic brain injury (rot-TBI) model, an increase of β-APP immunoreactivity was found in the corpus callosum and at the border zone of the gray-white matter illustrated in the upper row. This increase was time dependent, and showed a similar pattern as in the focal penetration traumatic brain injury (pen-TBI) model, and had its peak at 1–3 days. By day 5, β-APP immunoreactivity appeared to have declined and was more scattered. Positive immunohistochemistry for β-APP in the pen-TBI (lower row) was observed in the area surrounding the cavity. The β-APP immunoreactivity was substantial by day 3, and decreased by day 5 in the pen-TBI model. Scale bar=100 μm.
IHC: C3 protein and NeuN double labeling suggested neuronal expression
The C3 findings were also investigated by IHC, where injured axons, detected by APP accumulation, were double labeled with C3. There were few signs of co-localization in either pen-TBI (Fig. 5A) or rot-TBI (Fig. 5B). Double labeling of C3 with NeuN was also performed to investigate if C3 was specifically co-localized with neurons. In pen-TBI (Fig. 5C), several C3 immunoreactive positive cells were found in DG, and some of these were co-localized with neurons. In rot-TBI, there were only a few C3 immunoreactive positive cells found that were adjacent to neurons in DG.
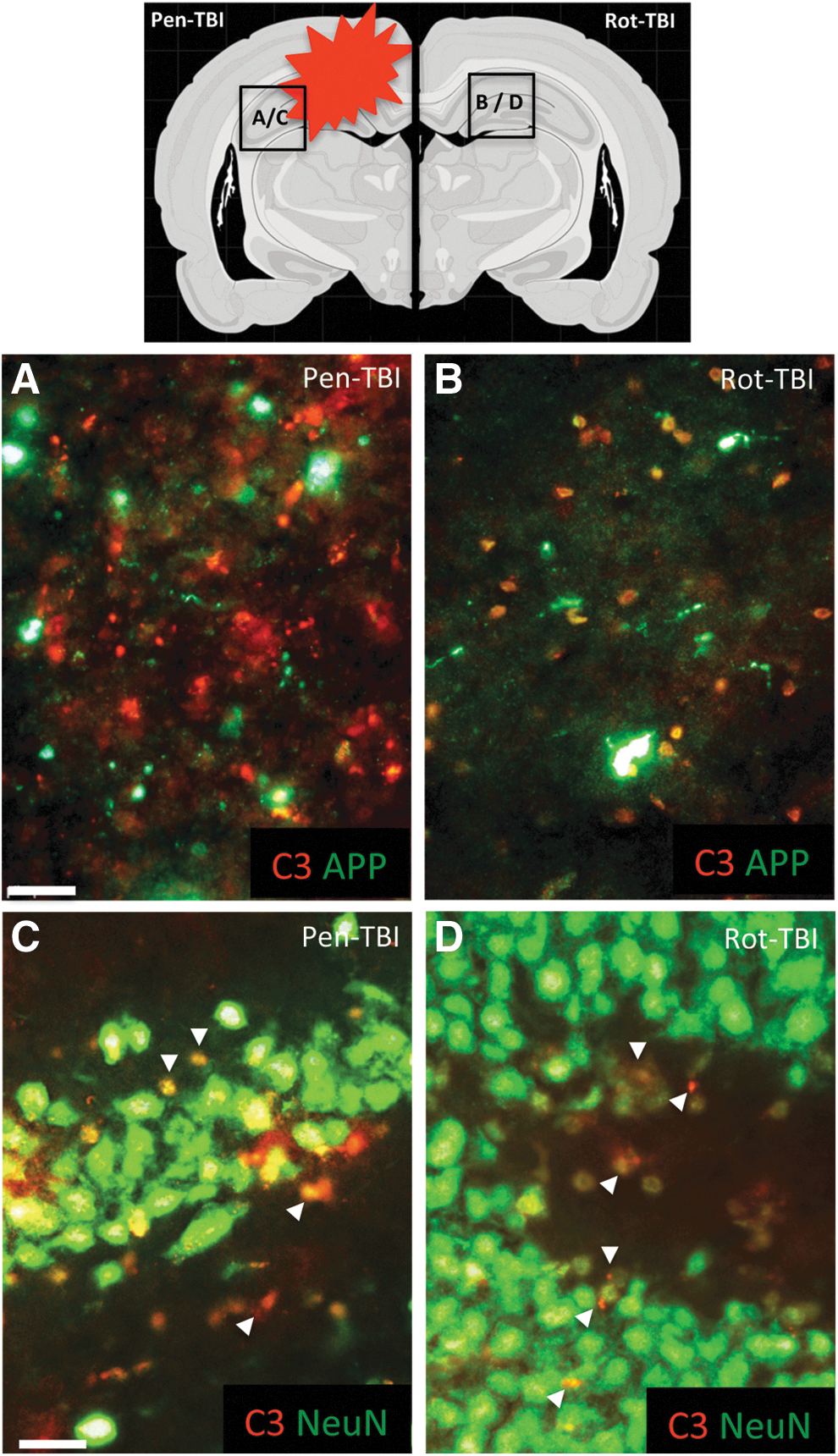
The upper picture illustrates a schematic picture to show the location of captured pictures. Injured axons were detected by amyloid precursor protein (APP) accumulation and double labeled with C3 to investigate complement proteins on injured axons. There were few signs of co-localization in either focal penetration traumatic brain injury (pen-TBI)
IHC: C5b-9 complex was detected in pen-TBI but not in the rot-TBI
Activation of the terminal complement pathway was assessed using an antibody targeting C5b-9 complex. There was a positive C5b-9 immunoreactivity in the area surrounding the cavity and in the remote area of the cavity at days 3 and 5 (Fig. 6A and B). However, no C5b-9 immunoreactivity was observed in the rot-TBI model at any time point (Fig. 6C). We investigated the expression of C5b-9 by Western blot, and could see positive bands in the pen-TBI model at day 1 and 3 but not in the rot-TBI model (Fig. 6D).
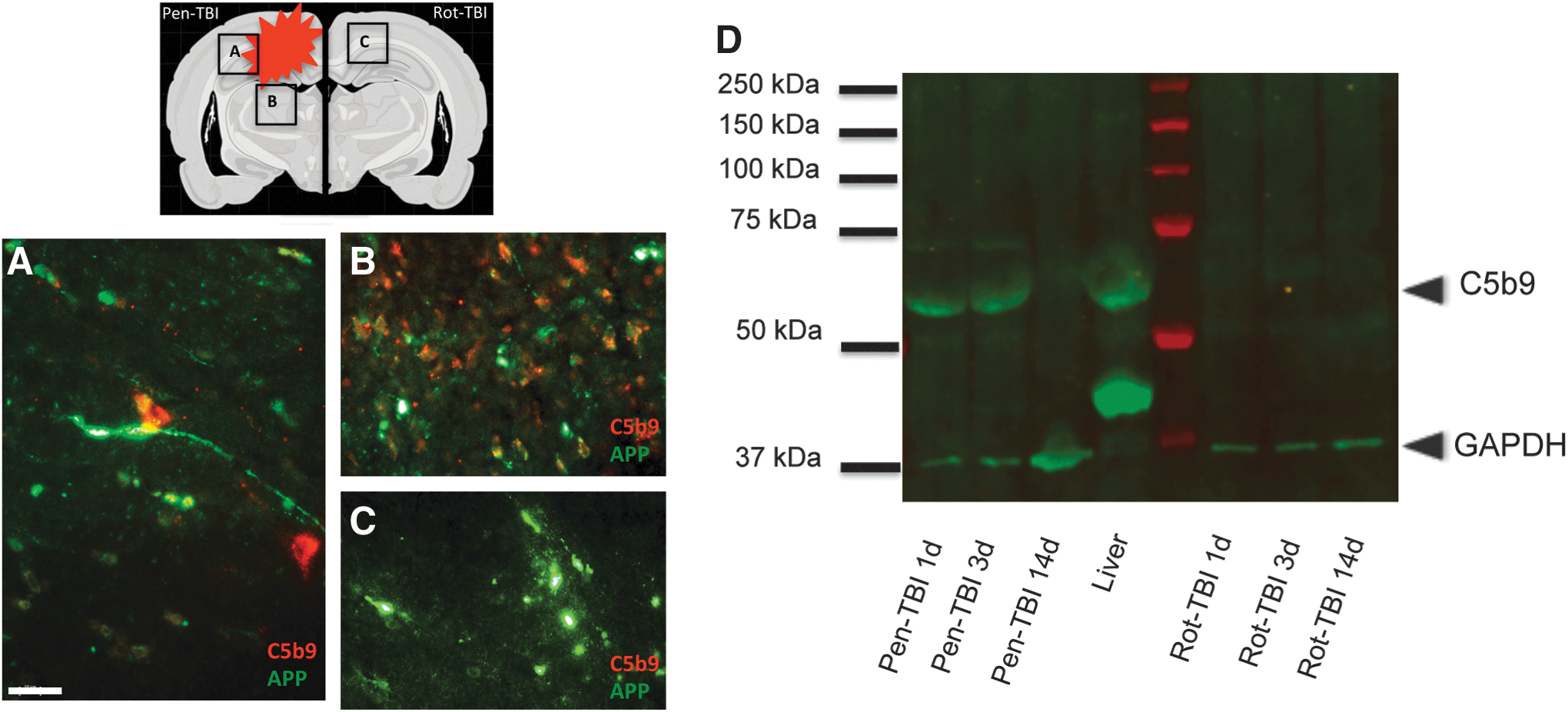
A schematic picture of the brain is illustrated in the upper left corner to demonstrate the areas of the confocal images below
We also investigated if there was a co-localization of APP and complement C3 protein and C5b-9 complex. By double labeling, we wanted to investigate if the complement protein and complex were co-localized with the injured axons (Fig. 6 A-C).
In summary, in the rot-TBI model APP was found in the corpus callosum and the hippocampus. The complement proteins C1q and C3 were expressed in the centroaxial lines, in addition to the hippocampus, amygdala, thalamus, and cingulated cortex. In the pen-TBI model there was a local expression of APP and the complement proteins C1q, C3, and C5b-9 complex in the border zone of the injury as well as in the thalamus and hippocampus ipsilateral to the injury. A summary of localization of APP and complement proteins and C5b-9 complex in both models is illustrated in Figure 7.
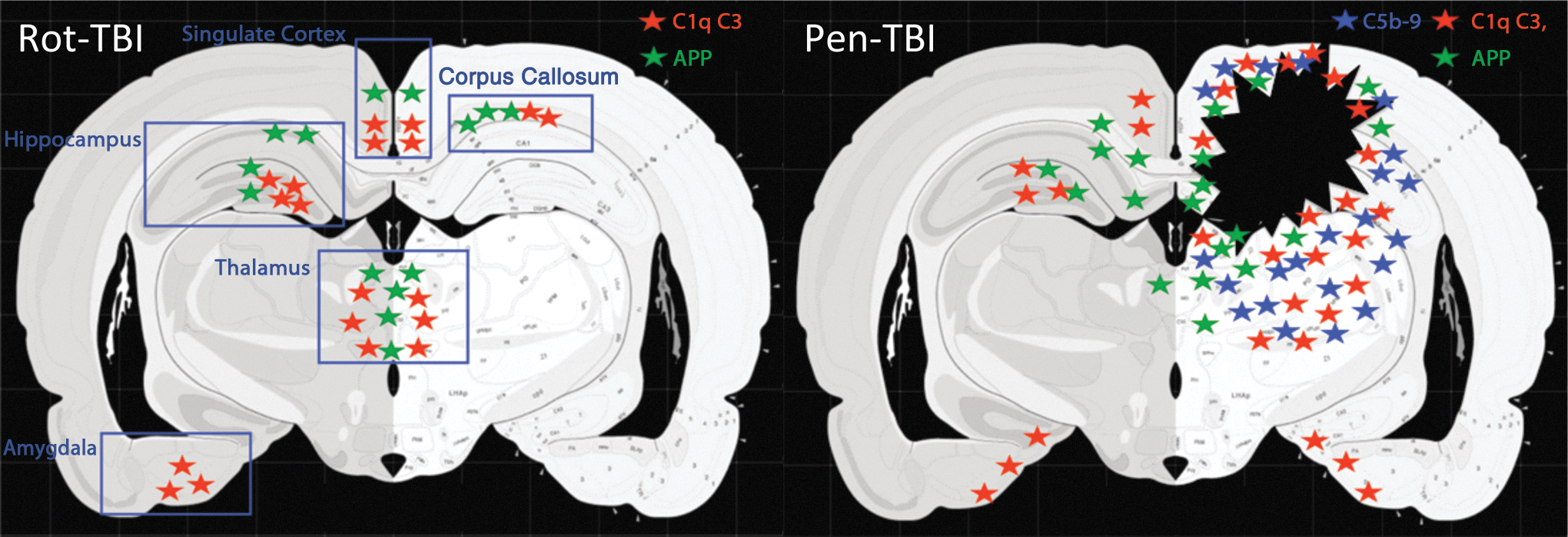
Localization of amyloid precursor protein (APP) and complement proteins. A summary of localization of APP and complement proteins in the rotational acceleration traumatic brain injury (rot-TBI) model (left) and the focal penetration traumatic brain injury (pen-TBI) model (right). Green stars represent APP, red stars represent complement proteins C1q and C3, and blue stars represent C5b-9 complex. In the rot-TBI model, APP was found in the corpus callosum and the hippocampus. The complement proteins C1q and C3 were expressed in the centroaxial lines, in addition to the hippocampus, amygdala, thalamus, and cingulated cortex. In the pen-TBI model there was a local expression of APP and the complement proteins C1q, C3, and C5b-9 in the border zone of the injury as well as in the thalamus and hippocampus ipsilateral to the injury.
IHC: Limited microglia activation in rot-TBI in contrast to pen-TBI
Currently, there are no specific markers that can distinguish macrophages from microglia by IHC. In addition, these markers stain leucocytes; however, it is believed that increased expression of ED-1 is more reflective of activated microglia/macrophages, whereas CD11b also stains to a greater extent for leukocytes. We used both of these markers in order to get a clearer picture of the activated microglia cells. Activation of microglial and/or invasion of macrophages were analyzed using CD11b and ED1 (Fig. 8). A few CD11b-positive cells were found at day 3 post-injury in the rot-TBI model. However, in the pen-TBI model, many positive cells were detected in the area surrounding the cavity already at day 1, and by day 3 there was a massive CD11b immunoreactivity. In the rot-TBI model, there were only a few cells with changed morphology and positive ED1 immunoreactivity found in the cortex. This was detected at day 1, and did not increase at day 5. The ED1 immunoreactivity in the pen-TBI model showed many positive cells at day 1, and a prominent labeling by day 3 in the area surrounding the cavity.
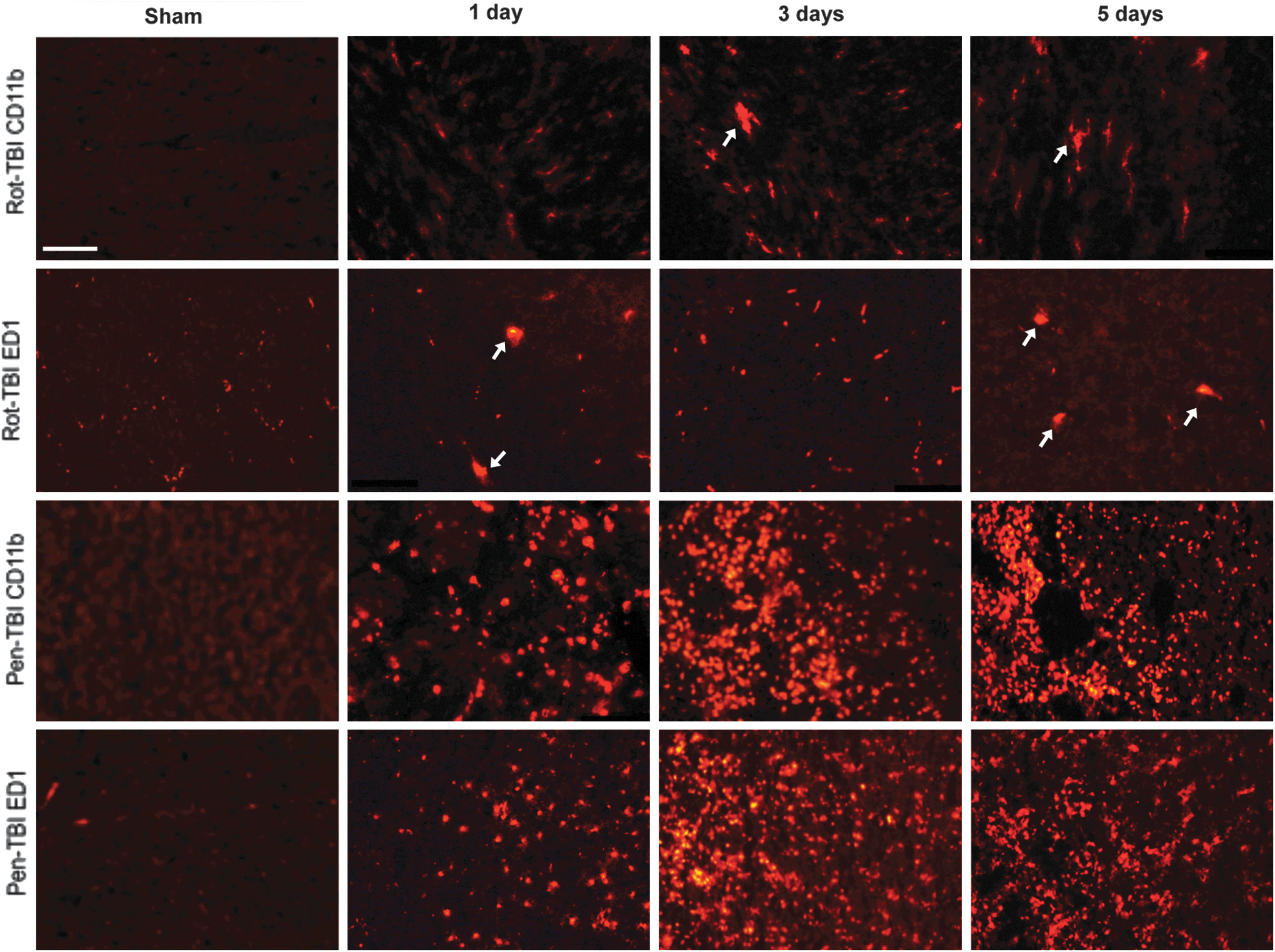
Activation of microglia/macrophages after injury using CD11b and ED1. A few CD11b positive cells were found in the cingulate cortex at day 3 post-injury in the rotational acceleration traumatic brain injury
Serum levels
No major systemic inflammatory response measured by serum CRP, C3, and C5b-9
The serum CRP measurements for all rodents were within the normal range. This indicated that there was no major systemic inflammatory reaction following the isolated TBIs. The results are presented in Fig. 9.
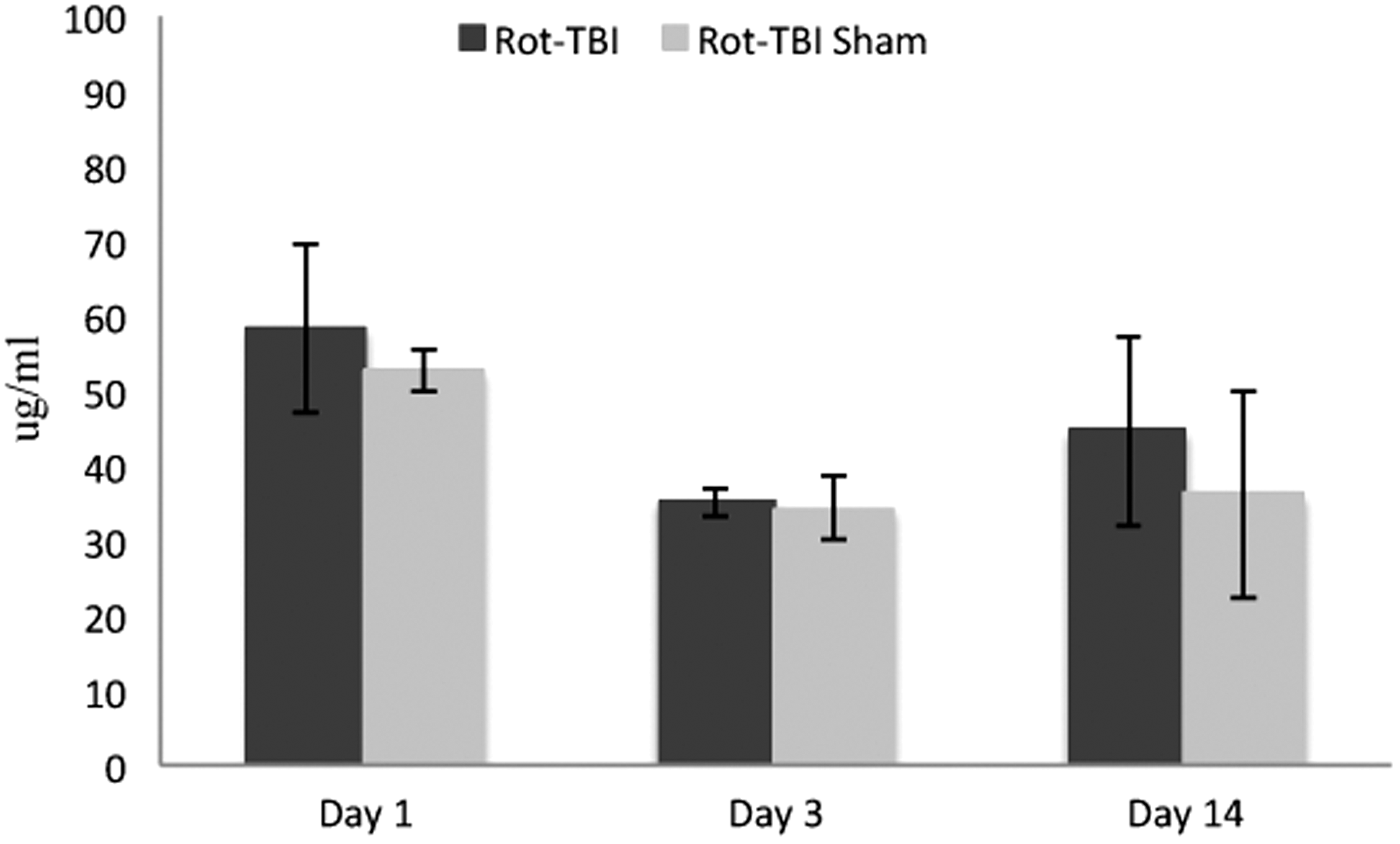
Serum C-reactive protein (CRP) measured by Aushon BioSystems multiplex immunoassay. The serum CRP measurements for all rodents were within the normal range (mean±SD). No significant differences were found in serum CRP levels between the sham and injured animals in any of the traumatic brain injury (TBI) models.
Using reverse phase protein microarray, no significant increase could be detected in serum C3 in animals exposed to trauma compared with sham-operated animals in any of the TBI models (rot-TBI p=0.051, pen-TBI p=0.913), although the p value in rot-TBI was close to significant. Serum levels of soluble C5b-9 (sC5b-9), on the other hand, showed significant increase in rot-TBI compared with sham-operated animals at all time points (p=< 0.001). No significant differences could be detected in pen-TBI (p=0.359). All the p values of comparison between the sham-operated and trauma-exposed animals for each time point following injury are illustrated in Table 2.
The results for each biomarker are given as the normalized Y-intercept values obtained from the RPPA analysis (mean±SD). The Y-intercept values are given in log10, hence the power of each fold change is vast. n for each group is 3. The p values are based on the follow-up analysis of ANOVA by pairwise comparison based on estimated marginal means. Bonferroni corrections were included in all analyzes. p values<0.005 were considered significant.
Pen-TBI, focal penetration traumatic brain injury; Rot-TBI, rotational acceleration traumatic brain injury.
Discussion
In the present two TBI models we can induce controlled and reproducible injuries. The morphological changes have been previously described. 30 –33 The pen-TBI is a focal injury with a vascular lesion, whereas the rot-TBI is characterized by diffuse TAI without apparent vascular lesions. Our previous studies show a transient disturbed integrity of the BBB in the pen-TBI model with a maximum at 1–3 days following injury, whereas no disturbed integrity of the BBB was found in the rot-TBI model. 30,32,33 This distinction between the models leads to recruitment of macrophages from the circulation into the injury in the pen-TBI, but not in the rot-TBI. As was demonstrated in the current study, we did not observe any evident microglia activation or macrophage infiltration in the rot-TBI, whereas there was a massive labeling of macrophages and/or microglia in the injured area in the pen-TBI. In addition, complement proteins from the circulation can enter the site of injury and may aggravate neuronal and myelin destruction following axonal injury. 36 –38 Activation of terminal pathway and formation of C5b-9/MAC on the injured neurons and axons could also accelerate secondary axotomy and cell death at the site of injury and other areas with compromised BBB.
Previously, we have demonstrated cell death in the injured area in the pen-TBI, but no apoptosis could be detected in rot-TBI. 30, 33 Furthermore, by using gene ontology in these two models, we also showed that gene families of inflammatory response and apoptosis showed greater alterations in the pen-TBI model than in the rot-TBI model. 32
Previous studies have revealed activated microglial cells surrounding the axonal injuries following closed skull TBI. 39 As microglial cells are activated in concert with the complement system, 15 we hypothesized that the secondary axotomy following DAI was the result of a complement attack. By targeting C1q, C3, and C5b-9 we investigated their role in the two different experimental models of TBI. In addition, we monitored the systemic inflammatory response to TBI by measuring CRP and the complement protein C3 and C5b-9 complex in serum. Here, we demonstrate that in the pen-TBI model, the initiation of the complement cascade leads to the activation of the terminal pathway, detected by C5b-9 using IHC and Western blot. This is in line with previous finding of C5b-9 in TBI with contusions. 10,15 However, C5b-9 could not be detected in the rot-TBI model. In this model, we induced a sagittal acceleration-deceleration force that generated TAI, and the severe level of the impact generated a distribution comparable to its clinical manifestation, DAI, in the corpus callosum, subcortical white matter, and brainstem. 30 Despite the findings of axonal injury, both with APP staining, silver staining and serum tau and neurofilament, there was no activation of the complement terminal pathway in the present rot-TBI model. This indicated that the axonal injuries per se did not activate the terminal pathway of the complement system and, furthermore, a role for C5b-9 in the development of the secondary axotomy seemed unlikely. These results and the current findings of C5b-9-positive cells in pen-TBI but not in the rot-TBI model altogether, indicated that the terminal pathway that leads to cell death was not activated in rot-TBI. It has been previously reported that not all traumatized axons undergo secondary axotomy or cell death, 40,41 and it has been suggested that some injured axons may recover. In addition, the Wallerian degeneration seen following axonal injury in the central nervous system (CNS) is very slow and may take months to years. One of the main reasons is the lack of extensive opening of the BBB in the CNS, which restricts entry of large numbers of peripheral opsonins and macrophages. 42 A BBB disruption as seen in pen-TBI may allow entry and involvement of macrophages and complement terminal pathway proteins that accelerate Wallerian degeneration and cell death.
In rot-TBI, there was an upregulation of C1q and C3 expression in the hippocampus. The C3-mRNA was found in the corpus callosum, cingulate cortex, hippocampus, thalamus, and amygdala, as well as at structures in the centroaxial line (data not shown for all areas). These areas coincide well with areas affected in patients with DAI, and are also known to be involved in memory disturbance, post-traumatic stress disorder, and cognitive dysfunctions seen in patients. 43,44 Components of the complement cascade, such as C1q and C3b, are known as opsonins that tag targeted cells to enhance recognition, recruitment, and phagocytosis by cells such as macrophages and microglia. 16 There is convincing evidence that complement activation plays an important role in the pathogenesis of brain injury and neurodegenerative diseases. 45,46 There are strong indications that an early event in neurodegenerative diseases is synapse loss. 47 Recent work has shown complement-mediated synapse loss in the developing and adult brain, 26 and it was suggested that C1q tags synapses for elimination. Our findings demonstrate the expression of C1q and C3 in the corpus callosum, the cingulate cortex, the hippocampus, the thalamus, and the amygdala, but without any further progression of the complement pathway. This generates a tantalizing postulation that rotational injury may cause complement-mediated synapse loss. As this process might be very slow because of the lack of entry of peripheral macrophages and little phagocytic activity of microglia in the CNS, as mentioned previously, the outcome of this synaptic loss may be detected a long time after injury. This synaptic loss could play a role in the manifested clinical symptoms in patients. However, further studies are needed to map the synaptic and dendritic density in this model of axonal injury and investigate the role of increased mRNA expression of C1q and C3.
We could not detect any increase in serum CRP in any of the models, indicating that there was no major systemic inflammatory reaction following the isolated TBIs. It should, however, be noted that negative CRP values do not exclude the release of other inflammatory markers such as cytokines. Neither could we detect significantly increased levels of C3 in the serum. However, there were significantly increased levels of C5b-9 in animals exposed to rot-TBI compared with sham-operated animals, despite the fact that no C5b-9 could be detected in the brain tissue of these animals. Although high levels of C5b-9 in CSF have been detected in patients with TBI 20 and are shown to correlate with secondary insults, 21 little is known about C5b-9 levels in the serum of TBI patients. A possible explanation for the peripheral increase of C5b-9 in the rot-TBI could be that the mechanical injuries of the neck muscles at the time of impact are the source of C5b-9 production.
Conclusion
In summary, this study shows that despite an evident axonal injury in this mild rot-TBI model, there are no signs of activation of the terminal complement pathway. In the pen-TBI model, the complement activation clearly is activated in the border zone of the injury. This suggests and endorses previous findings that complement proteins may be a possible target in treatment and aid of recovery after TBI, but indicates that the secondary axotomy following TAI is not complement mediated.
Footnotes
Acknowledgments
The study was funded by Karolinska Institutet and the Swedish Armed Forces and Vinnova, Sweden. We thank Viktoria Hammarstedt for editorial help and Dr Christine Mössinger for her generosity in contributing antibody GADPH.
Author Disclosure Statement
No competing financial interests exist.