
Abstract
Traumatic brain injury (TBI) initiates a neuroinflammatory cascade that contributes to neuronal damage and behavioral impairment. In the present study, we performed a widely used model of TBI to determine the neuroprotective propriety of palmitoylethanolamide (PEA) and the antioxidant effect of a flavonoid luteolin (Lut), given as a co-ultramicronized compound Co-ultraPEALut. We demonstrated that the treatment with Co-ultraPEALut resulted in a significant improvement of motor and cognitive recovery after controlled cortical impact, as well as markedly reducing lesion volumes. Moreover, our results revealed the ability of Co-ultraPEALut to reduce brain trauma through modulation of nuclear factor-κB activation. In addition, treatment with Co-ultraPEALut significantly enhanced the post-TBI expression of the neuroprotective neurotrophins glial cell line-derived neurotrophic factor compared with vehicle. Co-ultraPEALut at the dose of 1 mg/kg also modulated apoptosis, the release of cytokine and reactive oxygen species, the activation of chymase, tryptase, and nitrotyrosine, and inhibited autophagy. Thus, our data demonstrated that Co-ultraPEALut at a lower dose compared with PEA alone can exert neuroprotective effects and the combination of both could improve their ability to counteract the neurodegeneration and neuroinflammation induced by TBI.
Introduction
T
TBI induces a strong inflammatory response characterized by the recruitment of peripheral leukocytes and enhanced chymase, tryptase as well as nitrotyrosine activation into the cerebral parenchyma. 6 –8 Moreover, activated microglia migrate to injured sites and releases cytokines, chemotactic cytokines, reactive oxygen species (ROS), nitric oxide (NO), proteases, and other factors with cytotoxic effects, which may in turn exacerbate neuronal death. 9,10 Also, a close relationship exists between the degree of oxidative stress and the pathogenesis of TBI. 11 Enhanced production of ROS and reactive nitrogen species (RNS) causes oxidative/nitrosative stress leading to damage in lipids, 12,13 proteins, and nucleic acids. 14,15 In addition, Lai and colleagues 16 have demonstrated that oxidative stress contributes to overall autophagy in mice after TBI and that this pathway was partially inhibited by an antioxidant.
Autophagy, from Greek “self-eating,” is a major protein degradation system that targets primarily long-lived cytoplasmic proteins. 17 Autophagy involves a dynamic subcellular membrane rearrangement whereby cytoplasmic proteins and organelles are sequestered in double-membrane vesicular structures (autophagosomes) that fuse with the lysosome or vacuole where the material is catabolically degraded, recycled, and used as an energy source. 18 Several key molecular components participate in the initiation, progression, and completion of autophagy, such as the mammalian target of rapamycin (mTOR) that inhibits autophagy whereas Beclin 1 and light chain (LC)3 promote it.
In recent years, the pathophysiology of TBI has been a focus of extensive studies; animal models have been proved to be important tools in this field and are used to investigate the mechanisms of primary and secondary injury. Evaluation of improvement in neurological function using a clinically relevant TBI model such as controlled cortical impact (CCI) establishes efficacy and clinical relevance of an experimental therapy. 8 In the CCI model, the injury to the brain initially presents as necrotic cell death in the underlying tissue and white matter axonal injury, both reminiscent of the clinical TBI pathology, but also followed by apoptotic cell death in surrounding tissue because of multiple subsequent events such as edema, ischemia, excitotoxicity, and altered gene expression. 19 Our previous works clearly demonstrated that treatment with palmitoylethanolamide (PEA) at 10 mg/kg significantly reduced the inflammation process associated with experimental spinal cord injury (SCI) 20 and TBI mice models. 8
PEA is abundant in the central nervous system (CNS), and it is conspicuously produced by glial cells. 21 –23 It has been shown to inhibit peripheral inflammation and mast cell degranulation, 24 as well as exert antinociceptive effects in rats and mice. 25,26 PEA, however, lacks a direct antioxidant capacity to prevent the formation of free radicals and to counteract the damage of DNA, lipids, and proteins.
Luteolin (Lut), a common flavonoid present in many plants, has strong antioxidant and pharmacological activities, including a memory-improving effect. It displays excellent radical scavenging and cytoprotective properties, particularly when tested in complex biological systems where it can interact with other antioxidants, such as vitamins. 27 Lut displays specific anti-inflammatory effects, which are only partly explained by its antioxidant capacities. The anti-inflammatory activity of Lut includes activation of antioxidative enzymes, suppression of the nuclear factor (NF)-κB pathway and inhibition of pro-inflammatory substances. In vivo, Lut reduces increased vascular permeability and is effective in animal models of CNS inflammation. 28,29
In our study, we aimed to investigate the capability of Co-ultraPEALut, based on association of PEA with Lut in fixed doses of 10:1 in mass to counteract the secondary damage produced after TBI, by analyzing the neuroprotective properties of this new Co-ultramicronized combination formulation.
Methods
Animals
Male CD1 mice weighing 25–30 g were kept five per cage under a constant 12-h light/dark cycle, at room temperature (23°C). Food and water were available ad libitum. Animal care was in compliance with Italian regulations on protection of animals used for experimental and other scientific purposes (D.M.116192) as well as with the EEC regulations (O.J. of E.C. L 358/1 12/18/1986).
CCI experimental TBI
Surgical anesthesia was induced by ketamine and xylazine (2.6 and 0.16 mg/kg body weight, respectively) administered intraperitoneally (i.p.). After endotracheal intubation, the animals were secured in a stereotaxic frame and ventilated mechanically. A dab of sterile ophthalmic ointment was placed on each eye to compensate for the decrease in lacrimation during anesthesia. Using aseptic techniques, a midline scalp incision was made, and the skin and fascia were reflected to expose the skull. A craniotomy was made in the right hemisphere encompassing bregma and lambda and between the sagittal suture and the coronal ridge with a micro motor hand piece and drill (UGO Basile S.R.L., Comerio VA, Italy). The resulting bone flap was removed, and the craniotomy enlarged further with cranial rongeurs.
A cortical contusion was produced on the exposed cortex using a controlled impactor device Impact One™ Stereotaxic impactor for CCI (myNeurolab.com, Richmond, IL). Briefly, the impacting shaft was extended, and the impact tip was centered and lowered over the craniotomy site until it touched the dura mater. Then the rod was retracted and the impact tip was advanced farther to produce a brain injury of moderate severity for mice (tip diameter, 4 mm; cortical contusion depth, 3 mm; impact velocity, 1.5 m/sec). The impact tip was wiped clean with sterile alcohol after each impact and cleaned/disinfected further with Cidex® after surgery. Core temperature was maintained at 37±0.5°C with a heating pad during surgery and recorded with a rectal probe. Immediately after injury, the skin incision was closed with nylon sutures, and 2% lidocaine jelly was applied to the lesion site to minimize any possible discomfort.
Experimental groups
TBI was induced in mice (n=10 for group) by controlled cortical impactor.
Immediately after injury, the skin incision was closed with nylon sutures, and 2% lidocaine jelly was applied to the lesion site to minimize any possible discomfort.
Two sets of experiments were performed. The first set was to investigate the protective effects of PEA and Co-ultraPEALut treatment. All animals were randomized into one of six groups: 1. Sham+vehicle group: mice were subjected to the surgical procedures (anesthesia and craniotomy) except that the impact tip was not applied and vehicle was administered 1 h after craniotomy. 2. Sham+Co-ultraPEALut: mice were subjected to the surgical procedures as the group above (anesthesia and craniotomy) except that the impact tip was not applied and Co-ultraPEALut (1 mg/kg body weight, soluble 10% ethanol, i.p.) was administered 1 h after craniotomy. 3. Sham+PEA: mice were subjected to the surgical procedures as the group above (anesthesia and craniotomy) except that the impact tip was not applied and PEA (10 mg/kg body weight, soluble 10% ethanol, i.p.) was administered 1 h after craniotomy; 4. TBI+vehicle: mice were subjected to CCI and vehicle was administered 1 h after TBI. 5. TBI+Co-ultraPEALut: mice were subjected to CCI and Co-ultraPEALut (1 mg/kg body weight, soluble 10% ethanol, i.p.) was administered 1 h after TBI. 6. TBI+PEA: mice were subjected to CCI and Co-ultraPEALut (10 mg/kg body weight, soluble 10% ethanol, i.p.) was administered 1 h after TBI.
Mice (n=10 from each group for each parameters) were killed at 24 h after TBI to evaluate the various parameters. In a second set of experiments, other 10 animals for each group were observed after TBI to evaluate the behavioral testing.
Co-ultramicronization process of PEA and Lut
The Co-ultramicronization process was performed using jet mill equipment (Sturtevant Inc., Hanover, MA) with a chamber of 300 mm in diameter, operated with “spiral technology” and driven by compressed air at 10 to 12 bars. Crashing was determined by the high number of collisions that occurred among particles as a result of the high level of kinetic (not mechanical) energy. This process is effective not only in reducing product particle size, but also in modifying crystalline structure. Scanning electron microscopy showed an intimate intermixing of PEA and Lut, while analysis by differential scanning calorimetry and X-ray diffraction indicated transformation into a crystalline form different from the original two, definable as “a higher energy content form.” The composite showed the following particle size distribution: 96% <10 μm; 80% <5 μm; and 40% <2 μm. Co-ultraPEALut (Epitech Group s.r.l.) was dissolved in Pluronic F68 (Sigma-Aldrich, St Louis, MO) and used at a concentration of 1 mg/kg.
Behavioral testing
TBI mice display motor and cognitive deficits. Thus, the present behavioral tests that involved analyses of motor asymmetry were elevated biased swing test (EBST) and rotarod test. Training for the rotarod test was initiated at 1 week, respectively, before the CCI injury, whereas no training was needed for EBST. The retard treadmill (AccuScan, Inc., Columbus, OH) provided a motor balance and coordination assessment. Data were generated by averaging the scores (total time spent on treadmill divided by five trials) for each animal during training and testing days. Each animal was placed in a neutral position on a cylinder (3 cm and 1 cm diameter for rats and mice, respectively), then the rod was rotated with the speed accelerated linearly from 0 to 24 rpm within 60 sec, and the time spent on the retard was recorded automatically. The maximum score given to an animal was fixed to 60.
For training, animals were given five trials each day and declared having reached the criterion when they scored 60 in three consecutive trials. For testing, animals were given three trials, and the average score on these three trials was used as the individual rotarod score. The EBST provided a motor asymmetry parameter and involved handling the animal by its tail and recording the direction of the biased body swings. The EBST consisted of 20 trials with the number of swings ipsilateral and contralateral to the injured hemisphere recorded and expressed in percentage to determine the biased swing activity. 8
Measurement of edema (brain water content)
At 24 h after TBI, animals were euthanized to determine brain water content (edema). The cortices, excluding the cerebellum, were quickly removed, and the contralateral and ipsilateral hemispheres separately weighed. Each hemisphere was dried at 60°C for 72 h, and the dry weight was determined. Water content was calculated in ipsilateral hemisphere as: Water content (%)=(wet weight - dry weight) wet weight×100.
Evaluation of infarction using 2,3,5-triphenyltetrazolium chloride (TTC) staining
The mice were anesthetized with ketamine and decapitated. Their brains were carefully removed. The brains were cut into five coronal slices of 2 mm thickness. Slices were incubated in 2% solution of TTC at 37°C for 30 min and immersion fixed in 10% buffered formalin solution. TTC stains the viable brain tissue red, while infracted tissue remains unstained. 30 For quantification of infracted area and volumes, the brain slices were photographed using a digital camera (Canon 4x, Canon Inc, China) and then image analysis was performed on a personal computer with an image analysis software program (using an ImageJ for Windows). 31 To compensate for the effect of brain edema, the corrected infarct area equals left hemisphere area minus (right hemisphere area minus infarct area). 32 The corrected total infarct volume was calculated by summing the infarct area in each slice and multiplying it by slice thickness (2 mm).
Light microscopy
Tissue segments containing the lesion (1 cm on each side of the lesion) were paraffin embedded and cut into 5-μm–thick sections. Tissue sections were deparaffinized with xylene stained with hematoxylin and eosin (H&E) and studied using an Axiovision Zeiss (Milan, Italy) microscope. The segments of each brain that contained the lesion were evaluated by an experienced histopathologist. Damaged neurons were counted, and the histopathologic changes of the gray matter were scored on a 6-point scale 33 : 0, no lesion observed, 1, gray matter contained 1–5 eosinophilic neurons; 2, gray matter contained 5–10 eosinophilic neurons; 3, gray matter contained more than 10 eosinophilic neurons; 4, small infarction (less than one third of the gray matter area); 5, moderate infarction (one third to one half of the gray matter area); 6, large infarction (more than half of the gray matter area). The scores from all the sections from each brain were averaged to give a final score for individual mice. All the histological studies were performed in a blinded fashion.
Immunohistochemical localization of chymase, tryptase, and nitrotyrosine
At the end of the experiment, tissues were fixed in 10% (w/v) phosphate buffered saline (PBS)-buffered formalin, and 7 μm sections were prepared from paraffin embedded tissues. After deparaffinization, endogenous peroxidase was quenched with 0.3% (v/v) hydrogen peroxide in 60% (v/v) methanol for 30 min. The sections were permeabilized with 0.1% (v/v) Triton X-100 in PBS for 20 min. Nonspecific adsorption was minimized by incubating the section in 2% (v/v) normal horse serum in PBS for 20 min. Endogenous biotin or avidin binding sites were blocked by sequential incubation for 15 min with avidin and biotin (Vector Laboratories, Burlingame, CA). The sections were then incubated overnight with a dilution of primary anti-nitrotyrosine antibody (Millipore, 1:500 in PBS, v/v), anti-chymase antibody (Santa Cruz Biotechnology, 1:500 in PBS, v/v) and anti-tryptase antibody (Santa Cruz Biotechnology, 1:500 in PBS, v/v). Controls included buffer alone or nonspecific purified rabbit immunoglobulin G (IgG). Specific labeling was detected with a biotin-conjugated specific secondary anti-IgG and avidin–biotin peroxidase complex (Vector Laboratories, Burlingame, CA).
To verify the binding specificity for nitrotyrosine, chymase, and tryptase, some sections were also incubated with primary antibody only (no secondary antibody) or with secondary antibody only (no primary antibody). In these situations, no positive staining was found in the sections indicating that the immunoreactions were positive in all the experiments performed. Immunocytochemistry photographs (n=5) were assessed by densitometry. The assay was performed by using Axiovision software on a personal computer with Windows. All the immunocytochemistry analysis was performed without knowledge of the treatments.
Western blot analysis for P-JNK, glial cell line-derived neurotrophic factor (GDNF), Caspase 3, pIκBα, IκBα, NF-κB p65, nitric oxide synthase (iNOS), Bax, triptase, chymase, mTOR, p70S6K, Beclin-1, MAP LC3α, and p62.
Western blot was performed in the traumatic penumbra area from the ipsilateral injured brain and a similar area from the control and/or contralateral tissues. Cytosolic and nuclear extracts were prepared as described previously 34 with slight modifications. Brain tissue from each mouse was suspended in extraction Buffer A containing 0.2 mM phenylmethylsulfonyl fluoride (PMSF), 0.15 mM pepstatin A, 20 mM leupeptin, 1 mM sodium orthovanadate, homogenized at the highest setting for 2 min, and centrifuged at 12000×rpm for 4 min at 4°C. Supernatants represented the cytosolic fraction.
The pellets, containing enriched nuclei, were resuspended in Buffer B containing 1% Triton X-100, 150 mM NaCl, 10 mM Tris–HCl pH 7.4, 1 mM ethylene glycol tetraacetic acid (EGTA), 1 mM (ethylenediaminetetraacetic acid (EDTA), 0.2 mM PMSF, 20 mM leupeptin, 0.2 mM sodium orthovanadate. After centrifugation 10 min at 12,000 rpm at 4°C, the supernatants containing the nuclear protein were stored at −80 C for further analysis. The levels of P-JNK, GDNF, Caspase 3, IκBα, iNOS, and Bax were quantified in cytosolic fraction from brain tissue collected after 24 h after TBI, while NF-κB p65 levels were quantified in nuclear fraction.
The filters were blocked with 1x PBS, 5% (w/v) nonfat dried milk (PM) for 40 min at room temperature and subsequently probed with specific Abs anti-Bax (1:500; Santa Cruz Biotechnology) (D.B.A. s.r.l., Milan, Italy), anti-Bcl-2 (1:500; Santa Cruz Biotechnology) (D.B.A. s.r.l., Milan, Italy), anti-NF-κB p65 (1:1000; Santa Cruz Biotechnology) (D.B.A. s.r.l., Milan, Italy), anti-iNOS (1:1000; BD transduction) (Biogenerica s.r.l, Catania, Italy), anti-phospho-JNK (1:1000; Cell Signaling), anti-IκBα (1:500; Santa Cruz Biotechnology) (D.B.A. s.r.l., Milan, Italy), anti-pIκBα (1:1000; Cell Signaling), anti-GDNF (1:500; Santa Cruz Biotechnology) (D.B.A s.r.l., Milan, Italy), anti-Caspase 3 (1:1000; Cell Signaling), anti-tryptase (1:500; Santa Cruz Biotechnology) (D.B.A. s.r.l., Milan, Italy), anti-chymase (1:500; Santa Cruz Biotechnology) (D.B.A. s.r.l., Milan, Italy), anti-mTOR (1:1000; Cell Signaling), anti-p70S6K (1:1000; Cell Signaling), anti-p62 (1:1000; Cell Signaling), anti-Beclin-1 (1:500; Santa Cruz Biotechnology) and anti-MAP LC3α (1:500; Santa Cruz Biotechnology) in 1x PBS, 5% (w/v) nonfat dried milk, 0.1% Tween-20 (PMT) at 4°C overnight.
Membranes were incubated with peroxidase-conjugated bovine anti-mouse IgG secondary antibody or peroxidase-conjugated goat anti-rabbit IgG (1:2000, Jackson Immuno Research, West Grove, PA) for 1 h at room temperature. To ascertain that blots were loaded with equal amounts of proteic lysates, they were also incubated in the presence of the antibody against β-actin (1:5000; Santa Cruz Biotechnology) (D.B.A. s.r.l. Milan, Italy) or anti-laminin a/c (1:5000; Santa Cruz Biotechnology) (D.B.A. s.r.l. Milan, Italy). The relative expression of the protein bands of NF-κB p65 (65 kDa), iNOS (130 kDa), Bax (23 kDa), GDNF (15 kDa), IκBα (37 kDa), pIκBα (40 kDa), p-JNK (46 kDa), Caspase 3 (32 kDa), tryptase (34 kDa), chymase (37 kDa), mTOR (289 kDa), p70S6K (70 kDa), Beclin-1 (60 kDa), MAP LC3α (15–18 kDa), and p62 (62 kDa) was quantified by densitometric scanning of the X-ray films with GS-700 Imaging Densitometer (GS-700, Bio-Rad Laboratories, Milan, Italy) and a computer program (Molecular Analyst, IBM).
Immunofluorescence of glial fibrillary acidic protein (GFAP), tumor necrosis factor (TNF)a, interleukin (IL)1β, Iba-1, Neun, Beclin-1, and MAP LC3-α
After deparaffinization and rehydration, detection of GFAP, TNFα, IL1β, Iba-1, Neun, Beclin-1, and MAP LC3-α was performed after boiling in 0.1 M citrate buffer for 1 min. Nonspecific adsorption was minimized by incubating the section in 2% (v/v) normal goat serum in PBS for 20 min. Sections were incubated with mouse monoclonal anti-GFAP (1:100, v/v Santa Cruz Biotechnology), or with polyclonal rabbit anti-TNFα (1:100, v/v Santa Cruz, Biotechnology), or with rabbit anti-IL1β (1:100, v/v Santa Cruz Biotechnology), or with mouse monoclonal anti-Iba-1 (1:100, v/v Santa Cruz Biotechnology), or anti-Neun (1:100, v/v Santa Cruz, Biotechnology), or anti-Beclin-1 (1:100, v/v Santa Cruz, Biotechnology), or anti-MAP LC3-α (1:100, v/v Santa Cruz, Biotechnology) Abs in a humidified chamber O/N at 37°C. Sections were washed with PBS and were incubated with secondary antibody fluorescein isothiocyanate (FITC)-conjugated anti-mouse Alexa Fluor-488 antibody (1:2000 v/v Molecular Probes, UK) and with TEXAS RED-conjugated anti-rabbit Alexa Fluor-594 antibody (1:1000 in PBS, v/v Molecular Probes, UK) for 1 h at 37°C. Sections were washed and for nuclear staining 4′,6′-diamidino-2-phenylindole (DAPI; Hoechst, Frankfurt; Germany), 2 μg/mL in PBS was added.
Sections were observed and photographed using a Leica DM2000 microscope (Leica). Optical sections of fluorescence specimens were obtained using a HeNe laser (543 nm), a laser UV (361–365 nm) and an Ar laser (458 nm) at a 1-min, 2-sec scanning speed with up to eight averages; 1.5-μm sections were obtained using a pinhole of 250. Contrast and brightness were established by examining the most brightly labeled pixels and applying settings that allowed clear visualization of structural details while keeping the highest pixel intensities close to 200. The same settings were used for all images obtained from the other samples that had been processed in parallel. Digital images were cropped and figure montages prepared using Adobe Photoshop CS5 (Adobe Systems; Palo Alto, CA).
Biochemical assays for TNF-α and IL-1β
Each brain was weighed and homogenized in tissue protein extraction reagent (Pierce). After homogenization, samples were centrifuged at 4°C and 10,000 g for 10 min and the supernatant was collected as homogenate. The protein content of the supernatant was estimated using BioRad to ensure that an equal amount of protein from each sample was used for the assay. The levels of TNF-α and IL-1β were determined with enzyme-linked immunosorbent assay kits (R&D Systems Inc., Minneapolis, MN) according to the manufacturer's instruction. The concentration of the cytokines was quantified as picograms of antigen per milligram of protein.
Statistical methods
All values in the figures and text are expressed as mean±standard error of the mean (SEM) of N observations. For the in vivo studies, N represents the number of animals studied. In the experiments involving histology, the figures shown are representative of at least three experiments performed on different experimental days. A p value of less than 0.05 was considered significant. The results were analyzed by one-way analysis of variance followed by a Bonferroni post hoc test for multiple comparisons.
Results
Co-ultraPEALut decreased edema and brain infarctions and promoted recovery and improvement in behavioral function after TBI
Brain water content is a sensitive measure of cerebral edema. This measure indicates pathology associated with endothelial cell activation and endothelial dysfunction. Water content was significantly different between groups overall with levels significantly higher in animals subjected to TBI compared with Sham controls (78.57±1.45 vs. 70.47±1.54; p<0.05 vs. Sham). The increased water content in the ipsilateral brain induced by TBI was significantly decreased by Co-ultraPEALut treatment at 24 h post-injury (71.03±0.93 vs. 78.57±1.45; p<0.05 vs. TBI). This result indicates that Co-ultraPEALut could provide neurons and vessels protection.
Directly related to overall brain injury, measurement of brain infarctions is a standard method to evaluate brain injury after trauma. To evaluate the effect of a combination of Co-ultraPEALut on brain infarctions in the TBI, we performed TTC staining (Fig. A1, A2). As shown in Figure 1B and 1C, the infarction area and the infarct volume were significantly reduced after treatment with Co-ultraPEALut (1 mg/kg). Moreover, to investigate the relationship between neurological deficit and motor function in the setting of TBI, the mice were subjected, 24 h after TBI, to the EBST and the rotarod test. Mice subjected to moderate severe injury showed significant hippocampal damage and behavioral deficits but low mortality. CCI-injured mice displayed a range of impairments in locomotor tasks as showed in Figure 1D and 1E. Co-ultraPEALut treatment groups gradually and significantly improved latency compared with the TBI group and PEA group (data not shown).
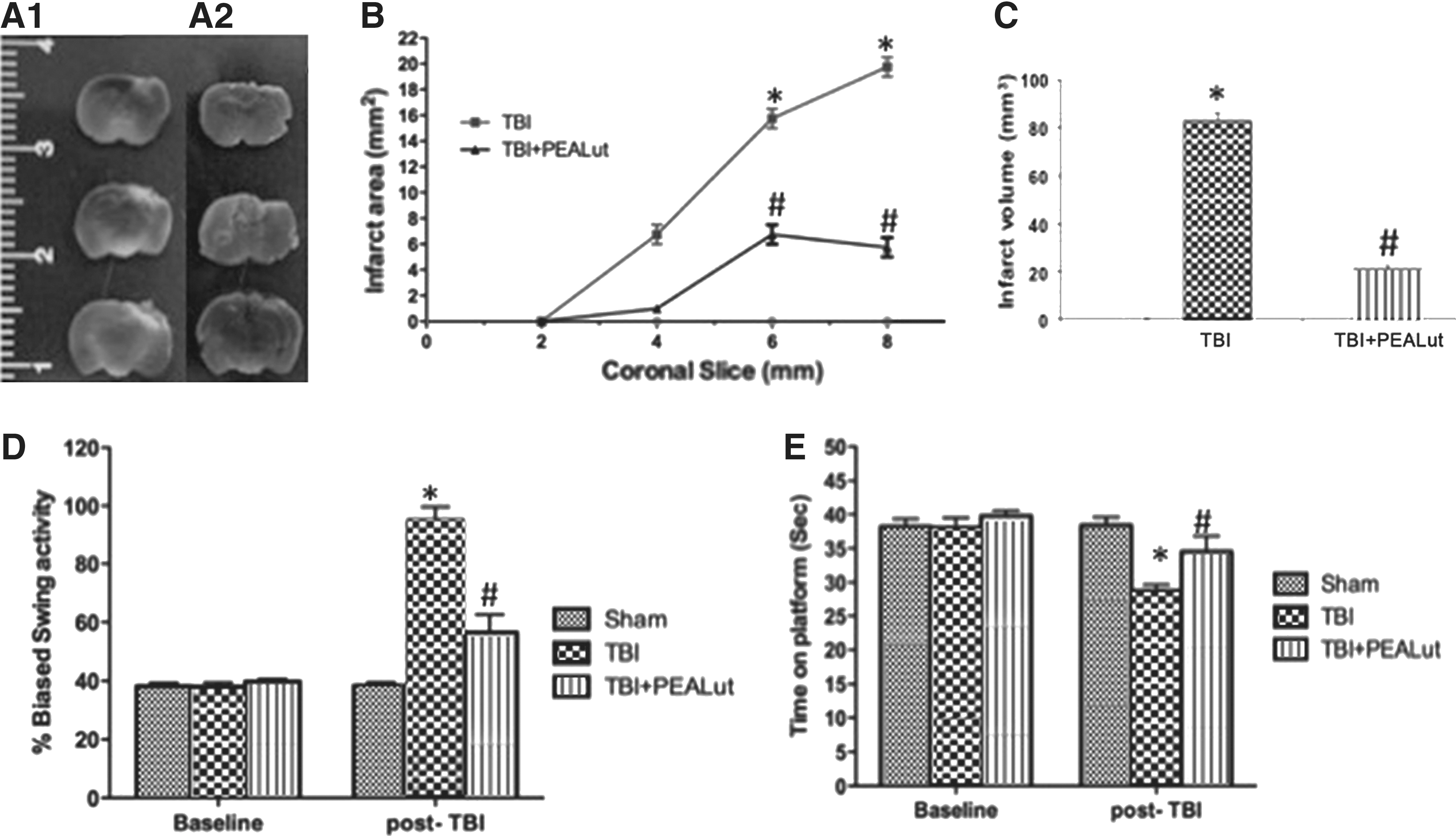
Co-ultraPEALut decreased edema and brain infarction area and volume and promoted recovery and improvement in behavioral function after traumatic brain injury (TBI). Representative 2,3,5-triphenyltetrazolium chloride (TTC) stained brain sections (three of the six consecutive sections from cranial to caudate region) corresponding to the largest infraction from the TBI group (
Effect of Co-ultraPEALut treatment on histological parameters
Twenty-four hours after TBI, the sections obtained from each group were stained with hematoxylin and eosin (H&E) for histologic assessment of contusion areas. Normal adult control mice showed no evidence of degenerating cells (Fig. 2A). Significant damage was observed in the brain tissue collected from TBI compared with sham-operated mice (Fig. 2B). Indeed, as shown in Figure 2C and relative quantification in Figure 2D, a significant and important decrease in the severity of trauma was observed in mice treated with Co-ultraPEALut (1 mg/kg). Treatment with the Co-ultraPEALut reduced histological alterations more effectively than the treatment with PEA administered alone (data not shown).
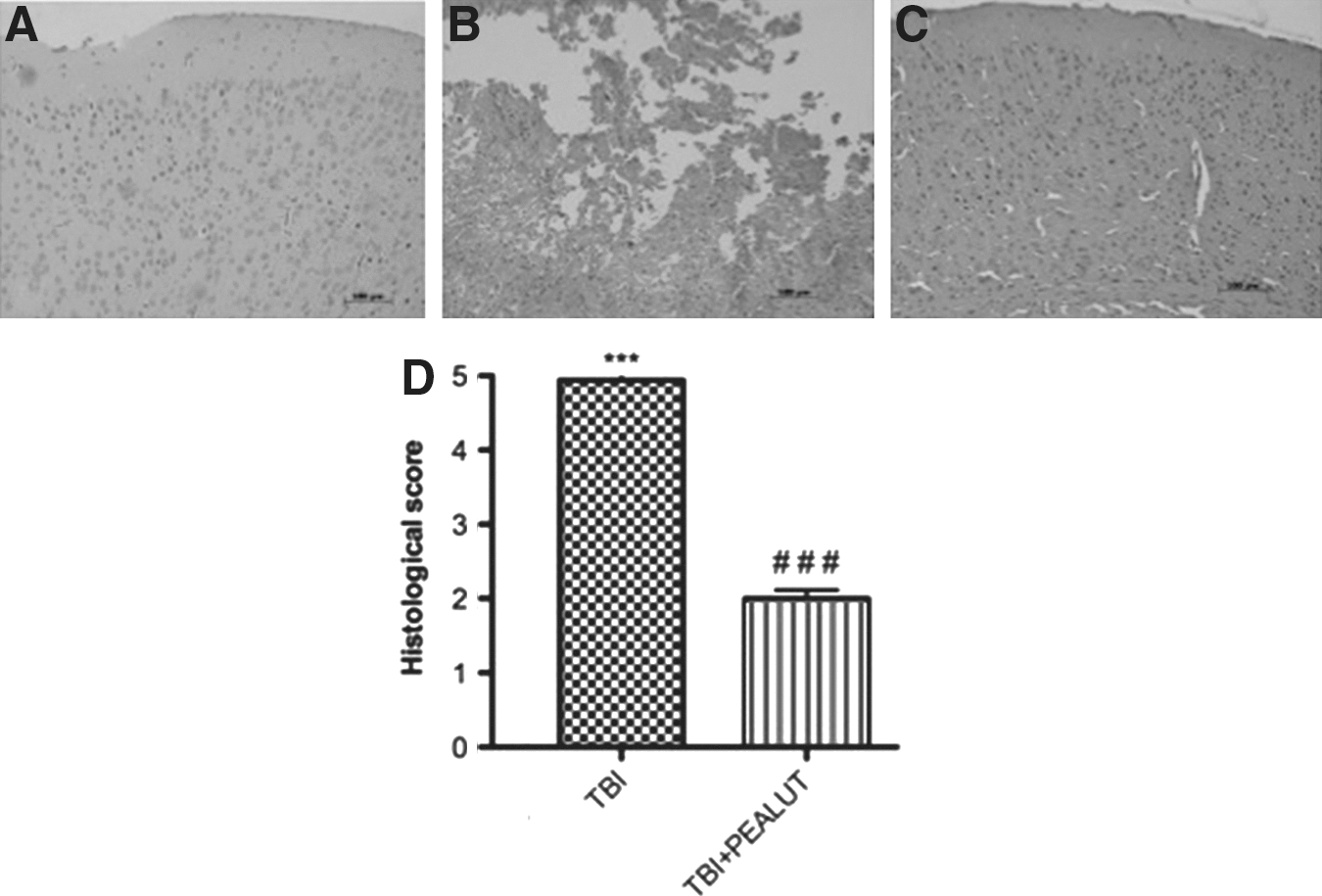
Effect of Co-ultraPEALut treatment on histological alterations of the brain tissue 24 h after injury. Brain samples were collected from the perilesional area 24 h after injury and stained with hematoxylin and eosin (H&E). Significant damage to the brain tissue was assessed in traumatic brain injury (TBI)-operated mice (
Co-ultraPEALut modulates IκBα phosphorylation, IκBα degradation, and NF-κBp65 nuclear translocation
To deepen the molecular mechanism by which Co-ultraPEALut attenuated inflammatory response, Western blot analysis was used to analyze the NF-κB pathway in terms of Iκκα and IκBα cytoplasmic levels and NF-κB nuclear translocation (NF-κBp65). 35,36 Activation of NF-κB requires that IκB be phosphorylated on specific serine residues (Ser 32 and 36), which results in targeted degradation of IκB. A basal expression of IκBα was detected in the Sham group, whereas TBI substantially decreased its expression (Fig. 3A). Treatment with Co-ultraPEALut prevented TBI-induced degradation of IκBα. Moreover, Western blot analysis revealed a low basal expression of pIκBα in the Sham group and TBI is able to induce the phosphorylation of IκBα (Fig. 3B).
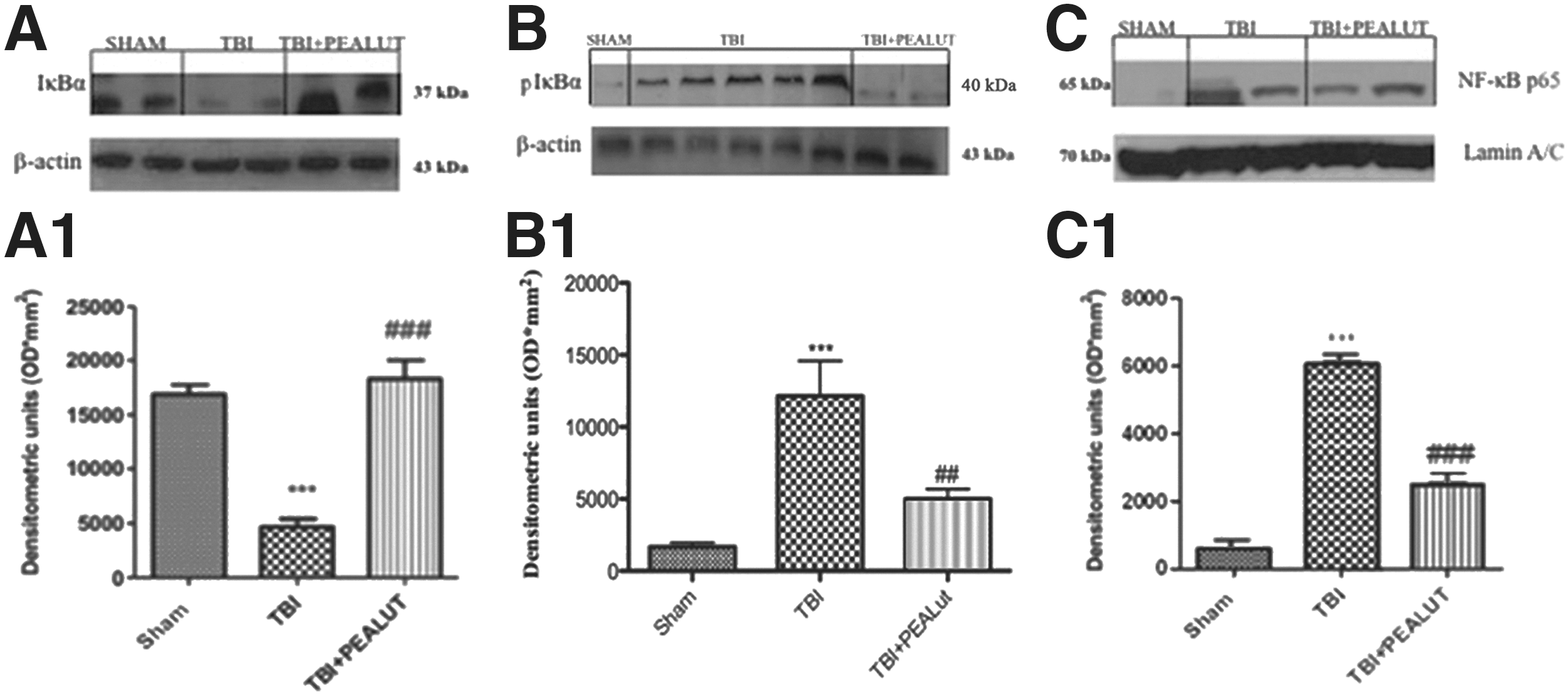
Co-ultraPEALut modulates IκBα degradation and NFκBp65 nuclear translocation. Traumatic brain injury (TBI) caused an important phosphorylation of Iκκ (
Treatment with Co-ultraPEALut 1 h after brain injury significantly decreased pIκBα. Consequently, translocation of p65 in the nuclear extracts from brain homogenates was also significantly increased 24 h after TBI compared with sham-operated mice (Fig. 3C). Co-ultraPEALut significantly prevents NF-κB nuclear translocation. The treatment with PEA administered alone was less effective in reducing pIκBα, IκBα degradation, and NFκBp65 nuclear translocation than the treatment with the Co-ultraPEALut (data not shown).
Colocalization of TNF-α and IL-1β with cell-specific markers by double immunofluorescent staining
To determine whether TBI causes microgliosis or astrogliosis, during the pro-inflammatory event in the brain, the protein expression levels of Iba1 and GFAP, microglial and astrocyte activation marker, respectively, were visualized by double immunofluorescence staining, with antibodies against TNF-α and IL-1β, both inflammation signs. Immunofluorescence staining revealed that TNF-α and IL-1β were significantly higher in the TBI group (Fig. 4G, M and Fig. 5G, M) than in the Sham group (Fig. 4A, M and Fig. 5A, M). Indeed, a significant and important decrease in the expression of TNF-α and IL-1β was observed in mice treated with Co-ultraPEALut (Fig. 4N, S and Fig. 5N, S) compared with Sham (Fig. 4N, S). Treatment with the Co-ultraPEALut reduced GFAP and Iba1 productions more effectively than the treatment with PEA administered alone (data not shown). The yellow arrow indicates the colocalization between GFAP and TNF-α (Fig/ 4G, I and Fig. 4N, P), Iba1 and TNF-α (Fig. 4J, M and Fig. 4Q, S), GFAP and IL-1β (Fig. 5G, I and Fig. 5N, P), and Iba1 and IL-1β (Fig. 5J, M and Fig. 5Q, S).
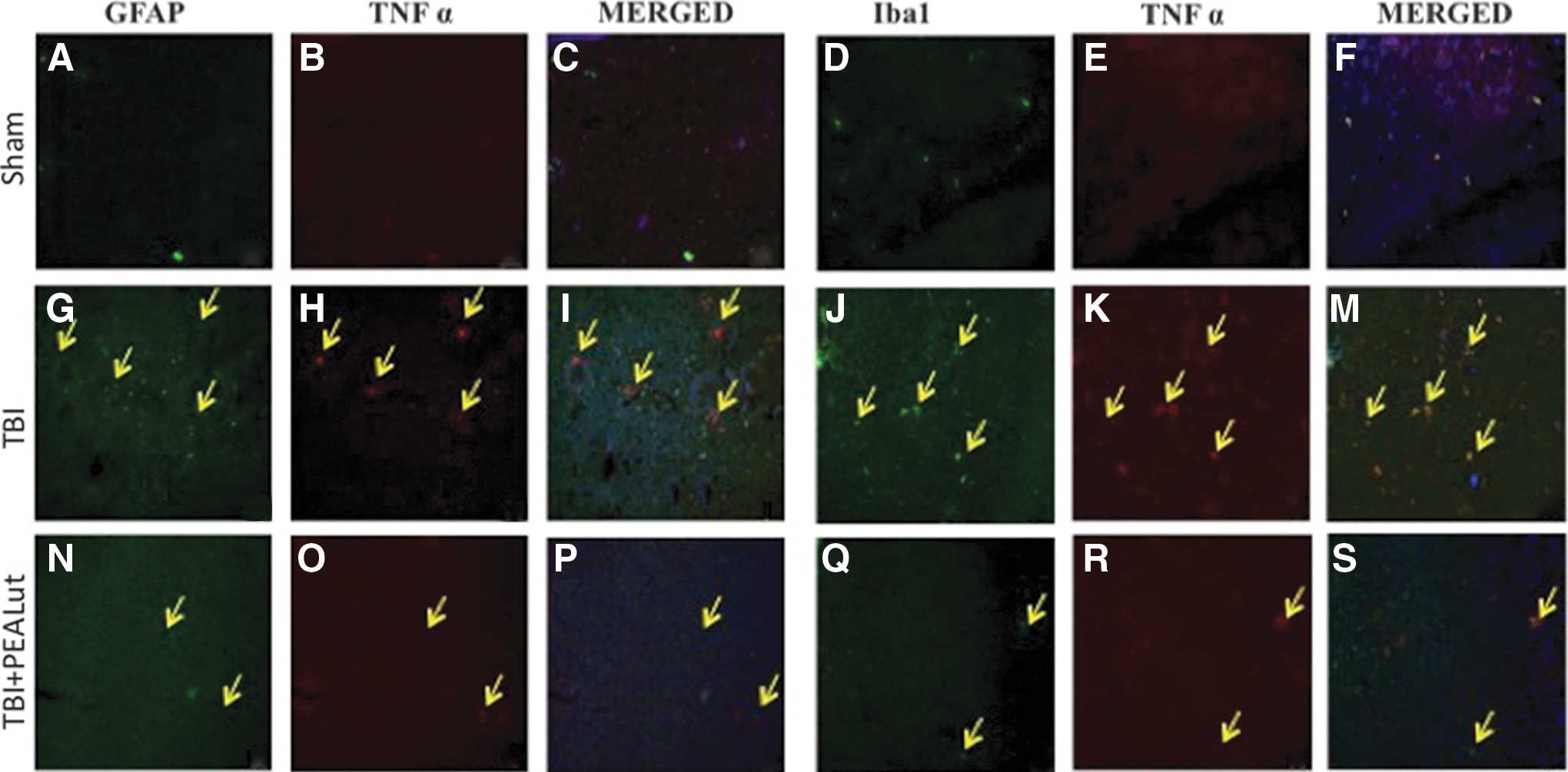
Colocalization of glial fibrillary acidic protein (GFAP)/tumor necrosis factor (TNF)α and Iba1/TNFα after traumatic brain injury (TBI). Results are shown for (
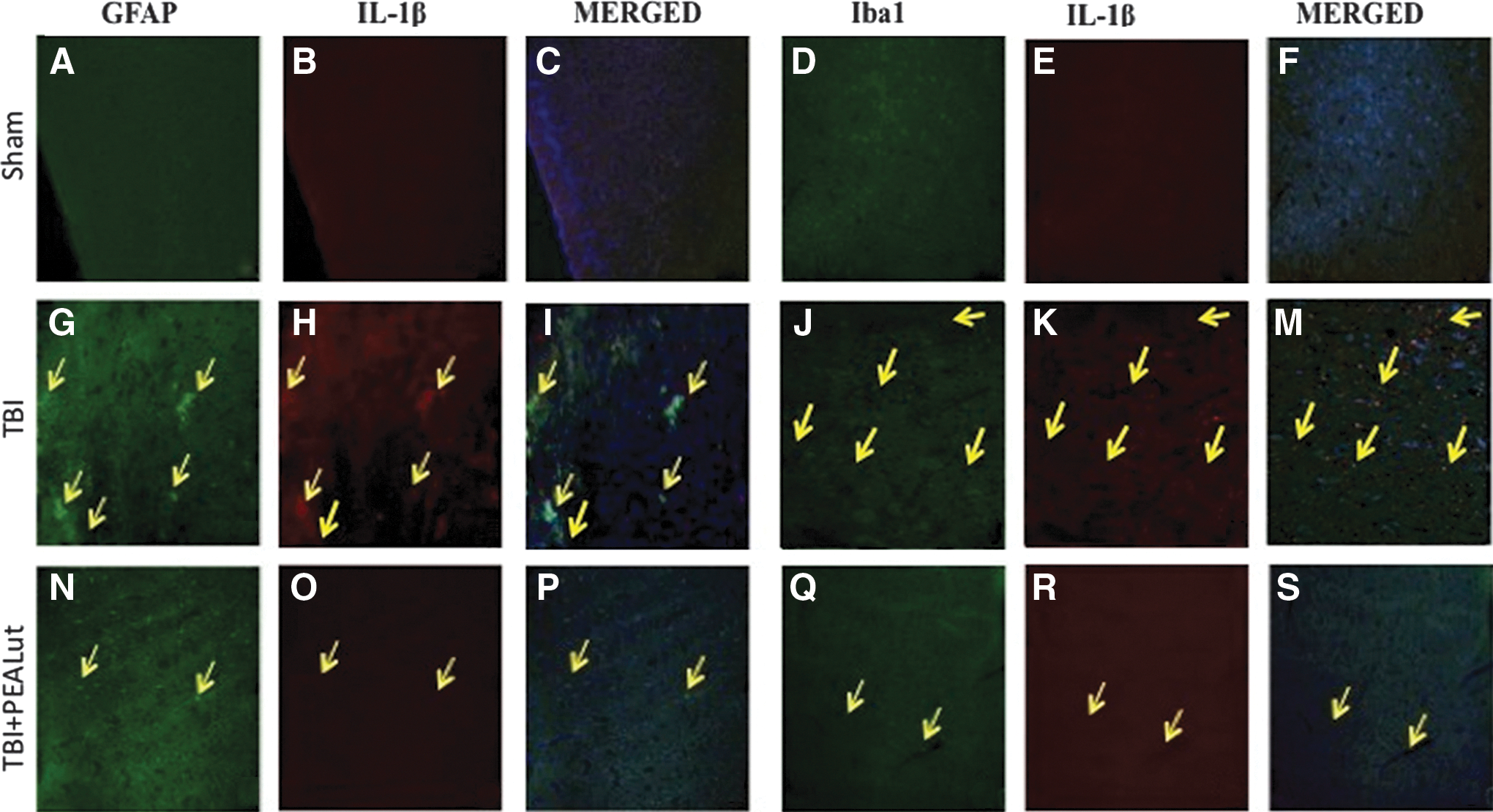
Colocalization of glial fibrillary acidic protein (GFAP)/IL1β and Iba1/interleukin (IL)1β after traumatic brain injury (TBI). Results are shown for (
As expected, the levels of IL-1β and TNF-α were significantly increased at 24 h after TBI in vehicle group versus sham-operated group (p<0.01). Co-ultraPEALut administration significantly reduced the levels of IL-1β and TNF-α compared with the vehicle group (p<0.01) (Fig. 6).
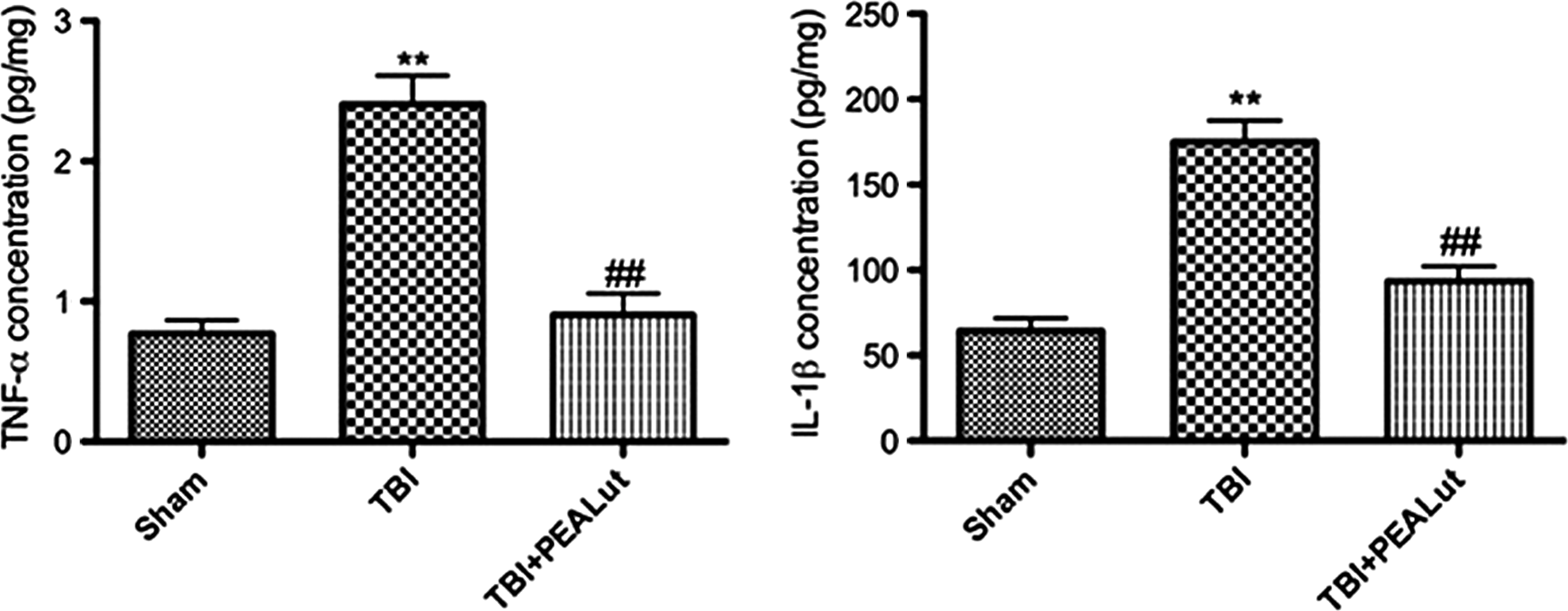
Co-ultraPEALut administration reduces tumor necrosis factor (TNF)-α and interleukin (IL)-1β expression. Co-ultraPEALut significantly reduced the expression of TNF-α (p<0.01) and IL-1β (p<0.01) in lesion boundary zone 24 h post-traumatic brain injury (TBI). **p<0.01 vs. Sham; ##p<0.01 vs. TBI+VEH. PEA, palmitoylethanolamide; Lut, luteolin; VEH, vehicle.
PEALut administration reduces chymase and tryptase expression during TBI
To test whether PEALut treatment may modulate and down-regulate the inflammatory response through the regulation of the serine peptidases, we analyzed by immunohistochemistry and Western blot the brain expression of chymase and tryptase. There was no staining for chymase and tryptase in the brain tissues obtained from the sham-operated mice (Fig. 7A, D). A substantial increase in chymase and tryptase expression was found in brain tissues collected at 24 h after TBI (Fig. 7B, E and relative densitometric analysis shown in Fig. 7G). Brain expression of chymase and tryptase were attenuated in the brain from mice that have received PEALut treatment (Fig. 7C, F and relative densitometric analysis shown in Fig. 7G) compared with the TBI group and PEA group (data not shown). Moreover, Western blot analysis revealed a marked increase of expression of tryptase and chymase in brain tissues collected at 24 h after TBI compared with the TBI group.
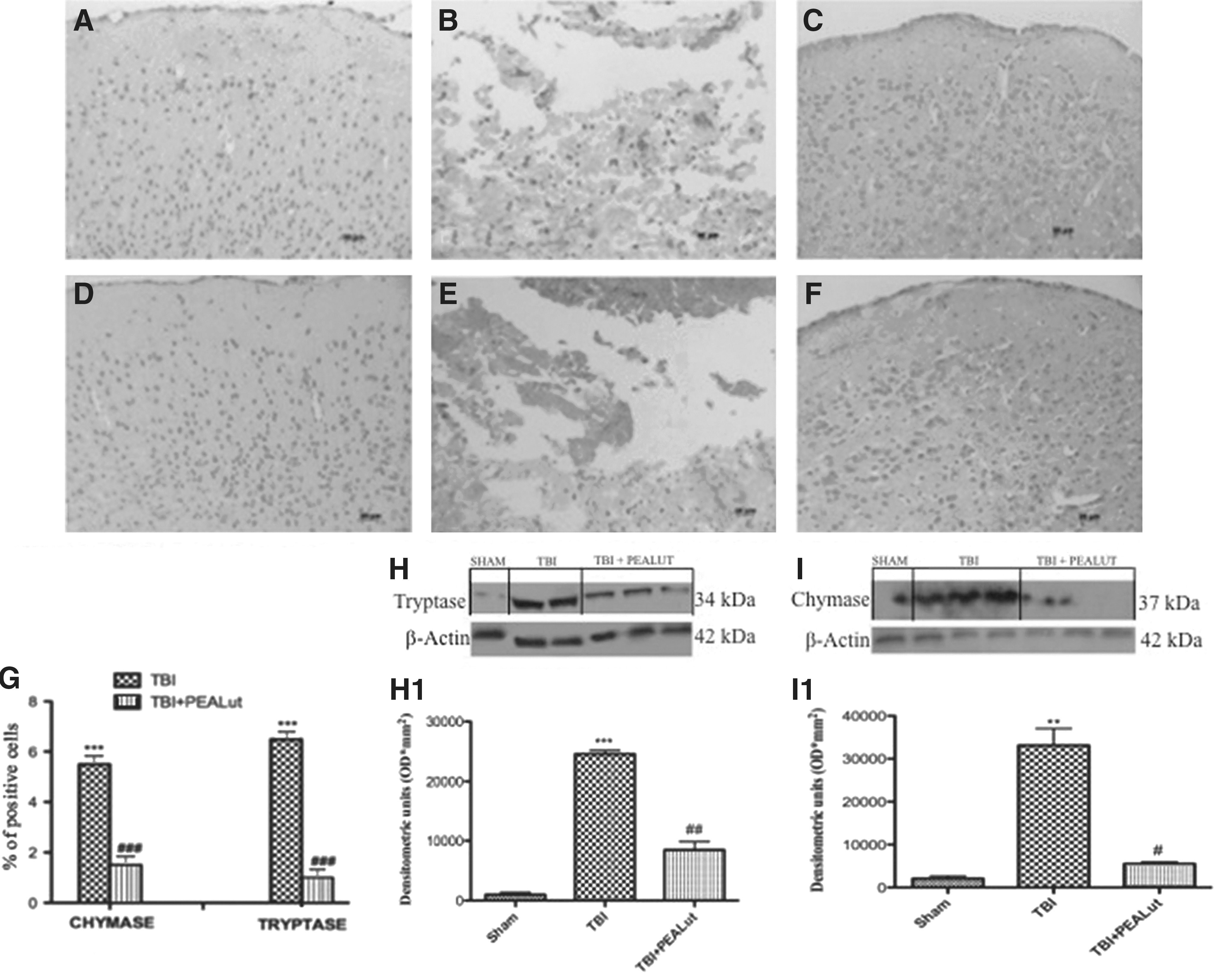
Co-ultraPEALut administration reduces chymase and tryptase expression during traumatic brain injury (TBI). There was no staining for chymase (
Co-ultraPEALut treatment significantly diminished the post-TBI expression of the chymase and tryptase (Fig. 7H, I and relative densitometric analysis shown in Fig. 7H1. I1). Co-ultraPEALut ameliorates chymase and tryptase expression more effectively than the treatment with PEA administered alone (data not shown).
Effect of Co-ultraPEALut on GDNF expression after TBI.
To test whether Co-ultraPEALut modulates the inflammatory process through regulation of the neutrophic factors levels, we have examined GDNF by Western blot analysis. As shown in Figure 8A, GDNF expression decreased at 24 h after TBI. As illustrated by densitometric analysis (Fig. 8B1) Co-ultraPEALut significantly enhanced the post-TBI expression of GDNF compared with TBI mice and PEA mice (data not shown).
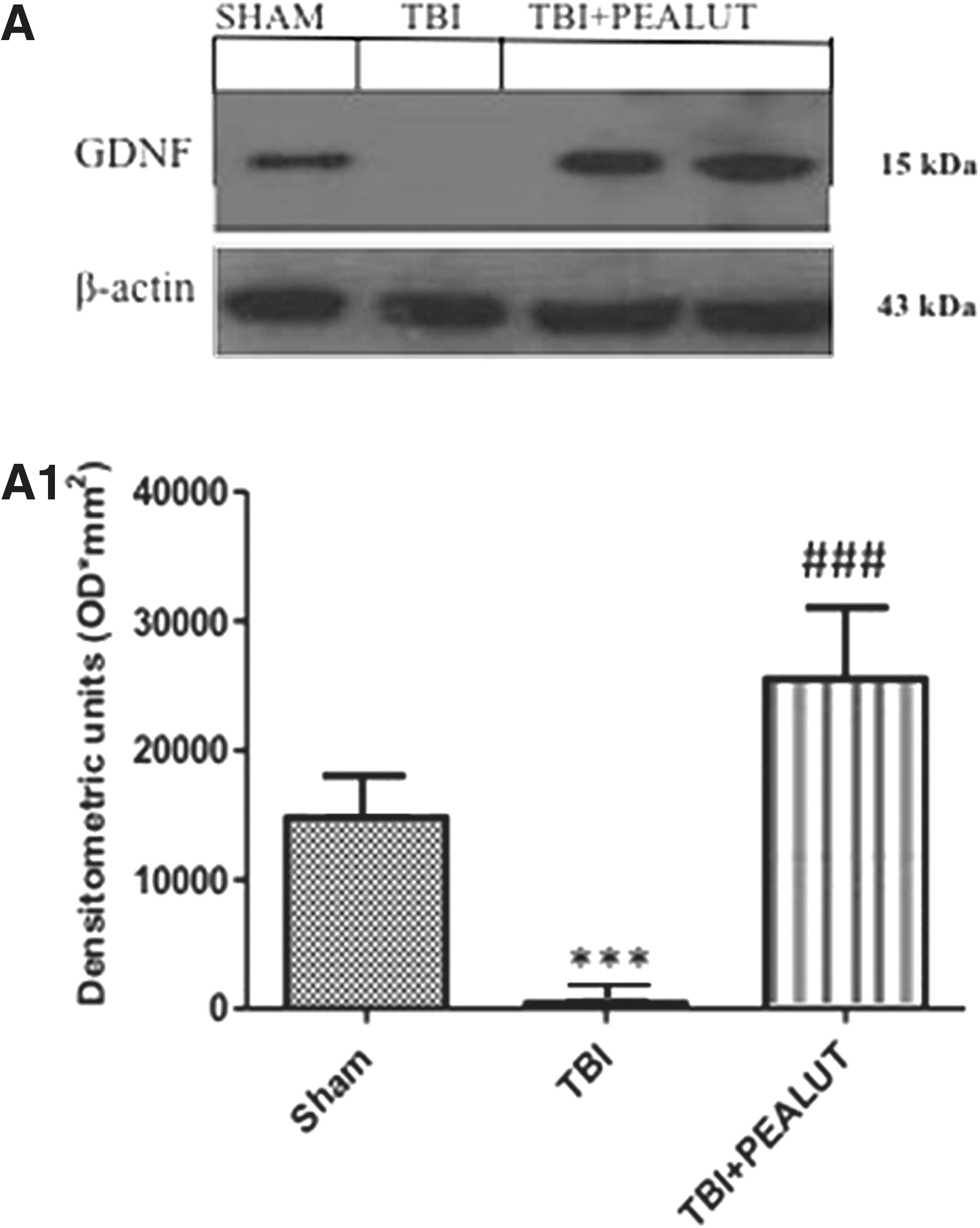
Effect of Co-ultraPEALut on glial cell line-derived neurotrophic factor GDNF. By Western blot analysis, a basal level of GDNF expression was detected in brain samples from controls. GDNF levels was significantly reduced in brain samples from TBI-vehicle mice. Co-ultraPEALut treatment significantly reduced the TBI-induced inhibition of GDNF expression. β-actin was used as internal control. A representative blot of lysates obtained from each group is shown in Figure 7
Co-ultraPEALut modulates expression of iNOS and P-JNK and nitrotyrosine after TBI.
To determine the role of NO produced during TBI, nitrotyrosine expression was evaluated by immunohistochemical analysis and iNOS and pJNK expression were evaluated by Western blot. Brain sections from sham-operated mice negatively stained for nitrotyrosine (Fig. 9A and relative densitometric analysis in Fig. 9D), whereas brain sections obtained from TBI mice exhibited positive staining for nitrotyrosine (Fig. 9B), mainly localized in the gray matter. Treatment with Co-ultraPEALut reduced the degree of positive staining for nitrotyrosine in brain tissues (Fig. 9C, see densitometric analysis Fig. 9D). Moreover, a low basal expression of iNOS and pJNK was detected in brain samples from sham-operated mice, whereas iNOS and pJNK levels were substantially increased in TBI mice (E, F). As showed by densitometric analysis (Fig. 9, E1, F1), Co-ultraPEALut drastically decreased iNOS and pJNK expression better than PEA administered alone (data not show).
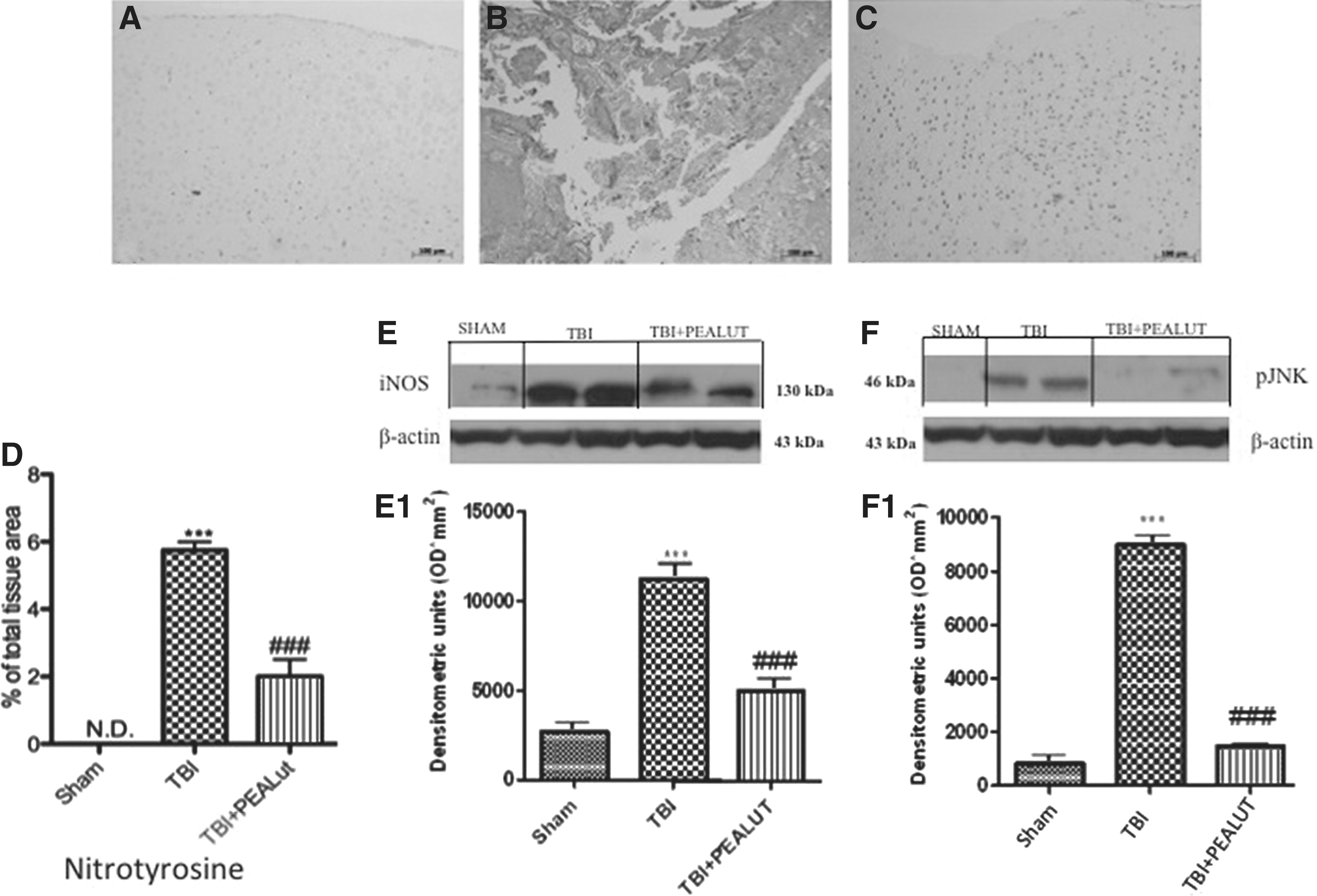
Effect of Co-ultraPEALut on nitrotyrosine formation and P-JNK and nitric oxide synthase (iNOS) expression. By Western blot analysis, a basal level of P-JNK and iNOS were detected in the brain from sham-operated animals, whereas P-JNK and iNOS levels were substantially increased in traumatic brain injury (TBI) mice (
Effects of Co-ultraPEALut on apoptosis in the brain after TBI
To test whether brain damage was associated with cell death by apoptosis 24 h after TBI, the appearance of apoptosis proteins such as Caspase-3, Bax, and Bcl-2 was investigated by Western blot. A significant increase in Caspase-3 was found in mice after TBI (Fig. 10A, B). TBI significantly increased Bax protein expression; however, it inhibited Bcl-2 protein expression, leading to an increase in the Bax/Bcl-2 ratio. (Fig. 10D). Co-ultraPEALut treatment decreased the post-TBI expression of Caspase-3 and Bax and restored Bcl-2 to Sham levels (Fig. 10A1, B1, C1). Co-ultraPEALut ameliorate Caspase-3, Bax, and Bcl-2 expression more effectively than the treatment with PEA administered alone (data not shown).
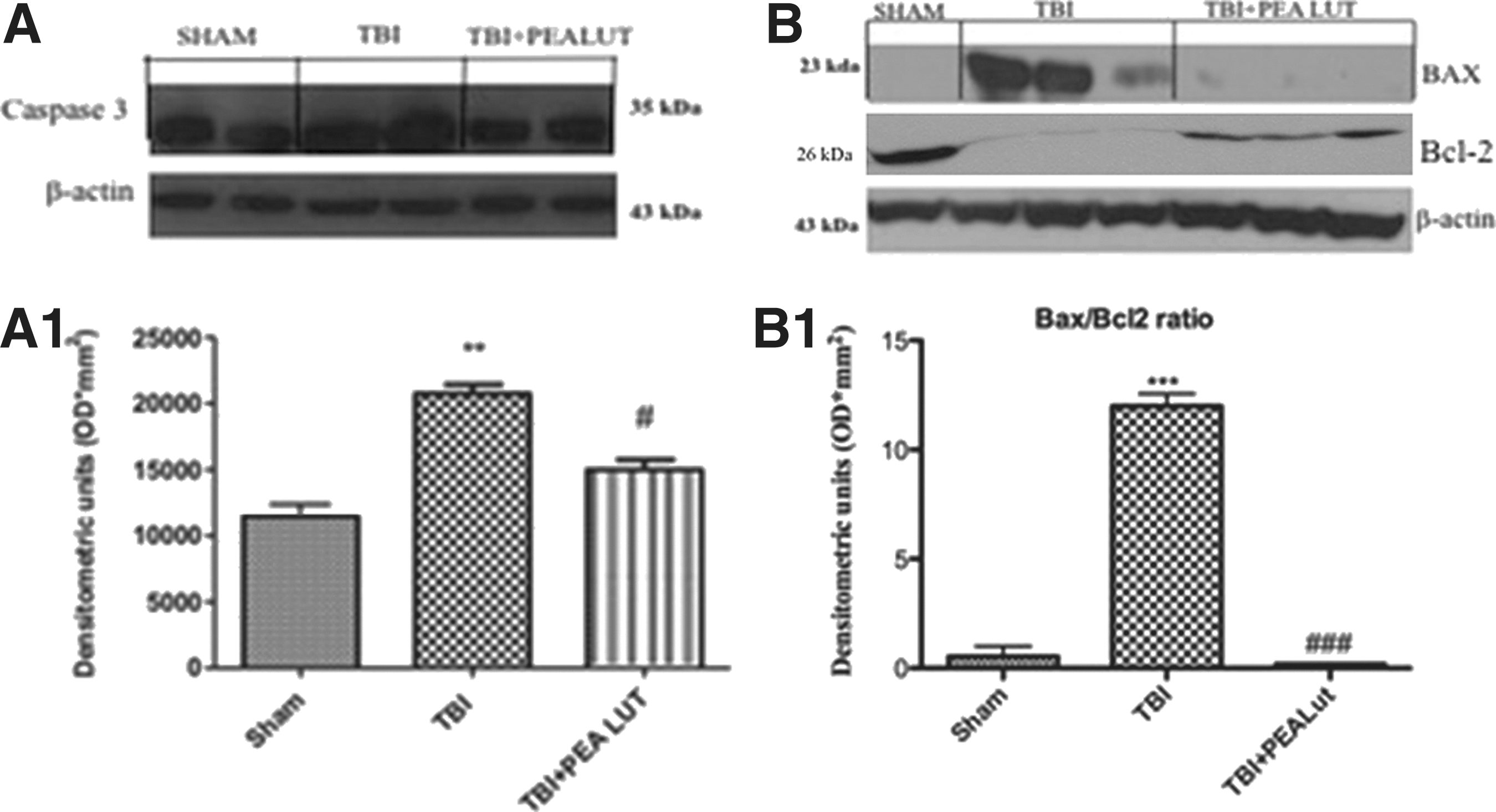
Effect of Co-ultraPEALut on apoptosis in brain tissue after traumatic brain injury (TBI). A significant increase in Caspase-3 and Bax expression was detected in the brain from mice subjected to TBI (Fig. 9A, A1, B, B1). Vice versa, brain trauma induced a significant decrease in Bcl-2 expression (Fig. 9B). Co-ultraPEALut treatment prevented TBI-induced Caspase-3 and Bax expression and restored Bcl-2 expression. β-actin was used as internal control. Co-ultraPEALut ameliorates Caspase-3, Bax, and Bcl-2 expression more effectively than the treatment with PEA traumatic brain injury (adm)inistered alone (data not shown). **p<0.01 vs. Sham; ***p<0.001 vs. Sham; #p<0.05 vs. TBI+VEH; ###p<0.001 vs TBI+VEH. PEA, palmitoylethanolamide; Lut, luteolin; VEH, vehicle.
Co-ultraPEAlut protects brain from autophagy after TBI
To determine how autophagic activity is altered after TBI and to understand if Co-UltraPEALut inhibits autophagy, the protein levels of mTOR and p70S6K, kinases regulator of autophagy, were performed by Western blot analysis. We found that mTOR and p70S6K expressions decreased after TBI (Fig. 11A, B) compared with the Sham group, while the treatment with Co-ultraPEALut significantly increases their expression.
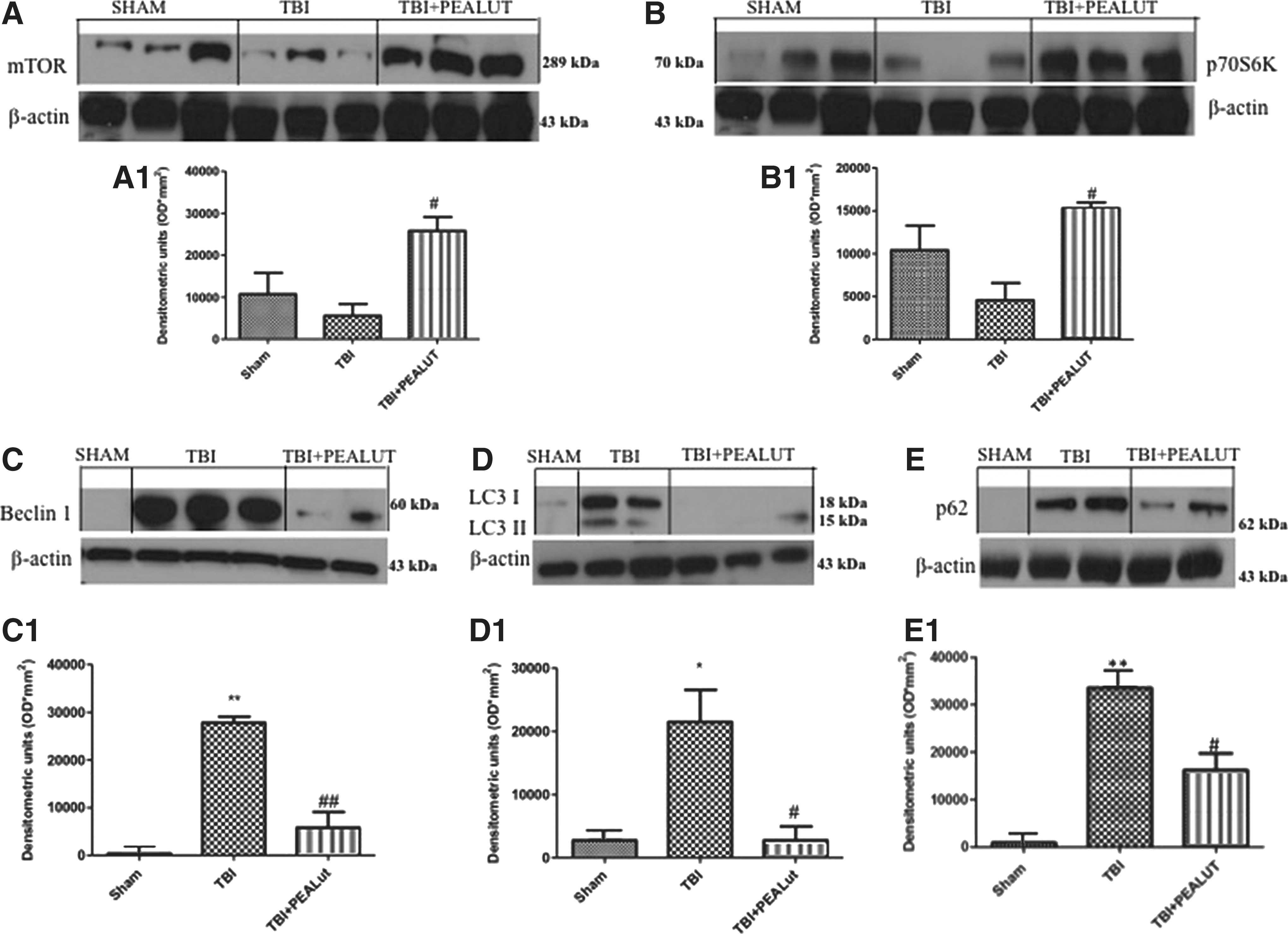
Co-ultraPEALut modulates autophagy. Western blot profiles from tissue collected from experimental animals 24 h after-TBI revealed that mammalian target of rapamycin (mTOR) and its downstream p70S6K levels decreased after injury (Fig. 11A, B), and are significantly increased after Co-ultraPEALut treatment. A representative blot of lysates obtained from each group is shown, and densitometry analysis of all animals is reported. Densitometric analysis of protein bands from three separated experiments is reported. #p<0.05 vs. TBI. Moreover, to quantify the amount of autophagy expression after TBI relative to the sham controls, we examined Beclin1, LC3, and p62 levels using Western blot analysis. Increased expression of all expression protein compared with sham-operated animals was evident in the TBI group; treatment with Co-ultraPEALt reduced this expression (Fig. 11 C, D, E). The treatment with PEA administered alone was less effective in modulating autophagy than the treatment with the Co-ultraPEALut (data not shown). *p<0.05 vs. Sham; **p<0.01 vs. Sham; #p<0.05 vs. TBI+VEH; ##p<0.01 vs. TBI+VEH. PEA, palmitoylethanolamide; Lut, luteolin; VEH, vehicle.
To establish how the autophagic machinery is altered after TBI and to confirm the ability of Co-ultraPEALut to inhibit autophagy, also the protein levels of Beclin-1 and LC3 II were determined by Western blot analysis. Beclin-1 (Atg6) is a key protein shown to be involved in the regulation of autophagy. 37 We showed that Beclin-1 expression dramatically increased after TBI compared with the Sham group, while the treatment with Co-ultraPEALut significantly reduced TBI-induced Beclin-1 expression (Fig. 11C, C1).
Formation of LC3 II is correlates with the extent of autophagosome formation. 38 The expression of LC3 II protein was significantly increased after TBI. Co-ultraPEALut drastically diminished TBI-induced LC3 expression (Fig. 11D, D1).
Recently, p62 protein has been suggested to interact with ubiquitinated proteins and LC3, which may regulate the selective autophagic clearance of protein aggregates. 39 After TBI, we showed an increased of p62 expression. Co-ultraPEALut significantly reduced p62 expression.
Moreover, to better characterize the Beclin-1 and LC3 expressing cells, TBI sections at 24 h after injury were double-stained with antibodies against Beclin-1 (red) or LC3 (red) with NeuN (green). In the double staining, the expression of Beclin1 or LC3 was observed in NeuN labeled cells of TBI group (Fig. 12G, M) compared with Sham (Fig. 12A, F); whereas a treatment with Co-ultraPEALut not only significantly decreased Beclin-1 and LC3 expression but also did not colocalize with NeuN (Fig. 12N, S). The yellow arrows indicate the colocalization between Beclin-1 and NeuN (Fig. 12G, I and Fig. 12N, P) as well as between LC3 and NeuN (Fig. 12J, M and Fig. 12Q, S). The treatment with PEA administered alone was less effective to modulates autophagy than the treatment with the Co-ultraPEALut (data not shown).
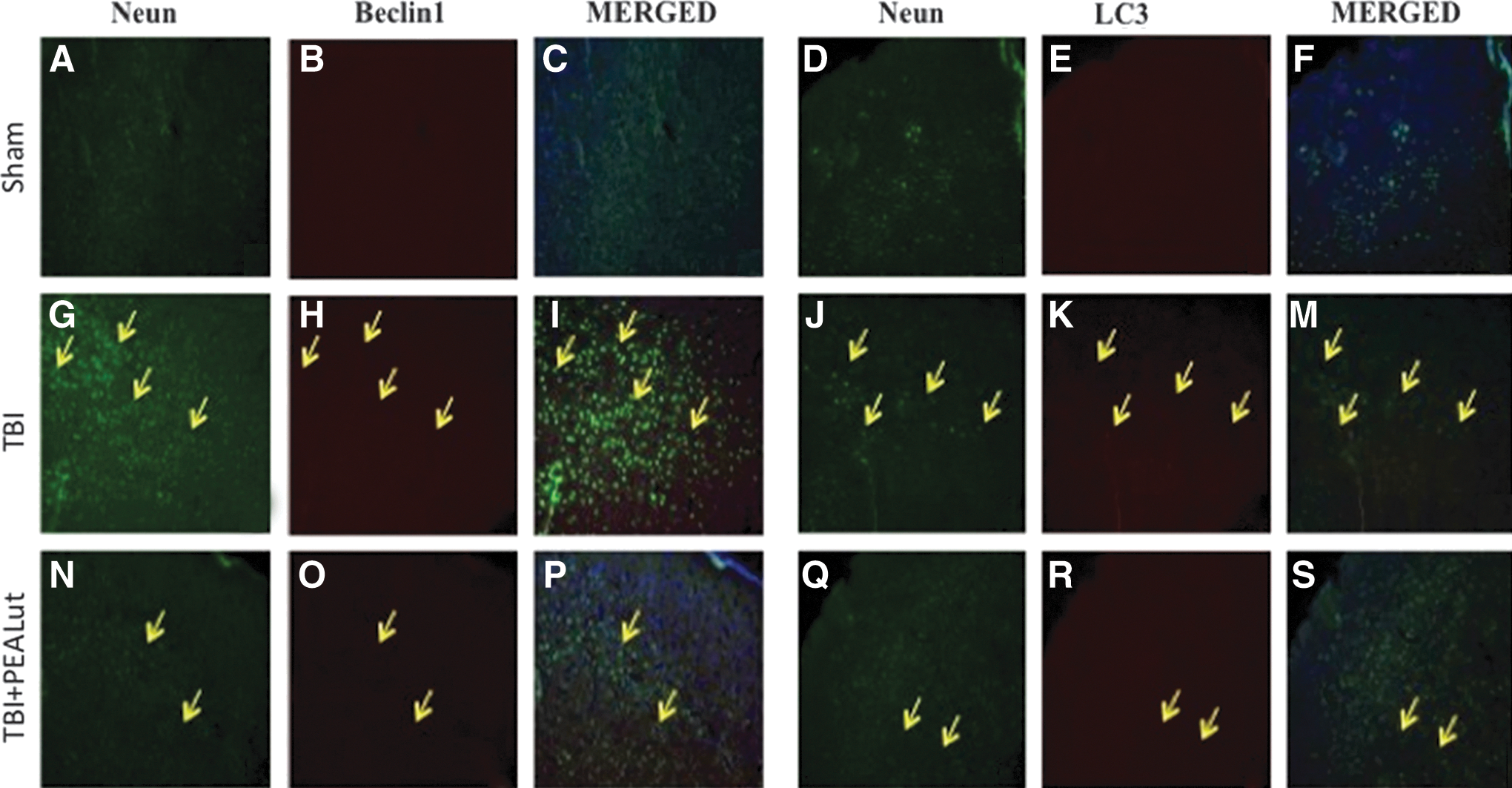
Colocalization of NeuN/Beclin1 and NeuN/LC3 after traumatic brain injury (TBI). Results are shown for (A–F) sham-operated mice, (G–M) mice with TBI, and (N–S) mice with TBI treated with Co-ultraPEALut. Brain sections were double stained with antibodies against NeuN (A, G, N green), Beclin1 (D, J, Q green), and LC3 (B, H, O, E, K, R red). Double-stained sections at 24 h post-injury indicated that both Beclin1 and NeuN were expressed in the same cells. Yellow arrows indicate colocalizations (G–S) and revealed a high colocalization between NeuN/Beclin1 and NeuN/LC3 double staining. Treatment with the Co-ultraPEALut reduced Beclin-1 and LC3 expression levels more effectively than the treatment with PEA administered alone (data not shown). All images were digitalized at a resolution of 8 bits into an array of 2048×2048 pixels. PEA, palmitoylethanolamide; Lut, luteolin.
Discussion
The inflammatory response after traumatic brain injury initiates a series of cellular and molecular events, evolving over the following hours and days, causes neuronal apoptosis, inflammation, autophagy and reactive gliosis, which contribute to secondary tissue loss, impaired regeneration, and associated functional disabilities. 40,41 Within minutes or hours after a traumatic event, a dramatic increase in free radical production occurs leading to the breakdown of membrane lipids, essential proteins, and DNA, ultimately leading to cell death. Factors that influence the outcome of brain injury are numerous and part of a complex network. Current treatment of acute TBI includes surgical intervention and supportive care therapies. Treatment of elevated intracranial pressure and optimizing cerebral perfusion are cornerstones of current therapy. These approaches do not directly address the secondary neurological sequelae that lead to continued brain injury after TBI. Consequently, no neuroprotective treatment options currently exist to improve neurological outcome after TBI.
In our previous study, 8 we examined the behavioral and biochemical changes elicited by the PEA treatment in TBI-injured mice. Briefly, we found that PEA improved neurobehavioral functions, reduced apoptotic cell death, inflammation, edema, improved tissue structure, microglial cells, astrocytes, and MCs in the lesioned area. Because oxidative stress is considered to play an important role in neuroinflammatory disorders and because PEA lacks any direct antioxidant activity, in the present work we studied a new formulation including PEA and the antioxidant compound Lut subjected to an ultramicronization process, “Co-ultraPEALut,” to understand whether the new formulation could ameliorate the secondary injury components of TBI.
The improvement in the neurological function was studied with two different assessments: EBST and rotarod test, behavioral tests widely used. 8 At 24 h after TBI, vehicle-treated animals showed significant impairments in motor deficits as revealed by significantly biased swing activity and shortened time to stay on rotarod. On the contrary, treatment with Co-ultraPEALut, 1 h after TBI, considerably enhanced the recovery and improved motor and behavioral function.
Moreover, to evaluate the effect of Co-ultraPEALut on brain infarctions in the TBI brain, we performed TTC staining. The infarction area and volume were dramatically reduced after treatment with PEA as we previously demonstrated, 8 compared with TBI brain while treatment with Co-ultraPEALut (at the dose of 1 mg/kg) significantly ameliorated the infarct volume and substantially recovered the neurological deficit compared with TBI brain and with PEA treatment alone (at the dose of 10 mg/kg). In addition, histological analysis showed that mice, after treatment with Co-ultraPEALut, presented a strong and important protection on the severity of trauma.
Further, we studied the expression of NF-κB, which is considered an important mediator of inflammation. NF-κ B is one of the major signaling pathways activated when cells are exposed to a variety of stimuli, including cytokines, such as TNF and IL-1, ultraviolet radiation, stress, and pathogenic assaults. In the quiescent state, NF-κB is sequestered in the cytoplasm by its inhibitor of NF-κB (IκB) molecules. On activation, IκB is phosphorylated by the IκB kinase (IKK) complex, composed of three major components: IKK1 (IKKα), IKK2 (IKKβ), and NF-κB essential modulator (NEMO) (IKKγ). 42 Phosphorylation of IκB leads to its degradation, and subsequently, nuclear transport of NF-κB proteins initiates the downstream transcription of target genes. 43 We report that brain trauma caused a significant IκB phosphorylation, IκBα degradation, and, as a consequence, the NF-κB p65 nuclear translocation in the brain tissues 24 h after TBI, whereas Co-ultraPEALut treatment significantly reduced the IκB phosphorylation, the NF-κB translocation, and prevented the IκBα degradation.
Moreover, various experimental evidences have clearly suggested that NF-κB plays a central role in the regulation of many genes responsible for the generation of mediators or proteins in inflammation, such as pro-inflammatory cytokines. 44,45
The expression of various types of cytokines such as the IL-1 family, IL-6, IL-10, and TNF-α increases after head trauma. 46 –48 In our results, IL-1β and TNF-α were significantly higher in the TBI group, but after Co-ultraPEALut treatment were significantly decreased. In particular, microglia were found to be strongly activated, as previously shown by Hernandez-Ontiveros and associates, 49 and thus can be considered a biomarker of TBI.
In addition, in accordance with our previous data, we found that after TBI, there was an enhanced activation of chymase and tryptase. 50 In this study, Co-ultraPEALut significantly decreased the activation of chymase and tryptase compared with TBI and PEA groups.
Despite the apoptotic event, astrocyte activation and proliferation after TBI seem to impair axonal regrowth, but these cells also release neurotrophic factors promoting tissue repair and neurogenesis. 51 In this study, protein analysis indicated that GDNF expression was down-regulated by TBI in mice, while a local, sustained, and significant increase in the perilesioned tissue after intraperitoneally administration of Co-ultraPEALut at minor doses, was more effective than PEA alone.
Several studies have shown that microglial cells activated in response to brain injury from inflammation produced ROS. 52 ROS has been characterized as an important secondary messenger and modulator for various mammalian intracellular signaling pathways, including the mitogen-activated protein kinase (MAPK) pathways—namely, ERK, JNK, and p38 MAPK. 53 Moreover, expression of nitrotyrosine, iNOS, and pJNK have been found near necrotic and inflammatory areas mainly in neutrophils/macrophages, where they play a crucial role in secondary brain damage subsequent to TBI in humans. 54 –56 In our study, we observed an increase of the expression of iNOS and pJNK after TBI that dramatically decreased after treatment with Co-ultraPEALut.
Cell apoptosis or programmed cell death is significant in the maintenance of the intrinsic stability of multicellular organisms. Members of the Bcl-2 family, such as Bcl-2 and Bax, play a significant role in the mitochondrial pathway of apoptosis. 57,58 Bax is a pro-apoptotic factor that is located in the mitochondrial matrix. Bcl-2 is an anti-apoptotic factor that is located in the outer layer of the mitochondrial membrane. Bax and Bcl-2 regulate apoptosis by controlling the activity of proteases and nucleases. Bax promotes apoptosis in response to certain mitochondrial stimuli by inducing the opening of the mitochondrial permeability transition pore to release cytochrome c. Further, because of mitochondrial dysfunction in the injured areas, TBI-induced apoptosis has been demonstrated (by TUNEL staining and Caspase-3 mRNA and protein expression) to be mostly located in areas at the site directly linked to the injury impact. 59 –61
Based on these evidences, we have identified in TBI animals apoptotic features, including a significant increase of Caspase-3 and Bax protein expression. Vice versa, we observed a significant decrease in Bcl-2 expression after TBI. Interestingly, we detected that Co-ultraPEALut treatment significantly reduced TBI-induced Caspase-3 and Bax levels and ameliorated Bcl-2 expression, thus suggesting that Co-ultraPEALut might prevent or diminish levels of apoptosis.
Programmed cell death, however, is not confined to apoptosis. Autophagy is a type of cell death differing from apoptosis by the presence of autophagosomes, autolysosomes, and an intact nucleus in the cells. 62,63
Akt/mTOR/p70S6K pathway in autophagy has been considered a central regulatory pathway of the protein translation involved in regulating cell proliferation, growth, differentiation, and survival. 64,65 mTOR is a central signal integrator that functions as a checkpoint, with upstream Akt and downstream p70S6K being the two most important mediators. 16 In this study, protein analysis indicated that TBI-reduced mTOR and p70S6K, whereas their levels were significantly increased by the treatment with Co-ultraPEALut.
Further, during autophagy initiation and autophagosome formation, Beclin-1 binds microtubule-associated protein-1 light chain 3 (LC3I) that is converted to its membrane-bound form (LC3II) and interacts with the ubiquitin-binding protein p62/sequestosome 1 (SQSTM1). 66 In the current study, we observed an increase of the expression of Beclin-1, LC3, and p62 after TBI that radically decreased after treatment with Co-ultraPEALut.
To confirm our data with fluorescence microscopic examination, we observed a strong redistribution of LC3 with bright punctuate fluorescence around the nucleus that reveals a strong colocalization of NeuN and LC3.
Conclusion
Taken together, our results suggest that Co-ultraPEALut at the dose of 1 mg/kg improved neurobehavioral functions, reduced apoptotic cell death, autophagy, inflammation, edema, improved tissue structure, and reduced microglial and astrocytes activation in the lesioned area better than PEA alone at the dose of 10 mg/kg. These observations indicate Co-ultraPEALut administration as a new therapeutic tool to ameliorate TBI 1 h post-injury.
Footnotes
Acknowledgments
The authors would like to thank Maria Antonietta Medici for her excellent technical assistance during this study and Mr Francesco Soraci for his secretarial and administrative assistance and Miss Valentina Malvagni for her editorial assistance with the manuscript.
Author Disclosure Statement
No competing financial interests exist.