
Abstract
Loss of olfactory function is an early indicator of traumatic brain injury (TBI). The regenerative capacity and well-defined neural maps of the mammalian olfactory system enable investigations into the degeneration and recovery of neural circuits after injury. Here, we introduce a unique olfactory-based model of TBI that reproduces many hallmarks associated with human brain trauma. We performed a unilateral penetrating impact to the mouse olfactory bulb and observed a significant loss of olfactory sensory neurons (OSNs) in the olfactory epithelium (OE) ipsilateral to the injury, but not contralateral. By comparison, we detected the injury markers p75NTR, β-APP, and activated caspase-3 in both the ipsi- and contralateral OE. In the olfactory bulb (OB), we observed a graded cell loss, with ipsilateral showing a greater reduction than contralateral and both significantly less than sham. Similar to OE, injury markers in the OB were primarily detected on the ipsilateral side, but also observed contralaterally. Behavioral experiments measured 4 days after impact also demonstrated loss of olfactory function, yet following a 30-day recovery period, we observed a significant improvement in olfactory function and partial recovery of olfactory circuitry, despite the persistence of TBI markers. Interestingly, by using the M71-IRES-tauLacZ reporter line to track OSN organization, we further determined that inducing neural activity during the recovery period with intense odor conditioning did not enhance the recovery process. Together, these data establish the mouse olfactory system as a new model to study TBI, serving as a platform to understand neural disruption and the potential for circuit restoration.
Introduction
T
Despite the growing data linking TBI to olfactory dysfunction, the precise portion of the olfactory circuitry that is involved is unclear. One model attributes TBI-associated olfactory loss to shearing of the olfactory nerve (cranial nerve 1) along the cribriform plate as the brain suddenly shifts in the cranial cavity during TBI. 8 It is also possible that a coup-contra-coup sequelae affect higher-level olfactory circuits, such as the olfactory bulb (OB) or downstream cortical areas, which is certainly consistent with the complexity of the data in studies of human TBI. 9 One recent study revealed that despite the regenerative ability of primary olfactory sensory neurons (OSNs), only a small portion (10%) of TBI patients diagnosed with anosmia show functional improvement after 1 year, suggesting that both the nature of the damage and potential for recovery varies. 10 Most animal TBI studies have focused principally on cortical and hippocampal regions, because they are central to the cognitive deficits experienced by many TBI sufferers. 11 –13 One limitation of these studies is the complexity of these brain regions, which makes it difficult to judge the subtle changes in neural circuitry that may result from TBI. Thus, a critical gap in TBI studies has been access to a simple model system with a known neural map that can be used to accurately study disruption and repair of neural circuitry after TBI and provide more clear anatomical and behavioral readouts.
To address this gap, we developed a new in vivo TBI model based upon the mammalian olfactory system that can be applied in studies of recovery from brain injury. We recognize that the cranial structure of the mouse and human are quite distinct, which makes it difficult to fully reproduce the consequences of human TBI in mice. However, we sought to focally recreate the olfactory aspects of a TBI, including OSN shearing, nerve compression, tissue damage, as well as the associated consequences to olfactory function. Many studies have demonstrated the experimental accessibility of the mammalian olfactory system, an organized circuit that gives rise to precise neural maps in the olfactory bulbs. 14 These stereotyped maps can be imaged using a variety of molecular and functional imaging techniques, 15,16 enabling direct determination of the effect of TBI on network function and organization. Because the sense of smell is intimately tied to innate behaviors, 17 we expanded this model with behavioral assays that measure functional loss and recovery of neural connections. In addition to its experimental accessibility, the olfactory system possesses life-long regenerative abilities, 18 presenting a novel platform to study recovery after injury. In sum, this new olfactory-based model broadens the range of questions that can be explored surrounding TBI-based neurodegeneration, neuroprotection, and neural repair.
Our olfactory-based TBI model exhibits many of the same molecular and anatomical features associated with other controlled cortical impact (CCI) studies, but these phenotypes are centered on olfactory circuitry. Functional deficits revealed through behavior assays also correlate with anatomical damage after injury, and recovery experiments show that olfactory circuitry and function may be partially restored despite the lingering persistence of some injury markers. Interestingly, whereas injury enhanced the number of some OSNs after recovery, strong odorant-induced stimulation suppressed that enhancement, suggesting a complex interaction exists between injury and subsequent neural activity.
Methods
Mice
Experiments were performed using the transgenic (Tg) line, OMP-GFP. 19 This knock-in mouse line expresses the green fluorescent protein (GFP) reporter in all mature OSNs in the nasal epithelium. This line is used to assess the anatomical integrity of the primary olfactory nerve (cranial nerve 1). Because the GFP also labels the OSN axons that synapse directly onto the brain through the OB, the presence or absence of OSN axons on the surface of the bulb is a direct indicator of TBI. We also used M71-IRES-tauLacZ mice, a line that has the tau-β-galactosidase (tau-βgal) fusion protein gene inserted directly downstream of the M71 odorant receptor coding region. 19,20 The M71-OSNs send axons that converge on the surface of the OB and allow for the select visualization of all neurons expressing the M71 receptor type through the use of X-Gal staining. Importantly, the odor ligand, acetophenone, has been shown to functionally and specifically activate these neurons. 21 All mice were bred in-house and all animal procedures conformed to National Institutes of Health guidelines and were approved by the National Institute of Neurological Disorders and Stroke/Animal Care and Use Committee.
Surgery procedure: Olfactory bulb impact
Mice at 7.5 weeks of age were anesthetized with an intraperitoneal (i.p.) injection of 100 mg/kg of ketamine hydrochloride and 10 mg/kg of xylazine hydrochloride and maintained with 100% oxygen supplemented with a 1.0–3.0% isofluorane gas inhalant. Mice were then secured in a small stereotaxic apparatus containing zygomatic cuffs (no ear bars) and placed under a low-magnification 10× Leica microscope (Leica Microsystems, Nussloch, Germany). The scalp was retracted over the dorsal surface of the OBs, a 2-mm craniotomy was made using a portable drill over the left OB, and the bone flap was removed. Mice were subjected to olfactory bulb impact (OBI) using the benchmark digital stereotaxic impactor (Impact One™; myNeuroLab.com). The 1-mm impactor tip accelerated down over a 1.0-mm distance, reaching the preset velocity of 3.5 m/sec, and the applied force remained there for a duration of 100 ms and then retracted automatically. Impact coordinates were preset at 4.28 mm anterior to bregma and 1.0 mm lateral to bregma (see Supplementary Fig. 4) (see online supplementary material at
Olfactory behavior assays
Naris occlusion
One day after OBI (or 22 days for recovery experiments), mice were anesthetized with 100% oxygen supplemented with 4.0% isofluorane gas inhalant. Mice were then positioned on their back and placed under a low-magnification 10× Leica MZFL3 microscope (Leica Microsystems). Visualizing the naris region, a polyethylene tube (Becton Dickinson, Mountain View, CA), 3–5 mm in length, coated with sterile ocular lubricant (Puralube Ointment; Fougera, Melville, NY), was inserted into the right nostril and mice quickly returned to consciousness after insertion.
Habituation/dishabituation assay
Four days after OBI (or 29 days for recovery tests), olfactory functioning was assessed using a version of the habituation/dishabituation assay. This simple test, which was first described in the 1980s, 22 is used to assess an animal's ability to detect odors. Animals were placed in a lidded, clean, bedding-free, empty mouse cage and left to acclimate to the new environment for 30 min to reduce the interference of exploration of a novel environment during the testing period. A solution of 0.001% amyl acetate (Thermo Fisher Scientific, Inc., Pittsburgh, PA) in MilliQ H20 was used to test the animal's ability to detect odors. The assay consisted of four 2-min trials, with a 30-sec interval between each trial. During the first three trials (habituation trials), a 1-square-inch (in 2 ) piece of filter paper saturated with 40 μL of MilliQ water (no odor) was dropped at the end of the cage and left for the animal to explore. (Animals are inherently curious and typically spend a lot of time exploring the paper on the first trial and less on subsequent trials as the object becomes familiar.) On the fourth trial, odor paper laced with amyl acetate was presented. Amy acetate typically activates a broad portion of the OB from ventral medial to ventral lateral OB, but very little activation occurs in the dorsal OB. The trials were timed with a stopwatch and video recorded to score the assay after administration of the test. Animals were scored based on the amount of time (in seconds) that they spent actively exploring the piece of filter paper. “Exploration” meant that their nose was 1 in or less from the paper and that they were clearly engaged in sniffing behavior (such as head bobbing).
Buried food assay
Four days after OBI (or 30 days for recovery tests), olfactory functioning was assessed using the buried food assay. This test was first described in the 1970s 23,24 and uses a simple paradigm to measure an animal's ability to smell. Animals were given three Pepperidge Farm cheddar goldfish per day for 3 days leading up to the assay to accustom them to the taste of the food. Twenty-four hours before the test, all food was removed from the cages to deprive the animals and ensure hunger motivation during the task. On the day of the test, animals were placed in a lidded, clean rat cage with 3-cm-deep bedding and left to acclimate to the environment for 30 min. The mouse was placed in a holder cage while the food was hidden under 2 cm of bedding at one end (but not corner) of the cage. Animals were placed at the opposite end of the cage and timed (10-min cut-off/failure point) for the latency to find (clasp in paws) the buried food.
Odor conditioning
M71-IRES-tauLacZ mice received 30 days of odor conditioning beginning 1 day after undergoing surgery. A 10% acetophenone (Sigma-Aldrich, St Louis, MO) in mineral oil solution was applied to the interior walls of the cage one time each day using cotton-tipped swabs that were then left on the cage floor bedding until the next conditioning day.
Immunohistochemistry
Four days after OBI (or 30 days for recovery experiments), mice were euthanized with an overdose injection of 200 mg/kg of ketamine hydrochloride and 20 mg/kg of xylazine hydrochloride i.p. and transcardially perfused with 1× phosphate-buffered saline, followed by 4% paraformaldehyde. OBs were removed and cryoprotected in 30% sucrose for 1 day, coronally sectioned on a cryostat at 40 μm, placed onto Superfrost slides (Fisher Scientific, Houston, TX) after antibody (Ab) incubation and washes, and mounted with Vectashield containing 4′,6′-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA). Immunohistochemistry (IHC) was performed on 40-μm floating sections at a 1:1000 dilution incubated overnight at 4°C. Primary Abs used were as follows: rabbit anti-β-amyloid precursor protein (APP; Covance, Princeton, NJ); rabbit anti-cleaved caspase-3 (Cell Signaling Technology, Danvers, MA); rabbit anti-growth-associated protein 43 (GAP-43; Novus, Littleton, CO); rabbit anti-p75NTR (Promega, Madison, WI); rabbit anti-glial fibrillary acidic protein (GFAP; Life Technologies, Grand Island, NY); and mouse anti-aquaporin 4 (AQP4; United States Biological, Salem, MA). Primary incubation was followed by a biotin-conjugated anti-rabbit or -mouse secondary at a 1:1000 dilution (Jackson ImmunoResearch, West Grove, PA) and a Cy-3-conjugated streptavidin reporter (Jackson ImmunoResearch) at a 1:500 dilution, each for 1 day at 23°C.
Image acquisition and laminar measures
Images for OMP-GFP mouse tissue were collected using a laser scanning confocal microscope Zeiss LSM 510 Meta attached to a Zeiss Axioscope 2 (excitation 543 nm, emission 570 nm; Carl Zeiss GmbH, Jena, Germany). OB and olfactory epithelium (OE) sections were imaged with a Plan-Neofluar 10×/0.30 objective, a Plan-Neofluar 25×/0, 88 Imm Korr differential interference contrast (DIC), and a Plan-Neofluar 40×/1, 3 oil DIC. Stacks (z-series) were acquired for these experiments using Zeiss LSM software (Carl Zeiss GmbH). For each picture, a z-series was collected from 30% and 50% of the anterior and posterior extent of the tissue. Five-micrometer z-stacks were collected at 1-μm intervals as 8 bit, 1024×1024 images. Single images located halfway through each z-stack were then used for TBI marker-panel pictures highlighting changes in protein expression and also for cell counting and OB area measurements. Quantification of OB area and OE GFP+ cells was done using Volocity Image Analysis Software (PerkinElmer Inc., Waltham, MA). A single section from each animal was used for quantification. DAPI counterstain was used to delineate laminar boundaries in the OB. The freeform tool was used to outline each layer. Measurements were collected adjacent to the impacted region between 500 and 1000 μm and ventral to the impacted region between 1000 and 1250 μm. The penumbra region was collected immediately anterior the impacted site. OE GFP+ cells were counted manually from 10× images from the dorsal septum spanning a region of 250 μm. Each image was expanded to a size of 8.5×11 in so that individual positive cells from the septum could be readily distinguished and counted.
X-Gal staining, cell quantification, and glomerular size measures
X-Gal staining procedures were based on and modified from Mombaerts and Brennan. 16,25 OBs and OE tissue was dissected out of each animal and incubated overnight at 37°C, shaking to achieve optimal staining. After staining, the epithelium was imaged with a microscope mounted digital camera at 6.3× magnification and M71-positive OSNs were counted by eye in turbinates 1 and 2. After whole-mount X-Gal staining, bulbs were cryoprotected and coronally sectioned at 30 μm on a sliding microtome, and floating sections were picked out for the visible presence of the M71 glomerulus. Sections containing the glomerulus were mounted on a Superfrost slide and imaged with a microscope mounted digital camera at 20× magnification. DAPI counterstain was used to delineate laminar boundaries. Quantification of the glomerulus area was done using Volocity Image Analysis Software (PerkinElmer), and these area measurements were summated for each glomerulus.
Statistical analysis
Comparisons were performed using the two-tailed t-test. p values of <0.05 were considered statistically significant and are noted in each figure.
Results
To mimic the olfactory circuit disruption associated with a human TBI, we targeted a distinct region of the mouse brain on the dorsal surface of the OB. Given the anatomical differences between mice and humans, this OBI generates a perpendicular stretching and tearing force on the OSNs that simulates the human condition. In Figure 1, we show the coordinates and parameters of the penetrating injury, which impacted the tissue and altered several layers of cells in the OB and OE. Because this injury has a pronounced influence on OSNs and their connections with the glomerular layer of the OB, we tested this model using the OMP-GFP mouse line, a transgenic line in which all mature OSNs are labeled with GFP. 19 This knock-in mouse line was used to assess the integrity of the primary olfactory nerve, and because the GFP also labels the OSN axons that terminate at the OB, the presence (or absence) of these cells is a direct indicator of TBI. Thus, in a mouse line that offers clear visualization of olfaction circuitry, we demonstrate that an OBI induces olfactory nerve damage and loss of OSNs; providing a new in vivo model for studies of TBI and recovery from injury.
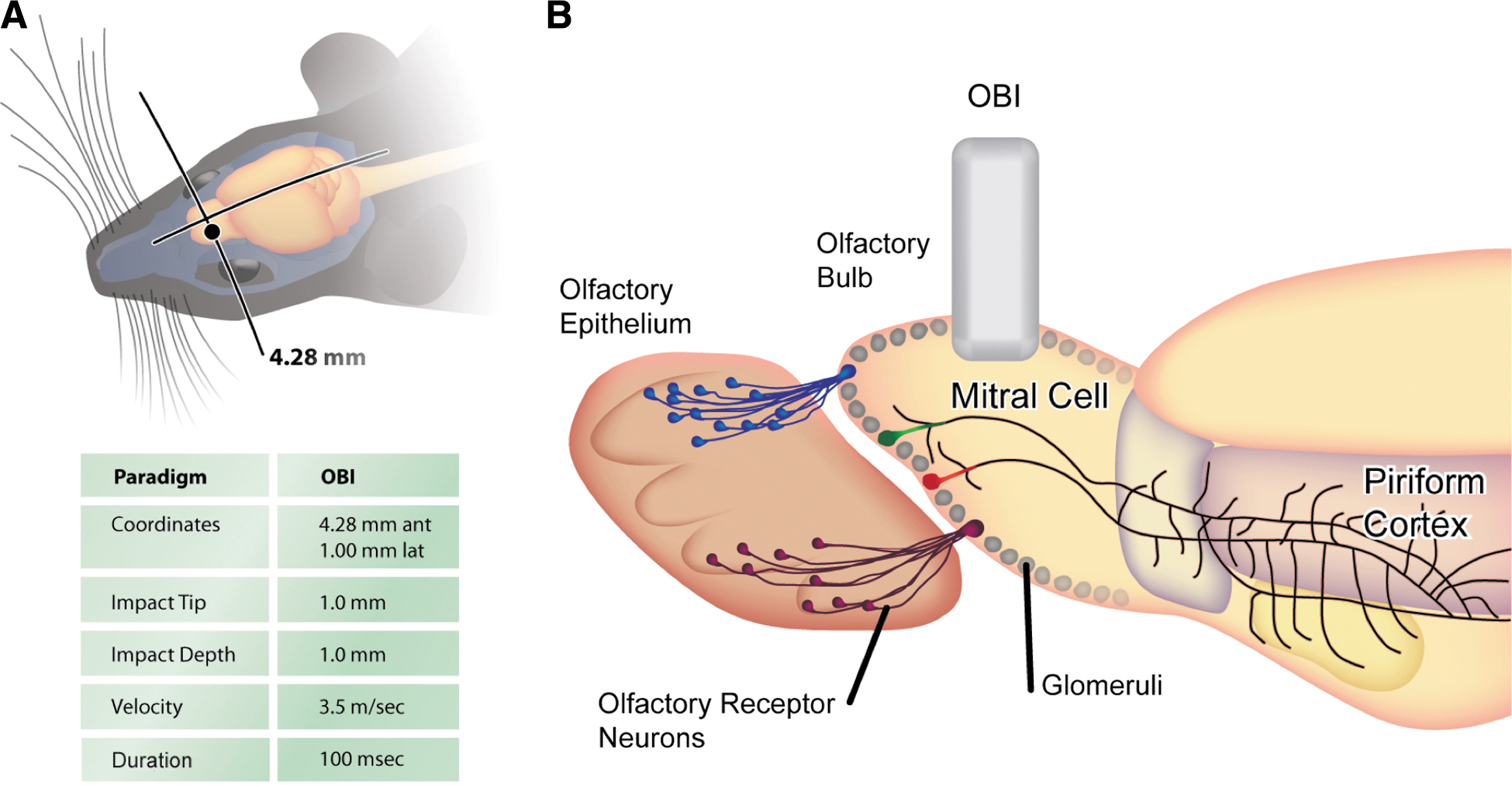
Schematic of experimental paradigm and injury parameters. (
We first sought to determine the degree of neural loss in the OE after an OBI. In Figure 2, we demonstrate loss of mature OSNs (GFP+) in a 250-μm region along the dorsal septum of the OE. OSN numbers were significantly reduced on the side projecting axons to the injured OB (ipsilateral) as compared to sham (p<0.00008), and also significantly less than the noninjured contralateral side (p<0.0000003). Although no significant difference was observed between the contralateral side of injured animals, and sham, some individuals appeared to have some cell loss. Low-power representative images of the OE show a robust reduction in the thickness of the mature OSN layer and reduced intensity of GFP signal on the ipsilateral side, as compared with the contralateral side (Supplementary Fig. 1) (see online supplementary material at
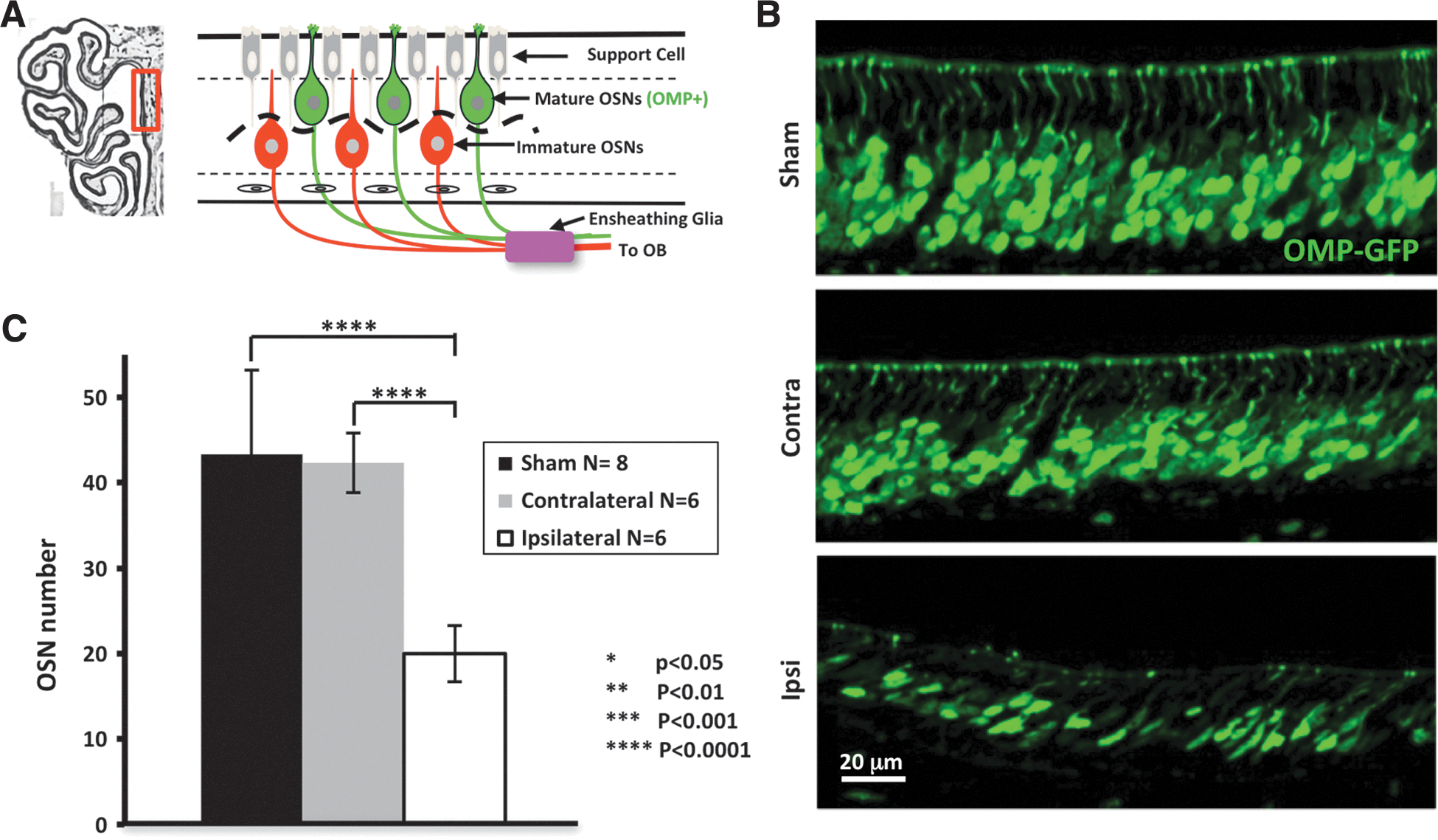
Olfactory sensory neuron loss after injury. (
TBI is characterized by cell death as a result of the initial mechanical insult followed by a cascade of secondary injury processes, such as excitotoxicity, OS, and inflammation. 3 To further evaluate the effect of OBI, we assessed the OE using a variety of molecular markers and associated neuronal processes. In Figure 3, we demonstrate the expression of various proteins associated with brain injury. Again, we consistently focused on an area of OE along the septum and compared marker expression in sham animals as well as the ipsi- and contralateral side of animals 4 days after undergoing an OBI.
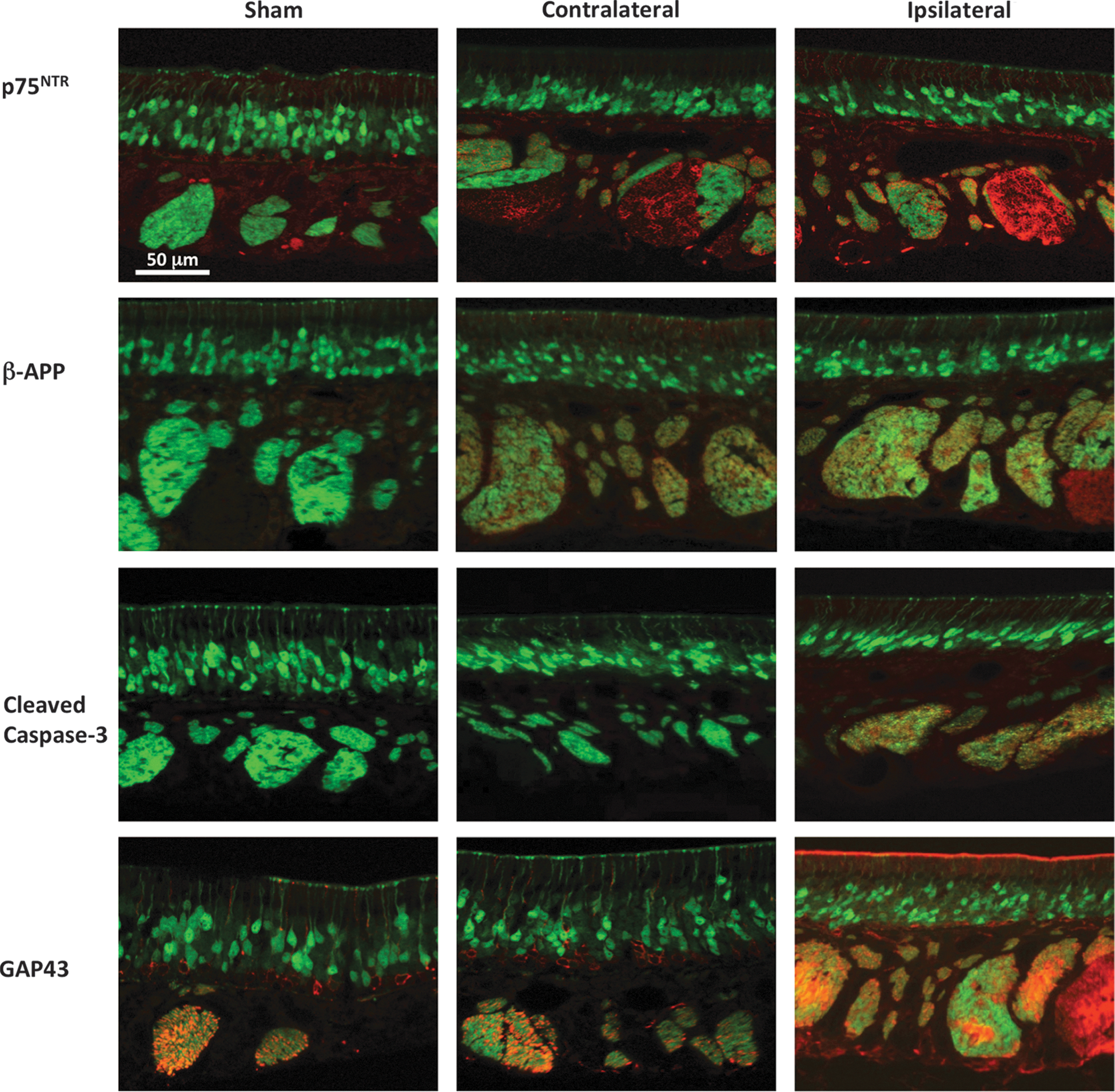
Markers of traumatic brain injury (TBI) in the olfactory epithelium 4 days post-OBI (olfactory bulb impact). Representative images from sham, contralateral, and ipsilateral olfactory epithelium (OE) are shown from left to right. p75NTR (
We performed IHC staining for the low-affinity neurotrophin receptor, P75 (p75NTR), which has been implicated in regulating neuronal survival. 26 As a proinflammatory cytokine tumor necrosis factor receptor, P75 has also been postulated to contribute to the neuropathology observed after TBI resulting from its up-regulation during inflammatory responses. 27 Blocking this pathway has also been postulated as a potential therapeutic target after TBI. 28 In Figure 3, we demonstrate p75NTR immunosignal in the OE, which was detectable in the nerve bundles and basal cells of OBI animals. Low levels of p75NTR were detected in the OE of sham animals.
The β-amyloid protein is formed after sequential cleavage of APP, a transmembrane glycoprotein. Evidence suggests that APP levels increase after TBI 29 and may stay elevated in certain regions of the brain, 30 potentially playing a role in subsequent neurodegeneration and delayed neural toxicity. As shown in Figure 3, we detected β-APP immunosignal in the nerve bundle layer of both the ipsi- and contralateral OE, whereas β-APP expression was not detected in the OE of sham animals.
Caspase-3 is a cystein protease that is activated by several different initiator pathways. Activation of caspase proteases has been strongly implicated in apoptotic processes after central nervous system (CNS) injuries, including TBI. 31,32 Not surprisingly, inhibition of caspase-3-mediated apoptotic pathways has been widely explored as a potential way to lessen degeneration after TBI. 33 As demonstrated in Figure 3, we detected cleaved caspase-3 in the nerve bundle layer of injured animals 4 days after OBI.
GAP-43, an intracellular membrane-associated phosphoprotein, is present throughout the entire population of olfactory receptor neurons in newborns and is found throughout the cells from their ciliated dendritic knobs to their axon terminals in the bulb. 34 With increasing age, this protein becomes progressively restricted to neural growth cones and presynaptic terminals, although it appears at elevated levels during development and regeneration. It has been implicated in neuronal growth, synaptic remodeling, and neuronal sprouting after injury, and this marker is often used as an indicator of neuronal plasticity. 35 In Figure 3, we show high levels of GAP-43 immunosignal 4 days after OBI. This marker was present in the nerve bundles and basal cells of injured, as well as sham, animals. On the ipsilateral side of OBI animals, the distribution of staining reverted back to a more newborn-like state, indicated by the intense GAP-43 signal in the dendritic knobs.
After determining the degree of cell loss and assessing protein expression in the OE after OBI, we examined the OB of animals after TBI and observed changes throughout the layers of the bulb. In Figure 4, we demonstrate a graded bilateral loss of OB neurons after injury, as determined by a loss of bulb area in distinct layers. Figure 4A shows the core site of the OBI, demonstrating tissue destruction in multiple layers of the ipsilateral OB after a 1-mm impact. We quantified cell loss in the glomerular layer (GL) and the exterior plexiform layer (EPL) of the OB by taking area measurements in the region directly adjacent to impact and the region ventral to impact. We found a graded cell loss in each area. In both layers adjacent to impact, cell loss was greatest in the ipsilateral OB (significantly more than both contralateral and sham), and cell loss in the contralateral OB was significantly greater than in sham animals. In both layers ventral to impact, cell loss was greatest in the ipsilateral OB (significantly more than both contralateral and sham), and cell loss in the contralateral OB was significantly more than in sham animals in the EPL, but not GL.
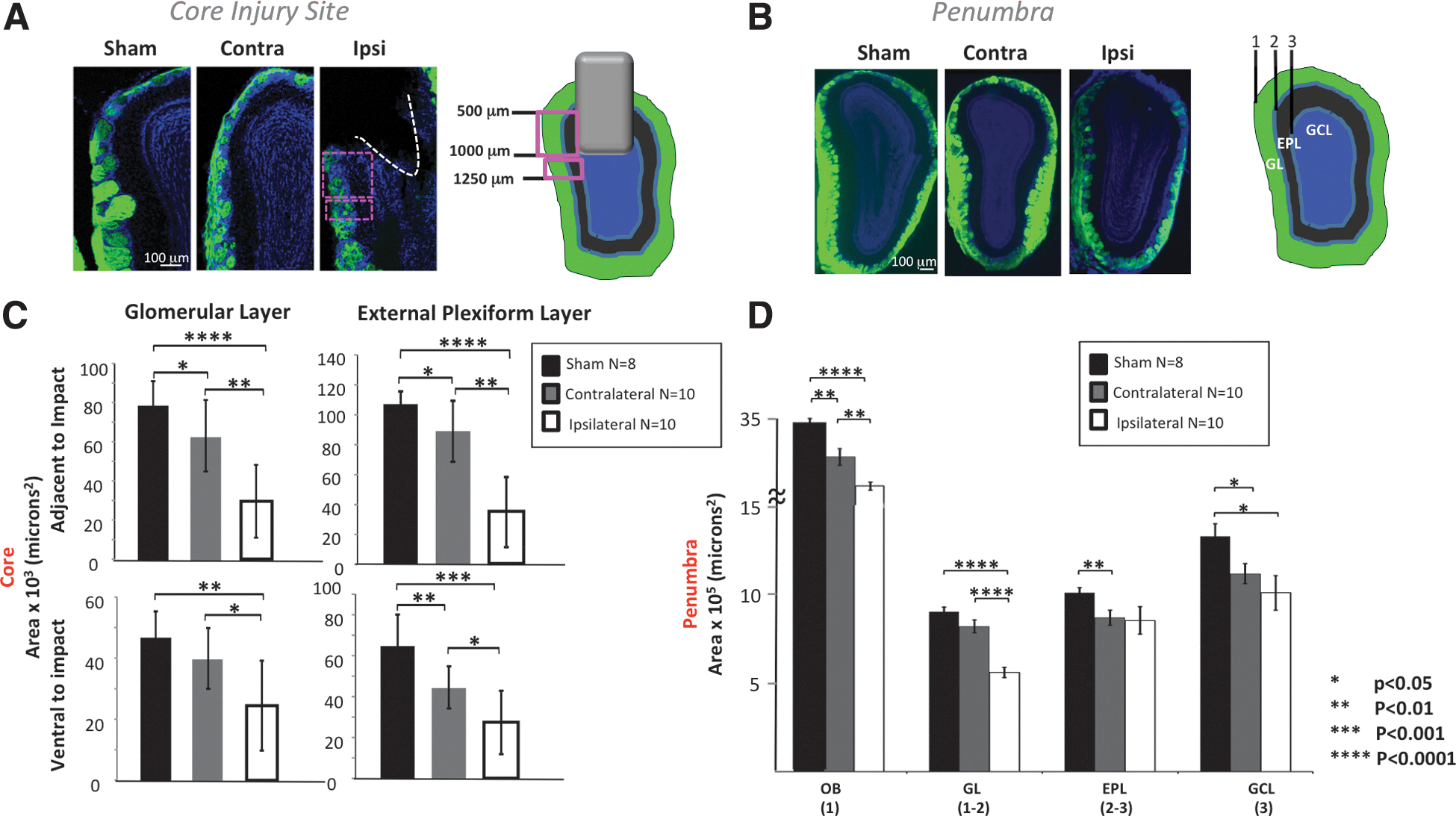
Bilateral loss of olfactory bulb (OB) neurons after injury. (
Figure 4B shows the penumbra, or region slightly anterior to the site (or path) of impact, where neuron loss was assessed by an area measurement in the overall OB, GL, EPL, and granule cell layer (GCL). We saw a graded cell loss in the OB (ipsilateral bulb area was significantly less than contralateral and sham, and contralateral bulb area was significantly less than sham). We saw a graded cell loss in the GL (ipsilateral bulb area was significantly less than contralateral and sham). We saw graded cell loss in the EPL (ipsilateral area had great variability, but was significantly less than sham, and although it trended toward less, contralateral EPL area was not significantly different than sham. We saw graded cell loss in the GCL (both injured OBs showed significant cell loss in this layer, compared to sham).
After demonstrating significant cell loss in multiple layers of the OB, we took a closer look by using various molecular markers associated with TBI. In Figure 5, we show changes in injury markers in the OB 4 days post-OBI. GFAP, an intermediate filament protein expressed mainly in astrocytes of the CNS, has been shown to be up-regulated in activated astrocytes and is an indicator of cell damage and inflammation.
36
We detected GFAP expression in both olfactory bulbs 4 days after OBI. On the contralateral side, GFAP signal was mainly found in the GCL (see also low-power representative bilateral image in Supplementary Fig. 2) (see online supplementary material at
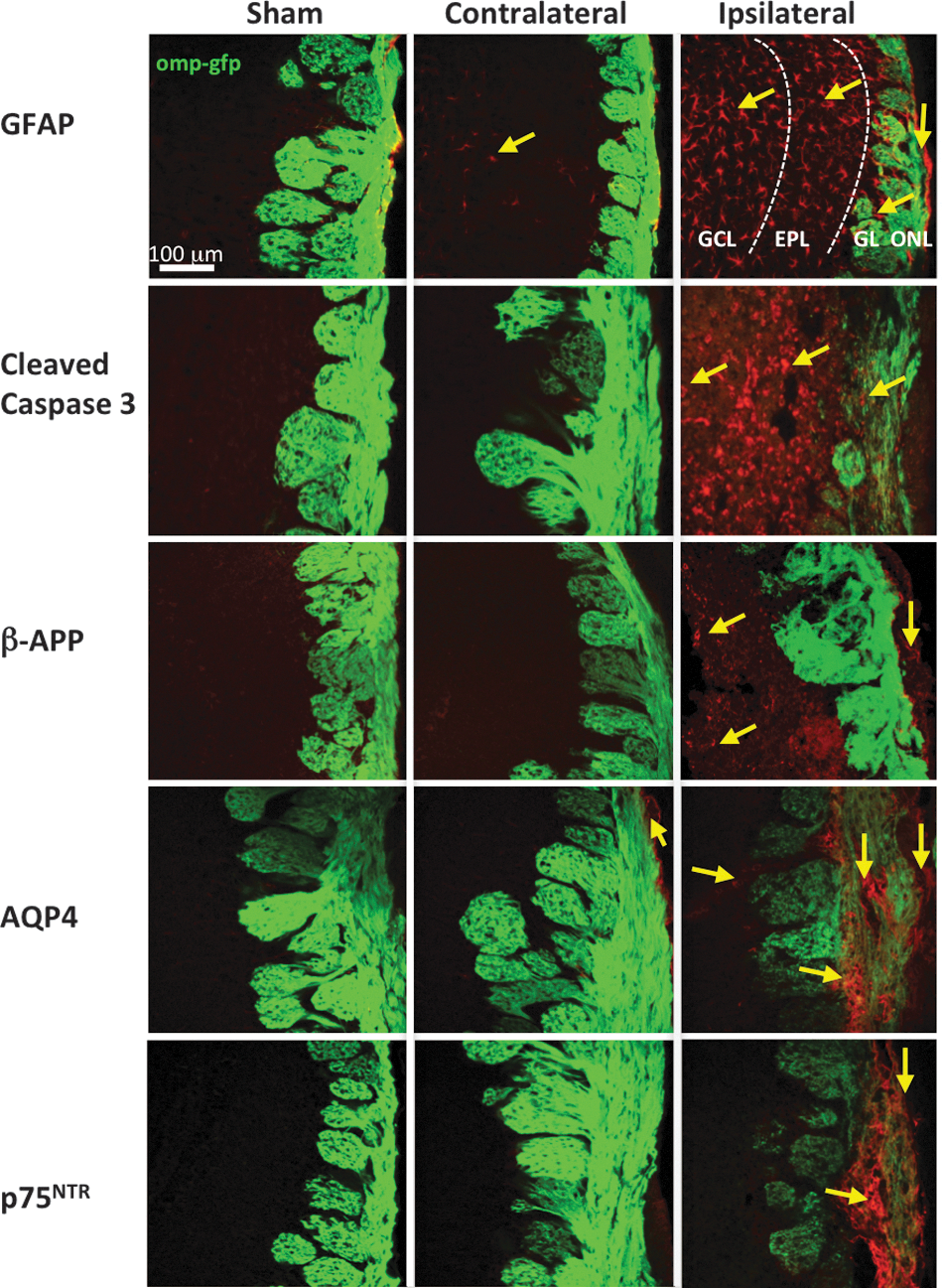
Markers of traumatic brain injury (TBI) in the olfactory bulb (OB) 4 days post-OBI (olfactory bulb impact). Representative images from sham, contralateral, and ipsilateral OB are shown from left to right. Glial fibrillary acidic protein (GFAP) (
We observed expression of the apoptosis signaling protein caspase-3 in OBI but not sham mice. Cleaved caspase-3 was detected in the GCL, EPL, and GL (see also low-power representative bilateral image in Supplementary Fig. 2) (see online supplementary material at
Recent studies suggest that AQP4, a water channel protein that facilitates the rapid transport of water, plays a significant role in the pathophysiology of brain edema after TBI.
38
AQP4 is usually found in astrocytic end-foot processes and ependymal cells in the brain and is up-regulated during edema.
39
To determine whether edema is also involved in olfactory system dysfunction associated with TBI, we assessed AQP4 expression in our OBI model. We observed AQP4 expression in the OB 4 days after OBI. The ONL of the contralateral bulb of impacted animals showed this marker. In the ipsilateral bulb, expression was detected in the GL and throughout the ONL (see also low-power representative bilateral image in Supplementary Fig. 2) (see online supplementary material at
Another marker of neural damage that was undetectable in olfactory neurons of sham animals was p75NTR. This molecular marker was highly expressed throughout the nerve layer of OBI mice (Fig. 5; see also low-power representative bilateral image in Supplementary Fig. 2) (see online supplementary material at
Given the significant loss of olfactory neurons after OBI and the presence of TBI protein markers, we suspected a correlated functional loss. Thus, to determine the effect of OBI on olfactory function, we used two different behavior assays. In the first, we took animals after undergoing OBI and performed a unilateral naris occlusion (right side), requiring them to use only the naris associate with the injured OB in behavior tests. In Figure 6A, we demonstrate a clear functional loss in mice 4 days after injury, as measured by the habituation/dishabituation assay (see Methods). Olfactory loss is illustrated by a significant difference in investigation time between sham and OBI animals on trial 4 of this assay (p<0.01). Thus, animals suffering a TBI did not dishabituate (no difference between trials 3 and 4) and showed a decreased sensitivity to odor (shorter investigation time between sham and injured animals). In Figure 6B, we used a buried food assay to further demonstrate olfactory functional loss in OBI animals, because they failed to locate the buried food after a 24-h period of food deprivation, indicating a decreased sensitivity to complex odors 4 days after injury (p<0.01). Taken together, these results demonstrate that the mouse olfactory system undergoes significant anatomical changes after TBI that cause a clear, measureable deficit in olfactory function.
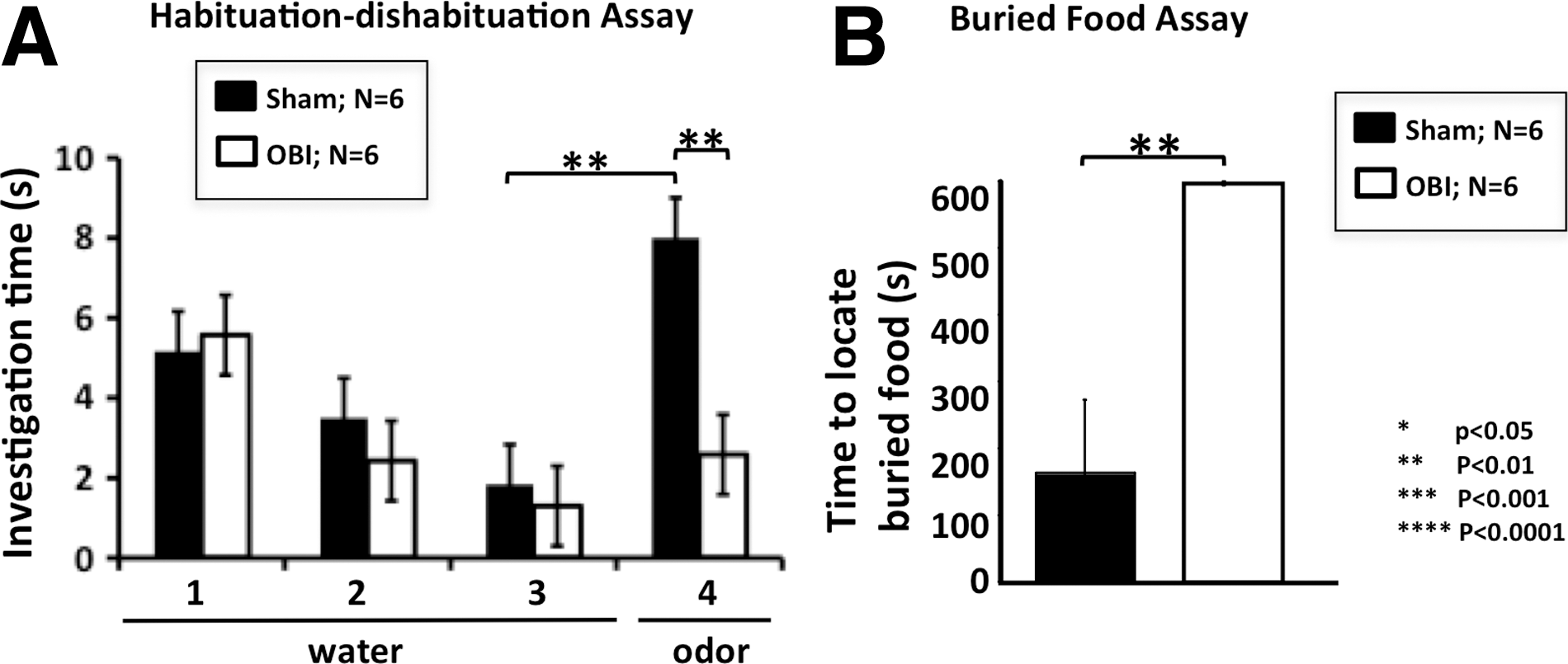
Olfactory functional loss 4 days post-OBI (olfactory bulb impact). Olfactory behavior assays indicate a clear loss of function after traumatic brain injury. (
Because neurons of the olfactory system are known to regenerate, we sought to determine whether a 30-day recovery period was sufficient to produce measureable restoration of the olfactory network after OBI. 40 We first assessed changes in the molecular markers that were previously observed 4 days after OBI. In Figure 7A, we show that p75NTR was still detectible at low levels in the OE. Similarly, β-APP expression was still detectible at low levels in the OE. By contrast, cleaved caspase-3 was no longer present in the OE 30 days after OBI, whereas GAP-43 was still highly detectable in both sides of the OE. In Figure 7B, we show changes in TBI marker expression in the OB after a 30-day recovery period. We observed that GFAP levels were observed on the contralateral OB of impacted animals, and GFAP expression shifted so that it was more detectable in outer layers of the ipsilateral bulb (glomerular layer and ONL). As with the OE, cleaved caspase-3 was not present in either ipsi- or contralateral OB after 30 days of recovery. AQP4 was still detected in both bulbs and was high in the ONL and core injury site on the ipsilateral side. We also found that p75NTR was still detectable in the ONL of the ipsilateral bulb. After observing these changes in protein expression level following this brief recovery period, we also examined recovery of circuitry in these animals by measuring OSN numbers and OB area at this same time point.
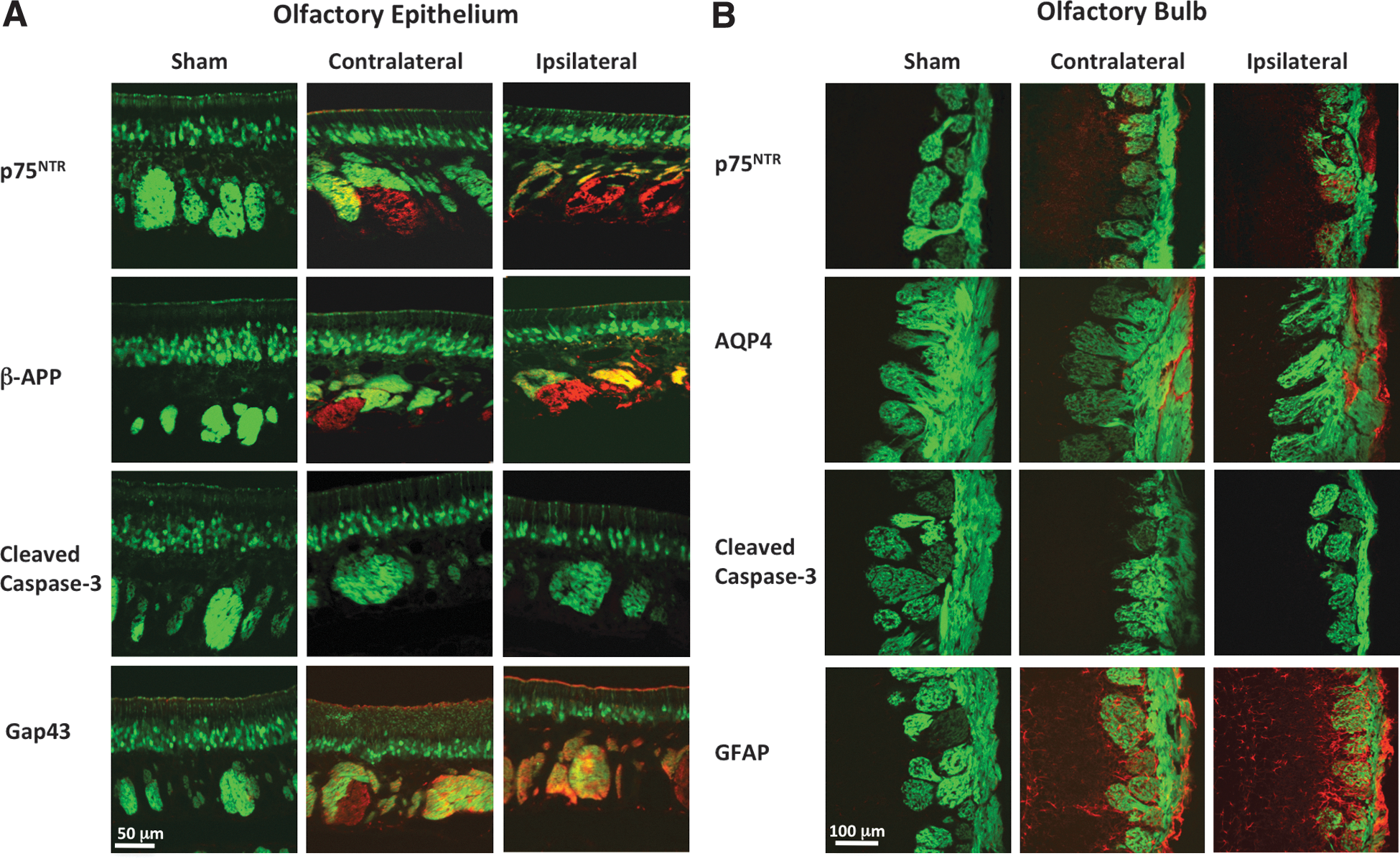
Markers of traumatic brain injury (TBI) in the olfactory epithelium (OE) and olfactory bulb (OB) after a 30-day recovery period. Representative images from sham, contralateral, and ipsilateral OB are shown from left to right. OE (
To measure the level of OE restoration, we counted OSNs (GFP+) along the dorsal septal region, revealing a clear increase in cell number after a 30-day recovery period from OBI. In Figure 8A, we show that both the contra- and ipsilateral sides have increased OSN numbers, with levels in the contralateral side even surpassing that of recovering sham animals. The ipsilateral side showed a 2-fold increase in the number of GFP-positive neurons, compared to 4-day post-OBI (see Fig. 2C). Interestingly, there was still a significant difference in OSN number between the ipsi- and contralateral side after 30 days of recovery (p<0.02). To quantify recovery in the OB after OBI, we took area measurements of the OB and glomerular layer, 30 days postinjury. In Figure 8B, we demonstrate a partial return of circuitry in the OB, as indicated by an increase in the overall areas of both the ipsi- and contralateral bulb. Despite this partial recovery (compare with Fig. 4D, OBI (1)), there was still a significant difference between the ipsilateral OB/contralateral OB and ipsilateral OB/sham animals (p<0.002; p<0.02). This partial recovery was also demonstrated by measuring the glomerular layer of the OB (compare with Fig. 4D, GL (1–2)), where we also still saw a significant difference between the ipsilateral OB/contralateral OB and ipsilateral OB/sham animals (p<0.0002; p<0.002).
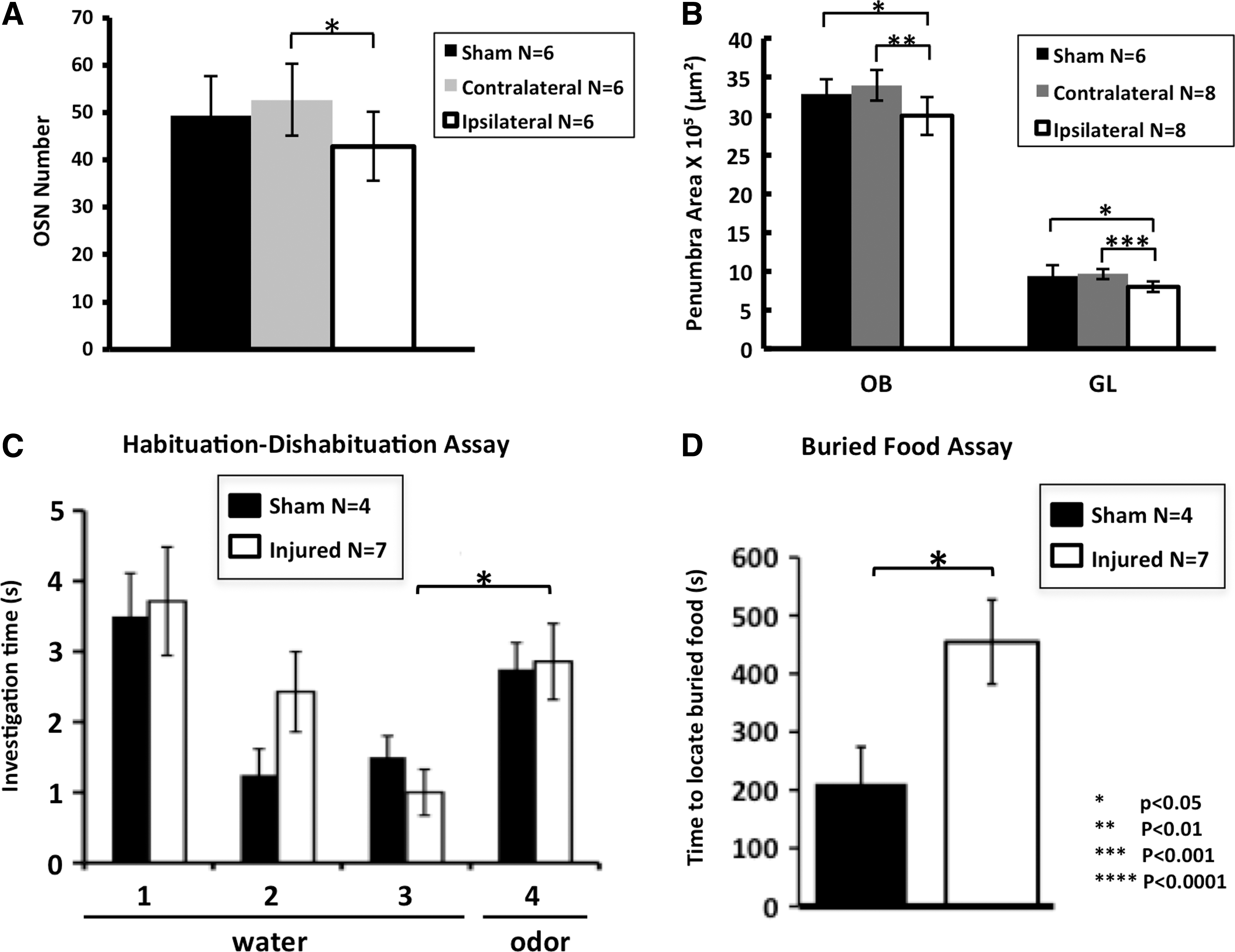
Partial return of olfactory circuitry and function after a 30-day recovery period. Histogram comparing olfactory sensory neuron (OSN) numbers of sham, contralateral side, and ipsilateral side (contra/Ipsi: p<0.02; compare with Fig. 2C). (
In order to examine functional recovery associated with these anatomical changes 30 days post-OBI, we again used the habituation/dishabituation assay and the buried food assay and found a partial return of olfactory function. In Figure 8C, we show that on trial 4 (trial in which novel odor is presented after period of habituation), sham and injured animals behaved nearly the same, exhibiting similar sensitivity to the odor stimulus after recovery (p<0.44; compare with Fig. 6A). In Figure 8D, we further demonstrate improvement in olfactory functioning using the buried food assay; though still significantly different than sham (p<0.03), some animals were able to locate the food after 30 days of recovery, whereas all animals failed in this task at 4 days after injury (see Fig. 6B). These experiments demonstrate a partial return of olfactory function that correlates with changes in olfactory circuitry after a brief recovery period.
Studies have shown that the olfactory system maintains a high level of neural plasticity throughout adulthood that is activity dependent and responds to environmental odors. 41 To investigate plasticity after TBI in the olfactory system, we utilized M71-IRES-tauLacZ reporter mice (Tg line that coexpresses β-galactosidase in OSNs that express the M71 odorant receptor) to test the effect of odorant-induced activity on olfactory circuitry recovery. Given that acetophenone has been identified as a ligand for the M71 receptor, 21 after unilateral OBI, we exposed M71-IRES-tauLacZ mice to 30 days of acetophenone odor conditioning. In Figure 9, we report differences in M71 OSN number and glomerular size. We chose to quantify changes in the M71 medial glomerulus on the contralateral (uninjured side) since the impacted bulb typically suffers extensive dorsal tissue damage and complete ablation of the M71 glomerulus, which was still absent after 30 days of recovery. We found that data trends for OSN number paralleled those for glomerular size among the four groups. Animals undergoing an OBI without odor conditioning had the largest glomerular size, as compared to all other groups, with a significantly greater size than sham animals that received 30 days of odor conditioning (p<0.02). Animals undergoing OBI without odor conditioning had a significantly greater OSN number than all other groups. The greatest differences were observed between the OBI/no-odor group and control animals (no injury or odor exposure; p<0.004) and between OBI/no-odor and sham/odor (p<0.001). Control and sham animals receiving odor conditioning had the smallest glomerular size as well as OSN number. Overall, these data demonstrates that OBI results in an increase of M71-positive OSNs and M71 glomerulus size on the side opposite the impact after 30 days of recovery, and this increase may be inhibited or suppressed by strong odor-induced activity.
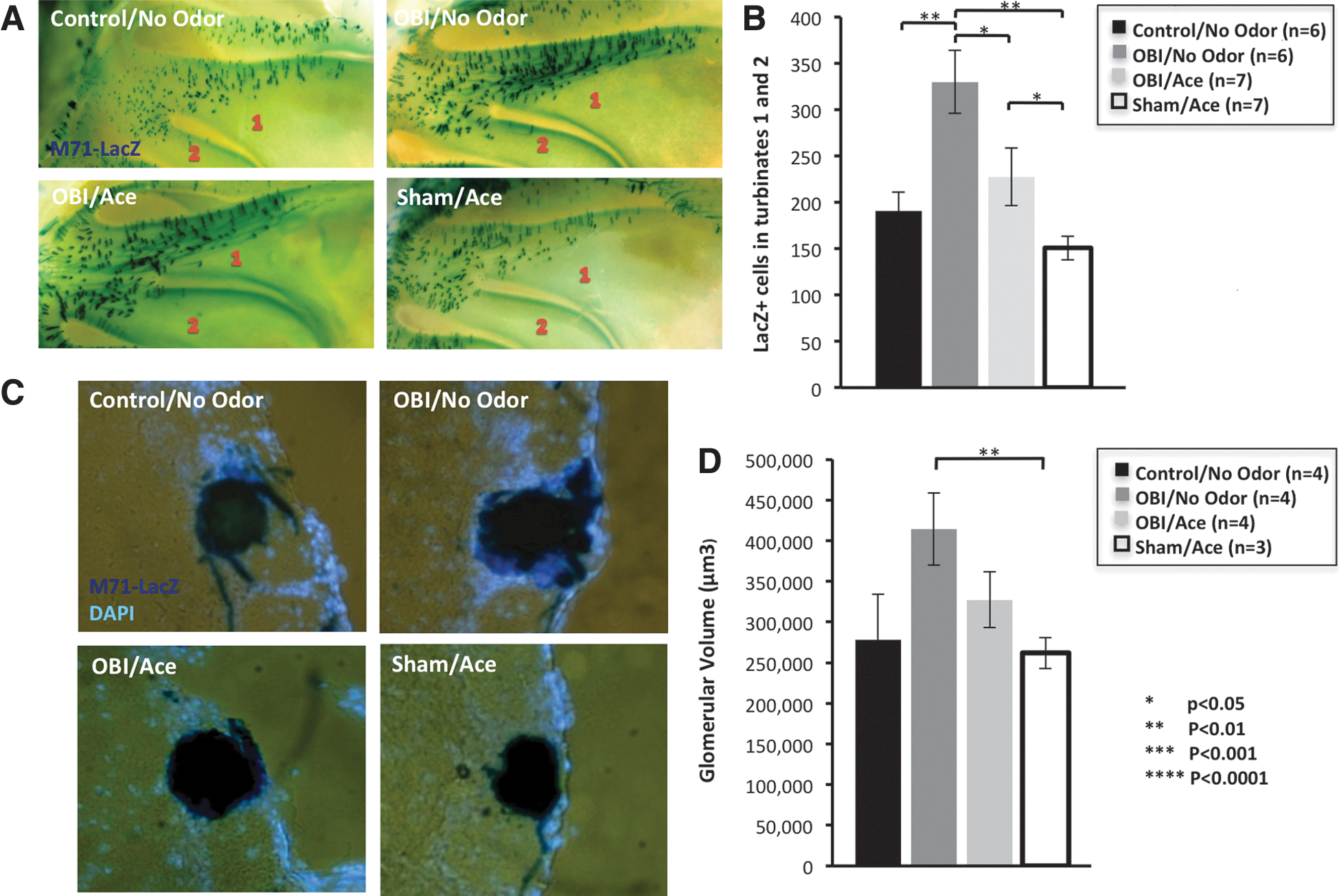
Effect of odor-induced activity on olfactory system recovery after TBI. Images show X-Gal staining of M71+ olfactory sensory neurons (OSNs) in turbinates 1 and 2 of M71-IRES-tauLacZ mice after 30 days of 10% acetophenone conditioning after injury. (
Taken together, the results of this study establish the olfactory system as a useful model to study TBI. We have shown that, 1) a penetrating unilateral OBI significantly alters olfactory circuitry, causing widespread changes in protein expression and associated deficits in olfactory functioning, 2) changes in TBI molecular markers and improvement in olfactory functioning after a 30-day recovery period, and 3) introducing odor activity during this recovery period may not be beneficial to plasticity processes that occur after TBI in the olfactory system.
Discussion
In the present study, we establish the mouse olfactory system as a new model to study TBI. We directly impacted the dorsal surface of the mouse olfactory bulb and found that this penetrating injury resulted in a loss of mature OSNs on the ipsilateral side and a graded loss in adult cells in several layers of the OB, both at the core injury site as well as in the penumbra region. Various nerve injury models have been used to investigate neural recovery and regeneration of the olfactory system after brain injury. Mild and Severe TBI has been examined by passing a blade through nerve fibers between the OB and cribriform plate. 42 Whereas this model employs a direct surgical transection and ours uses an indirect shearing of the nerves resulting from a shifting force applied to the dorsal surface of the OB, we both find functional recovery despite incomplete anatomical restoration. This common finding through complementary models strengthens the interpretation that persistent olfactory dysfunction may not be solely the result of loss of OSN connectivity.
Regardless of the specific nature of the injury, these olfactory models make it possible to measure specific molecular and anatomical changes to a defined circuitry after TBI and then assess the functionality of those circuits through innate behaviors. We examined changes in protein expression throughout the OB and OE and used two innate olfactory behavior assays to show a loss in olfactory functioning after OBI, with injured animals displaying significantly diminished odor sensitivity. We examined changes in olfactory circuitry after a brief recovery period and found that some molecular markers were still detectable, and some shifted to different parts of the OB or OE. We demonstrated a partial restoration of olfactory circuitry and functioning after 30 days of recovery that is hindered by odor exposure during this time period.
TBI can result from an abrupt acceleration and deceleration of the brain (a rapid change in direction causing the brain to collide twice with the skull, known as a coup/contra-coup injury), blunt force trauma (such as a fall), pressure waves (blast-induced injury), or a penetrating force on the brain tissue (such as a bullet). The current general model of TBI posits that after an injury, neuronal degeneration occurs in a biphasic manner with primary and secondary processes defining the pathology. 43,44 The primary process reflects the initial mechanical insult to the brain and is characterized by a deformation of tissue and degradation of cell membranes. 45 A secondary process involves diverse biochemical and molecular cascades that further cell death, damage surrounding structures, and impair functionality. Progressing from hours to days after the initial trauma, this is largely a result of injury of the cerebral vasculature. 46 Also, multi-organ damage in trauma patients can lead to elevated circulatory levels of inflammatory cytokines that may contribute to the postinjury pathogenesis of the brain. 47 The specific nature of these events is dependent on the location and severity of the injury, but it is often accompanied by vascular damage, 48 alterations in neurotransmitter release, such as calcium- and acetylcholine-signaling changes, 49,50 activation of microglia and macrophages through inflammatory responses, 51 blood–brain barrier disruption, 52 and edema. 53
Although circuit disruption, functional deficits and symptomology after TBI vary based on the location and severity of the injury, anosmia and hyposmia have been found to be early and fairly common indicators of head trauma. Several group studies have found that a majority of people suffering from TBI exhibit anosmia (ranging from 56% to 67%), whereas others show hyposmia (20–23%). 6,7 Interestingly, there are recorded cases of very mild TBIs that cause complete and permanent anosmia, whereas some severe injuries have resulted in no smell deficit or a brief case of anosmia. 5 This variance suggests that many factors, such as injury location, severity, and postinjury environment, interact to determine whether a person will suffer from anosmia or hyposmia after head trauma. Further, some studies have revealed that among TBI patients suffering from anosmia, only a small portion (10%) show functional improvement within 1 year. 10,54 Although the likelihood of recovery has been observed to decrease with time after injury, there was a reported case of a patient that recovered from complete anosmia 9 years after a severe TBI. 55 Taken together, these studies of anosmia prevalence and recovery rates suggest that the nature of olfactory-deficit–inducing damage varies greatly and should be studied more extensively to implement effective diagnostic and treatment strategies after TBI.
More specifically, a better understanding of the specific time course of up-regulation and clearance of injury markers would be useful to identify the window of opportunity for various pharmacological and behavioral treatments. The aforementioned protein markers that can be observed in the OE, OB, and throughout several other regions of the brain after injury are dynamic, with levels changing rapidly in the brain after injury. 56,57 Further, several of these markers are detectible in the blood and cerebrospinal fluid of TBI sufferers. 58 Specifically, several studies have suggested that TBI patient GFAP and S100B serum levels are useful indicators of neuronal damage as well as predictors of outcome in clinical trials. 59 Using the olfactory system model to study these and other biomarkers that may be detectable in human fluids, in combination with refined olfactory function assays, could lead to great advances in the diagnosis and prognosis of TBI.
To date, most animal TBI studies have focused on cortical and hippocampal regions of the brain, because these areas are implicated in many of the learning, memory, and motor dysfunctions that follow brain injuries. Studies have shown that, in a CCI model, injury results in elevated caspase activation within the first hours after TBI, 60 and subsequent increased β-APP levels have been linked with a heightened risk for the development of Alzheimer's disease. 61 Further, studies examining the role of molecular changes associated with TBI have highlighted that pharmacologically or genetically altering expression of multiple proteins and signaling pathways could benefit long-term TBI outcome. For example, one study found that blocking caspase signaling suppresses apoptosis and the subsequent β-APP accumulation that has been found to play a role in dementia. 62 Another study demonstrated that modulation of p75NTR signaling constituted a potent therapeutic strategy for TBI. 63
Water channel proteins (specifically aquaporins) and associated edema after TBI have been examined as a therapeutic target in the prevention of brain swelling 64,65 to aid in recovery. AQP4, expressed mainly in the astrocytic end-feet processes of the brain, was examined in the present study and found to be present in the OB for up to 30 days after impact. Some studies have shown that inhibiting AQP4 expression leads to reduced cerebral infarction and better outcomes after neural insult. 66 One study found that intranasal administration of nerve growth factor after TBI attenuated the edema response and decreased cell death and associated this with a marked decrease in AQP4 content. 67 Other studies suggest a detrimental effect of AQP4 inhibition, 38 and a critical role in clearance of interstitial solutes, including amyloid-β from injured brains. 68 Thus, the specific type of injury, postinjury environment, and nature of the edema itself should be closely examined in the implementation of aquaporin-targeted interventions.
GAP43 is expressed in immature neurons and has been examined in several CCI-TBI studies as an indicator of axonal plasticity and regeneration. 69 It has been found to be up-regulated (peaking at 24–48 h) in the hippocampus and cortex after moderate, but not severe, TBI 70 ; therefore, this protein not only has the potential for use as an indicator of the severity of trauma, but also illuminates the capacity for recovery after injury. Given that we saw a dramatic and long-lasting up-regulation of GAP43 in our OBI model, this also shows that the olfactory system possesses great potential for studies of regeneration after TBI. More broadly, our 30-day recovery studies, showing partial circuit restoration through quantification of OMP-GFP OSNs and OB layers, demonstrate that different parts of this circuitry have varying capacities for recovery after TBI, but changes in this highly dynamic system can be observed through protein marker changes as well as OSN and OB layer regeneration
As previously mentioned, most TBI studies have focused on hippocampal and cortical regions of the brain. Even given the neurogenesis observed in regions of the hippocampus, use of this structure is somewhat limited by the complexity of its neural maps. Alternatively, the mammalian olfactory system possesses clear, anatomically distinct structural layers in the OB, simple molecular maps based upon odorant receptor expression, and odor columns that define an intrabulbar map. 18 Additionally, this system possesses great plasticity and a unique ability to regenerate relatively rapidly in an activity-dependent manner. 71,72 It has also been found that olfactory map plasticity has no critical period and extends throughout adulthood, allowing for TBI recovery and regeneration studies in animals of all ages. 71 Importantly, the olfactory system's restorative ability offers an advantage for repair studies, but the findings of these studies may not be limited to areas that regenerate.
Despite the increased recognition and prevalence of TBI, the pathogenesis of TBI-induced brain injury is still poorly understood and we lack effective treatments. The complex nature and progression of TBI-induced pathology limits treatment options to either blocking the secondary injury phase or facilitating plasticity and repair at certain optimal time points. In studying the effectiveness of various treatments, the olfactory system may lend unique benefits. To gain a better understanding of the time course of secondary phase progression and the interplay of injury biomarkers, we utilized the simple circuitry of the olfactory system with its well-defined layers that allow for good visualization of these processes. Because olfaction is a dynamic sensory system in which new cells are continually incorporated into the circuitry in an activity-dependent manner, 72 studying the mechanisms by which this plasticity is enhanced could provide broad insight that extends to other areas of the brain.
Additionally, simple smell assays, such as the tests performed in the present study, could be conducted to assess return of functioning after TBI as various pharmacological treatments are implemented. For example, studies have shown that nicotinamide, a B-group vitamin, is effective in both reducing cortical infarct size and facilitating recovery of functioning when administered both shortly after CCI and at longer time points. 73,74 Whereas these studies utilize working memory tasks to assess behavior improvements induced by administration of the drug, the simple olfactory tests that our model uses could track functional improvements and shed more light on the ideal window for administration of neuroprotective drugs to maximize outcome.
Other neuroprotective drugs found to facilitate anatomical and behavioral improvements after TBI target detrimental processes in the neurovasculature. Secondary injury processes in endothelial cells exacerbate damage; specifically, as the BBB is broken down, excessive superoxide reacts with nitric oxide to form peroxynitrite, which hinders recovery. 75,76 Treatment with S-nitrosoglutathione (GSNO), a nitrosylation-based signaling molecule, has been shown to reduce brain levels of peroxynitrite and improve behavioral outcome, as indicated by improved performance on the sensorimotor deficit behavioral test and rotarod test. 76 Again, our olfactory system model would be useful in similar studies of recovery from TBI-induced neurovascular damage. These studies demonstrate that long-term treatment of TBI with a low dose of GSNO promotes plasticity and enhances the expression of various neurotrophic factors leading to functional improvement. In the olfactory system, damaged axons can regenerate to form new connections on OB glomeruli, showing easily traceable targeting fibers and associated changes in glomerular size. This could be used as a simple measure of drug-enhanced plasticity in the CNS after TBI, accompanying easily measurable olfactory functional improvements.
The subventricular zone (SVZ) along the walls of the lateral ventricles continuously produces new neurons in adulthood,
77,78
and these cells travel along the rostral migratory steam (RMS) to reach the OB. Further, peripheral OSNs are continuously replaced in adulthood by new OSNs generated from local progenitors in the epithelium. These dynamic processes are influenced by sensory experience and activity; the continuous addition of new cells provides a level of plasticity that allows circuits to adjust to changing environments.
78,79
TBI drastically changes the physiology of the brain; the initiation of apoptotic processes that lead to cell death after injury is accompanied by up-regulating of signaling molecules that aid in regeneration and repair. Previous studies have shown that lesions produced by an injection of N-methyl-
Some studies have reported that activity-dependent plasticity is related to an increased activation of various neurotrophic factors. Specifically, brain injury has been shown to increase growth-promoting molecules, which may lead to enhanced cell proliferation in a compromised system. One study showed that a fluid percussion brain injury results in significant increases in several intracellular signaling proteins, such as synapsin 1, calcium-calmodulin–dependent protein kinase 2, and phosphorylated mitogen-activated protein kinase 1 and 2. 84 Interestingly, this study also demonstrated that acute voluntary exercise decreased expression of these proteins (including cyclic adenosine monophosphate response-element-binding protein), failed to increase brain-derived neurotrophic factor levels (as exercise does in an uninjured brain) and severely worsened outcome for rats undergoing TBI. This suggests that the injured brain experiences a period after insult during which activation of the system may be detrimental to recovery.
As previously mentioned, inducing activity with odor conditioning has been shown to dynamically change the circuitry of the olfactory system. We showed that intense odor conditioning with acetophenone for 30 days after OBI results in an inhibition of the up-regulated plasticity in the olfactory system that appears to follow TBI, as demonstrated by fewer OSNs and smaller glomerular size in odor-conditioned animals. Our results are in line with the concept that TBI induces plasticity signals that aid in recovery, but that the injured system may have difficulty repairing itself with the addition of heightened activity soon after injury. Various other interventions have been studied extensively in an effort to shed light on the best way and time to facilitate anatomical and functional recovery from TBI. As such, dietary changes/nutritional supplementation 85,86 and exercise regimens 87,88 are further examples of possibilities that may be promising to enhance plasticity and repair in the CNS after trauma, but are sensitive to the specific nature of the injury and time windows of implementation. This posits that further research is important to determine the proper type of activity and time window during which intervention aids in recovery.
In summary, we show that the overall experimental accessibility of the olfactory system offers great potential for studies that can shed light on pharmacological and behavioral therapeutics for TBI. Ultimately, our olfactory system model presents three unique opportunities for TBI studies: changes in protein expression within easily accessed and well-defined neural maps; olfactory functionality deficits that can be assessed using simple smell tests; and a regenerative capacity not seen anywhere else in the mammalian brain that opens the door for studies of recovery from TBI.
Footnotes
Acknowledgments
This work was supported by the Center for Neuroscience and Regenerative Medicine (HJF# 60855–CNRM) and the National Institute of Neurological Disorders and Stroke Intramural Research Program (ZIANS003116-02).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.