
Abstract
Traumatic brain injury (TBI) is a heterogeneous disease, and the discovery of diagnostic and prognostic TBI biomarkers is highly desirable in order to individualize patient care. We have previously published a study in which we identified possible TBI biomarkers by mass spectrometry 24 h after injury in a cell culture model. Ezrin-radixin-moesin (ERM) proteins were found abundantly in the medium after trauma, and in the present study we have identified extracellular ezrin as a possible biomarker for brain trauma by analyzing cell culture medium from injured primary neurons and glia and by measuring ezrin in cerebrospinal fluid (CSF) from both rats and humans. Our results show that extracellular ezrin concentration was substantially increased in cell culture medium after injury, but that the intracellular expression of the protein remained stable over time. Controlled cortical impact injured rats showed an increased amount of ezrin in CSF at both day 3 and day 7 after trauma. Moreover, ezrin was present in all ventricular CSF samples from seven humans with severe TBI. In contrast to intracellular ezrin, which is distinctly activated following TBI, extracellular ezrin is nonphosphorylated. This is the first report of extracellular ERM proteins in human and experimental models of TBI, providing a scientific foundation for further assessment of ezrin as a potential biomarker.
Introduction
T
The most promising biomarkers for TBI can be found in interstitial fluid, in blood, or in cerebrospinal fluid (CSF). Blood samples are easily obtained, but TBI is often complicated by multitrauma, which can compromise the biomarker profile. Hence, CSF may be the better option because of its proximity to the brain and the injury itself. Many severe TBI patients have ventricular drainage implanted in order to control the intracranial pressure, making CSF samples readily available. 4,5
We have previously published data on possible biomarkers found 24 h after injury in a cell culture model of TBI. By mass spectrometry identification, we found the actin-interacting proteins ezrin and moesin to be of special interest because of their high score. 6 Ezrin and moesin both belong to a family of highly homologous proteins called the ezrin-radixin-moesin (ERM) proteins. The ERM proteins are known to bridge the actin cytoskeleton to the cell membrane and play an important role in most actin-dependent mechanisms; for example, cell motility, cell division, phagocytosis, phagosome maturation, cell signaling, secretion, and relocation of membrane proteins. 7 –9 The activity of the ERM proteins is regulated by phosphorylation of a conserved threonine residue that enables binding to filamentous actin (F-actin), as well as to other proteins in the plasma membrane. 10 –13
Although the intracellular functions of ezrin, moesin, and radixin have been well characterized, the extracellular role of the proteins is unknown. In the present study, we have focused on the extracellular fraction of ezrin and moesin at certain time points after TBI. The investigation includes analysis of primary cell cultures and tissue samples from mice and CSF from both rats and humans, in order to elucidate the scientific foundation for further assessment of ezrin and moesin as potential biomarkers for TBI.
Materials and Methods
Animals
Fifteen Sprague–Dawley male rats, initial weight 310–350 g and 30 C57BL/6 male mice, initial weight 24–27 g (Taconic AS, Denmark) were used for the in vivo experiments in the study. All procedures described herein were approved by the Uppsala County Animal Ethics Board and followed the rules and regulations of the Swedish Board of Agriculture.
Cell culture experiments
Cortices from E14 mice were expanded as neurospheres before being seeded as single cells on coated coverslips as previously described. 14 The cells differentiated in serum-free, B-27 supplemented medium for a period of 8 days before the start of the experiments. A scalpel cutting 30 times through the differentiated neural culture, 10 times perpendicularly in either direction, ∼2 mm apart, followed by 10 cuts diagonally, was used to injure the cells, and uninjured cultures served as controls (n=3). For medium analysis, a total of 800 μL per well was used and medium was collected at 2 h and 2 days from three independent cultures. A protease inhibitor was added to the samples according to manufacturer's instruction (Roche). The tubes were centrifuged for 10 min at 10,000g at 4°C to clear any remaining dead cells or debris from the medium, and the supernatants were then stored in −70°C until analysis.
For preparation of total cell lysates, cell cultures from three independent mice were seeded and injured or left uninjured. The cells were incubated for 1, 3, 5, 7, and 14 days before the cells were washed twice, put on ice, and lysed with lysis buffer (20 mM Tris pH 7.5, 0.5% Triton-X-100, 0.5% deoxycholic acid, 150 mM NaCl, 10 mM ethylenediaminetetraacetic acid (EDTA), 30 mM Na Pyro P) supplemented with protease inhibitor cocktail according to manufacturer's instruction (Roche) and 0.1 M sodium orthovanadate (Na3VO4) (Sigma-Aldrich). The cells were scraped off the glass cover-slips with a cell lifter, and collected in Eppendorf tubes. The tubes were kept on ice for 30 min prior to being cleared of any unlysed material by centrifugation at 4°C, 10,000g for 10 min. The cleared lysates were transferred to new Eppendorf tubes and stored at −70°C until analysis.
Controlled cortical impact (CCI) brain injury and whole-cell lysate preparation from mice
The CCI is a predominately focal TBI model, 15,16 and was performed identically in mice and rats with the exception of the size of the craniotomy (6×6 mm for rats and 4×4 mm for mice), the diameter of the tip of the impactor, and the depth of the impact. The animal was put into a plastic box and anesthesia was induced with 4% isoflurane in air for 2 min, after which the animals were placed in a stereotaxic frame. Anesthesia was maintained using 1.4% isoflurane in a mixture of nitrous oxide and oxygen (70/30%) administered through a nose cone. Core body temperature was maintained at 37±0.3°C using a heating lamp and a heating pad coupled to a rectal probe (CMA150, CMA, Stockholm, Sweden). The scalp was opened by a midline incision after application of bupivacaine as local anesthesia (Marcaine®, AstraZeneca, Sweden). The craniotomy was performed over the left parietal cortex, 1 mm posterior to bregma, with the lateral margin at the crista temporalis. The stereotaxic frame was moved to the CCI-device (VCU Biomedical Engineering Facility, Richmond, VA) and a moderate CCI was performed according to the following parameters; the diameter of the tip of the impactor was 4.5 mm for rats and 3 mm for mice, and the compression depth was 2.0 mm for rats and 0.5 mm for mice. The animals recovered from anesthesia under a heating lamp, and when sufficiently awake they were returned to their home cage. Naïve animals were used as controls.
Tissue lysates were made from mice injured by CCI at 1, 3, 7, 14, or 30 days post-trauma or from naïve mice (controls). Samples of cortical material in close proximity of the injury site, or corresponding cortical portions of naïve animals, were gathered and homogenized manually in 300 μL of lysis buffer. Lysates were centrifuged at 10.000g for 10 min and stored in −70°C until analysis.
CSF extraction in rats
The rats were injected intraperitoneally with a terminal dose of pentobarbital (150 mg/kg) and placed in a stereotaxic frame after becoming unconscious. The skin of the back of the neck was cut, and using fine forceps, the muscles were dissected through to get access to the cisterna magna. Using a 30G syringe, the thin bone above the cisterna magna was pierced, and 50–100 μL of CSF drawn. After extraction, the samples were centrifuged at 4°C at 10,000g for 10 min to clear any cells, and the supernatant was transferred to new tubes and kept at −70°C until analysis.
CSF collection in humans
Human ventricular CSF was obtained from a total of seven severe TBI patients, four patients at two separate time points, one at three separate time points, and two patients with CSF on one occasion while they were being admitted to the neuro-ICU at Uppsala University Hospital, Uppsala, Sweden. Control ventricular CSF was collected from two patients with posterior fossa tumors requiring intracranial pressure monitoring by external ventriculostomy in the neuro-ICU. All samples were taken by external ventriculostomy, and a volume of ∼1 mL per sample was collected in an Eppendorf tube and centrifuged at 4°C for 10 min at 2020g to remove any cells. Samples were stored at −70°C until analysis. The participants or the next of kin, caretaker, or guardian provided their written, informed consent to participate in this study, as mandated by the Regional Ethics Committee of Uppsala University.
Western blot (WB) analysis
Medium and CSF samples (either from rat or human) were normalized by volume and loaded on pre-cast 10 well NuPAGE® Novex® 4–12% Bis-Tris Gels, 1.5 mm. The gels were run under reduced conditions (NuPAGE Reducing agent, 10×) in NuPAGE LDS loading buffer (4×), in NuPAGE MES running buffer supplemented with NuPAGE Antioxidant. LDS sample buffer (×4), reducing agent (×10), and sample were mixed and heated at 70°C for 10 min before loading. A total volume of 35 μL was loaded per well (total sample volume was 22.75 μL per well). Samples were run according to manufactures instruction for 2–3 h at 175 V to ensure proper protein separation. Transfer onto polyvinylidene difluoride (PVDF) pre-cut blotting membranes (0.2 μm) was performed in NuPAGE Transfer buffer with 10 % methanol for 60 min at 30 V. Blocking was performed in 5% Bovine Serum Albumin (BSA, Sigma-Aldrich) in phosphate-buffered saline (PBS) with 0.2 % Tween® 20 (Sigma-Aldrich), hereafter referred to as PBS/Tween. Tissue and whole cell lysates were measured for protein content with Pierce® BCA Protein Assay kit (Thermo Scientific) at 595 nm with a plate reader. Fifteen μg per sample were run on pre-cast NuPAGE Novex 4–12% Bis-Tris Gels 1.5 mm, 15 well, and run as described previously. Amersham ECL™ Prime detection agent and Hyperfilm™ ECL were used throughout the study (GE Healthcare). All materials were purchased from Life Technologies if not otherwise stated.
Immunohistochemistry
Cell cultures were fixed at room temperature (RT) for 15 min in 4% paraformaldehyde (Sigma-Aldrich) in PBS. Blocking and permeabilization was performed in 5% normal goat serum (NGS) in 0.1% Triton X-100 in PBS (PBS/Triton) for 30 min. Cover-slips were incubated with primary antibodies either overnight in 4°C or at RT for 1–4 h. Secondary antibodies were incubated at RT for 1 h, and nuclei were counterstained with 0.5 μg/mL−1 4′,6-diamidino-2-phenylindole (DAPI) in PBS for 5 min, before being mounted with EverBrite (Biotium). All antibodies were diluted in PBS/Triton with 0.5% NGS at concentrations stated subsequently.
Antibodies
Primary antibodies used for WB were all made in rabbit against the following targets: ezrin (#3142), ERM (#3145), moesin (#3146), phospho-ezrin (Thr567)/radixin (Thr564)/moesin (Thr558) (41A3) (phospho-ERM [pERM], #3149), and radixin (#2636), all from Cell Signaling Technology, and all diluted 1:1000. Mouse anti-ezrin (sc-58758, Santa Cruz Biotechnology) diluted 1:500. β-actin antibodies (Imgenex, #IMG-5142A) were diluted 1:2000. HPR-linked anti-rabbit or anti-mouse secondary antibodies (GE Healthcare) diluted 1:25000, were used for WB. For the negative control, only secondary HPR-linked anti-rabbit antibodies were used at 1:25000. Primary antibodies were used for immunostainings against the following targets: rabbit-glial fibrillary acidic protein (GFAP, 1:400) (DAKO, #Z0334), mouse-neuronal class III β-tubulin, (β-tubulin, 1:200) (Covance, #MMS-435P), mouse-2′,3′-cyclic nucleotide-3-phosphodiesterase (CNPase) (1:500) (Sigma-Aldrich, #C5922), rabbit-ERM (1:200) (Abcam, #ab76247), and rabbit-pERM (1:200, see previous description). The following secondary antibodies for stainings were all from Life Technologies and diluted 1:400: Alexa Fluor® 488 goat anti-rabbit IgG (#A-21206), Alexa Fluor 555 goat anti-rabbit IgG (#A-21428), Alexa Fluor 488 goat anti-mouse IgG (#A-11001), and Alexa Fluor 555 goat anti-mouse IgG (#A-21422).
Statistical analysis
Data were analyzed using the Statistica® (StatSoft, Tulsa, OK) software. After testing for normality using the Shapiro–Wilk test, and finding that data were normally distributed, we used parametric tests. Student's t test was used to determine differences in protein levels between injured and uninjured cell culture medium. One way ANOVA with Fisher's least significant difference (LSD) post-hoc test was used to find differences over time for the different proteins in lysates from mice. A p<0.05 was considered statistically significant.
Results
Accumulation of extracellular, but not intracellular, ezrin after TBI in vitro
In order to study extracellular and intracellular expression of ezrin, moesin, pERM and β-actin following TBI, we used a highly reproducible scratch injury model 6 and performed WB analysis of both medium samples and cell lysates. Two hours after injury, there was an almost fourfold increase in ezrin expression in medium compared with the uninjured cell cultures, although the levels varied (3.73±2.9 p=0.15). Two days after injury, the extracellular ezrin levels had increased further in the injured cultures and reached significance (4.16±0.7 p=0.003) (Fig. 1A and B). Moesin content on the other hand, was very low throughout the experiment and remained unchanged after injury (1.09±0.3 p=0.97) (Fig. 1A and B). Similarly, WB showed extremely low concentrations of pERM in both injured and uninjured medium (0.83±0.2 p=0.36), indicating that the fluid-phase ezrin released after injury was unable to bind actin. The extracellular levels of actin showed a nonsignificant increase in β-actin in medium after injury (1.23±0.07 p=0.060). The long-time effect on intracellular ERM expression was studied in whole-cell lysates after injury and compared with parallel uninjured controls. Interestingly, the intracellular levels of all the ERM proteins, phosphorylation of ERM, and β-actin remained stable after injury compared to controls (Fig. 1C–H). Taken together, these results demonstrate that nonphosphorylated ezrin is actively secreted after injury, because a passive leakage would increase the levels of all the proteins.
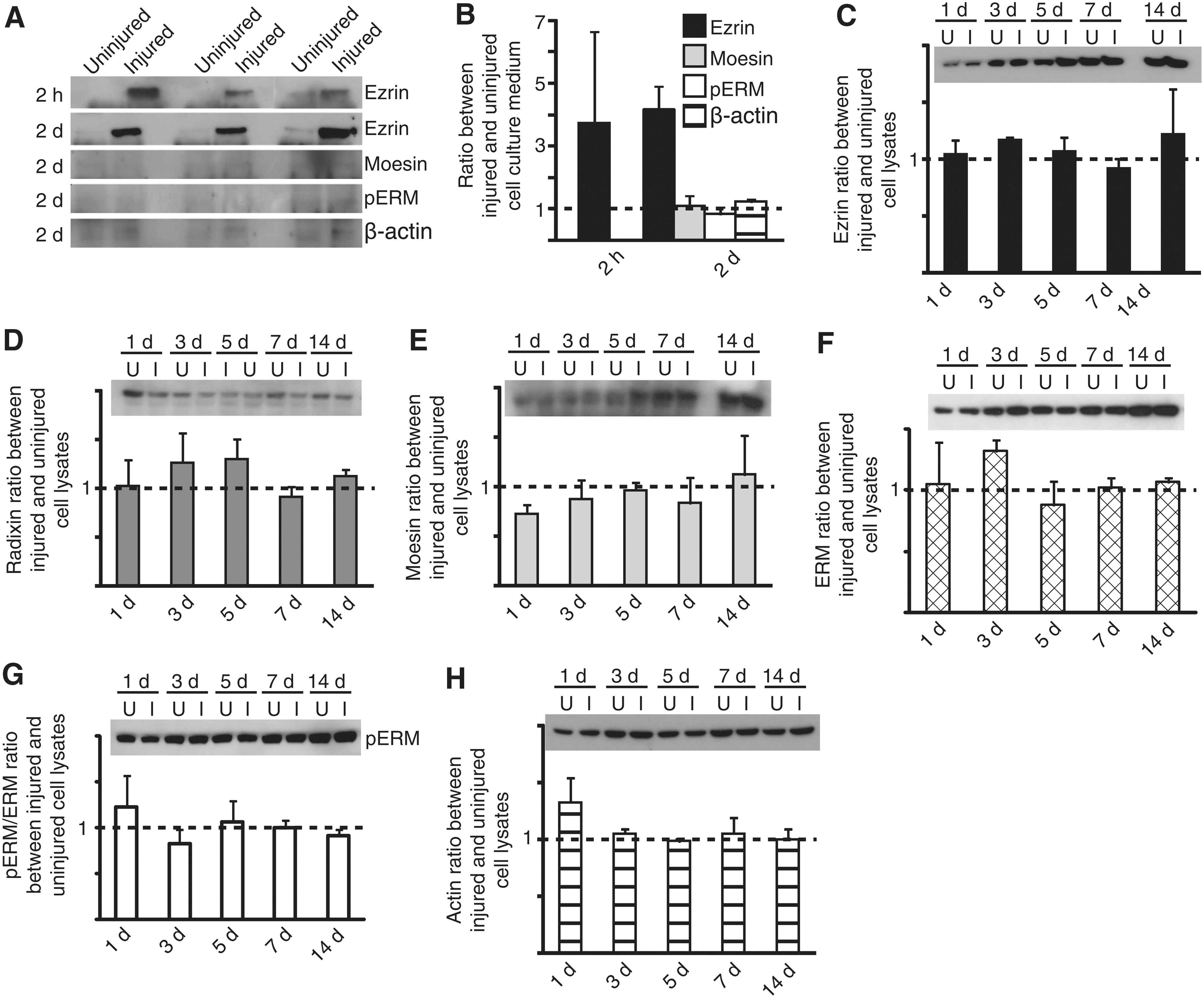
Extracellular ezrin is significantly increased after trauma in vitro. Medium from either uninjured or injured cell cultures were analyzed for ezrin content at 2 h and 2 day
The ezrin content in CSF increases after TBI
To confirm the validity of ezrin as a possible biomarker for TBI, we analyzed CSF from rats and brain tissue lysates from mice following CCI. CSF was collected from rats 3 days (n=5) and 7 days (n=5) post-injury and from naïve animals (n=5). WB showed a significant increase in ezrin at 3 days after experimental TBI (p=0.006) that decreased slightly by 7 days after injury, but was still significantly higher than in controls (p=0.04) (Fig. 2A). In contrast to ezrin, no statistical significant changes were found in extracellular moesin, in injured rats compared with controls, at either time point (p=0.568 and p=0.362 for 3 and 7 days post-CCI, respectively) (Fig. 2B). No pERM bands of the predicted size were detected at either time point.
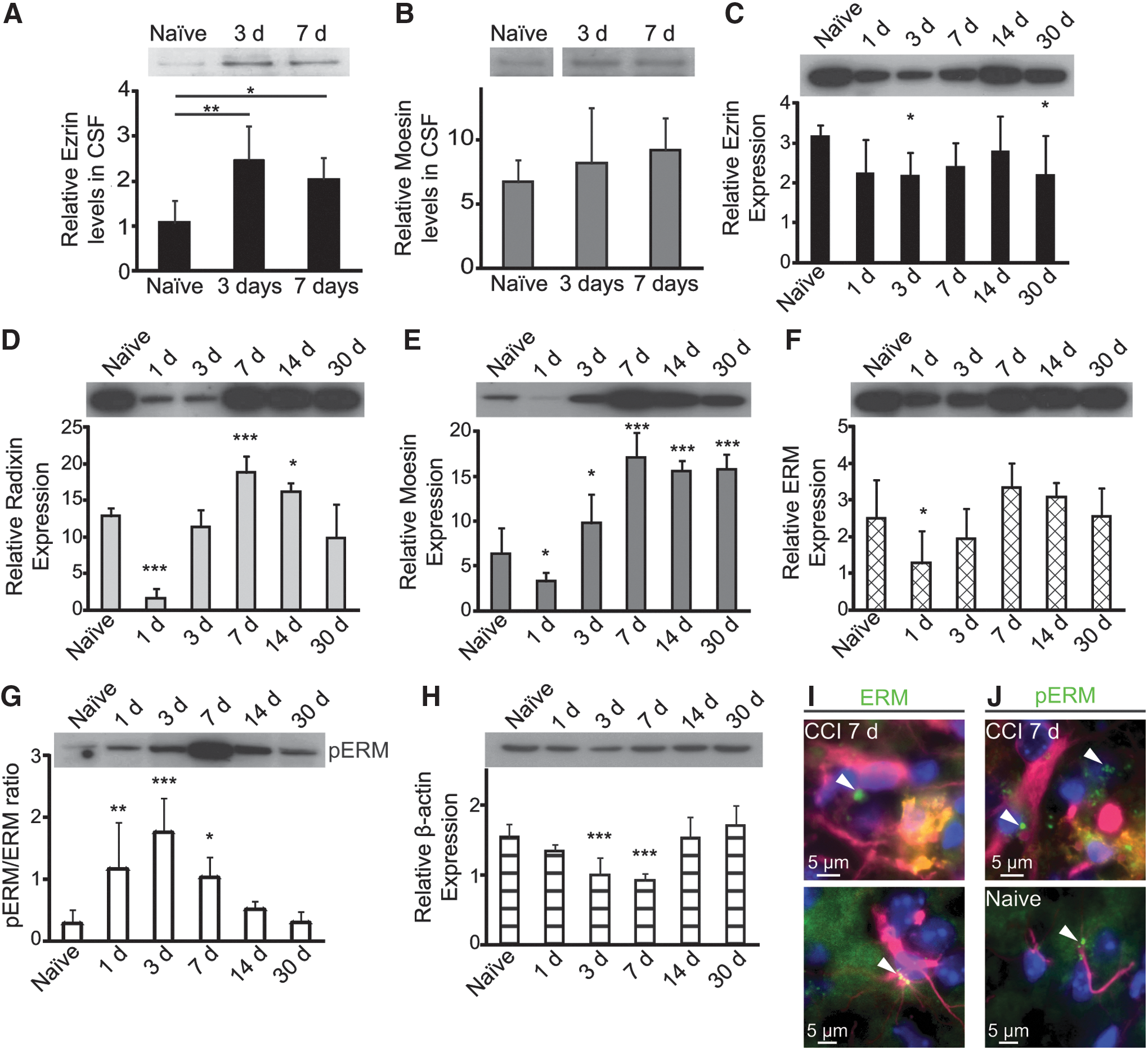
Ezrin is released into the cerebrospinal fluid (CSF) in response to injury, but the intracellular levels remain stable. Ezrin is significantly upregulated in rat CSF 3 and 7 days after injury compared with CSF from naïve rats
Whole-cell, WB analysis after CCI in mice showed a significant downregulation by day 3 of ezrin levels compared with controls (p=0.04) indicating that intracellular ezrin is decreasing after injury in contrast to extracellular ezrin. We also found a significant downregulation 30 days after injury (p=0.04) that might reflect another secondary injury mechanism (Fig. 2C). Radixin, which was not found extracellularly in medium after injury, 6 did show significant changes in cell lysates after CCI in mice. One day after injury, the levels of radixin were highly downregulated (p<0.0001), but the expression increased over time and on day 7, it was instead upregulated (p=0.0008). The levels began to decline again by day 14, but the levels of radixin were still significantly higher in injured than in naïve mice (p=0.04) (Fig. 2D). Similarly to radixin, the levels of moesin were downregulated by day 1 (p=0.04) and then upregulated by day 3 (p=0.03). Moesin levels continued to increase and were highly upregulated by 7, 14, and 30 days after CCI in mice (p<0.0001, for all time points) (Fig. 2E). ERM antibody recognizes all the ERM homologs independently of their activation (phosphorylation) status. The whole-cell analyses of ERM levels demonstrated that the total ERM content was rather stable over time, and only samples from day 1 showed a significant decrease after CCI compared with naïve mice (p=0.02) (Fig. 2F). Although the total level of ERM was decreased 1 day after CCI, the activity of the ERM proteins was significantly upregulated (p=0.003). The ratios between pERM and ERM continued to increase over time to peak at day 3 after injury in mice (p<0.0001) (Fig. 2G). ERM proteins are known to interact with actin when activated, but WB analyses revealed an inverse relationship between the activation status of ERM proteins and β-actin in whole-cell lysates from mice after injury (Fig. 2H). The activity of ERM peaked at day 3, whereas β-actin was highly downregulated (p<0.001) at that time as well as at day 7 (p<0.001) (Fig. 3H). Immunohistochemistry showed that both microglia/macrophages and astrocytes expressed ERM and pERM in vesicle structures after TBI (Fig. 2I–J), and that they could be responsible for the ezrin secretion. In naïve mice, the ERM expression was more diffuse, but could occasionally be found in concentrated, vesicular structures (Fig. 2I–J). Taken together, our studies of whole cell lysates from mouse cortex after TBI showed that ezrin had a very different expression pattern than moesin and radixin, indicating that the proteins may have different intracellular functions.
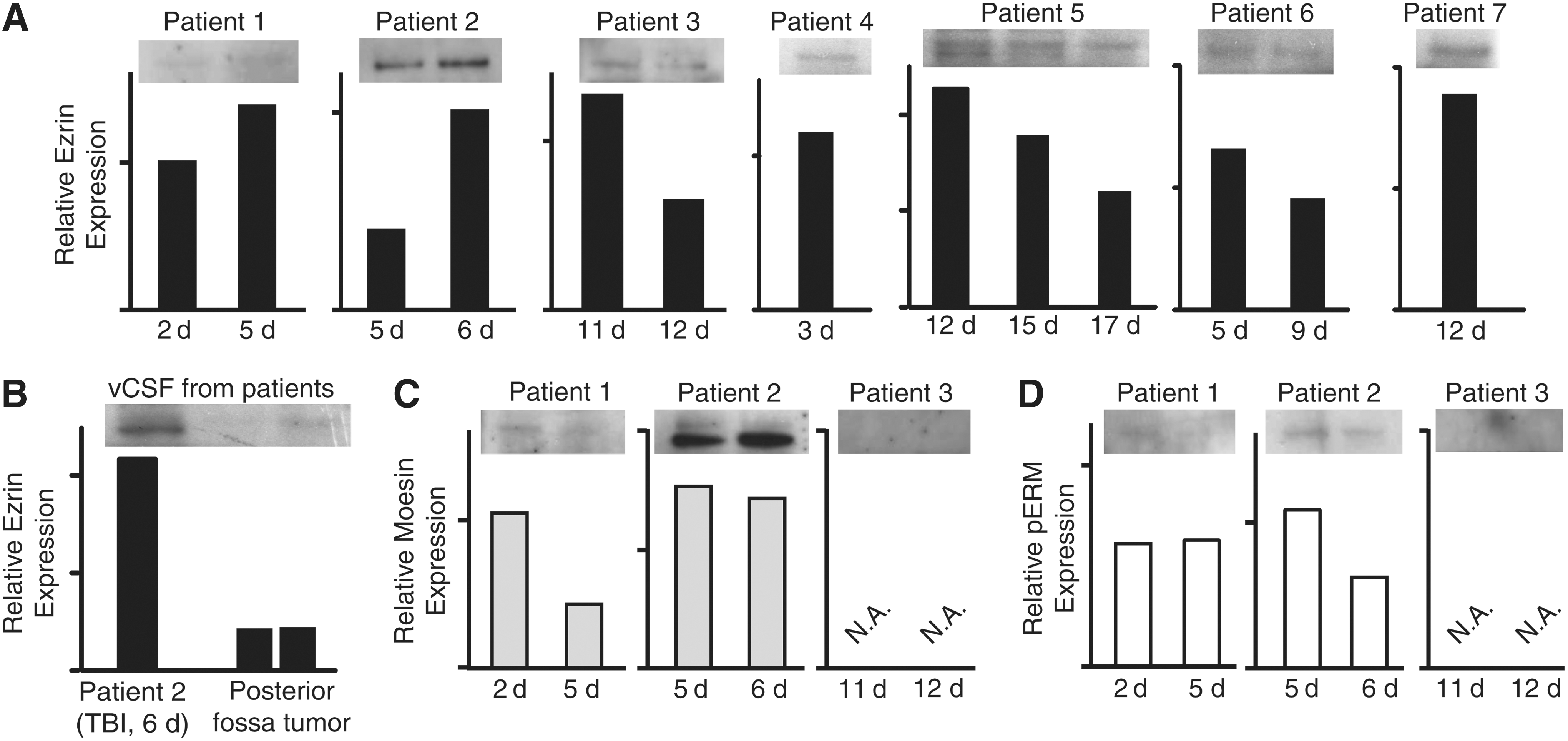
Traumatic brain injured (TBI) patients have ezrin in the cerebrospinal fluid (CSF). TBI patients (n=7) have varying levels of ezrin in ventricular CSF, with temporal patterns with individual variations
Ezrin is present in human CSF after TBI
The next step was to confirm our findings in human samples, and for this purpose, we used ventricular CSF from seven patients with severe TBI at different times after injury. The results show that all patients' CSF contained ezrin, but that the levels varied a lot (Fig. 3A). Patient 1 had very low levels at 2 days post-injury, which increased slightly to day 5. Patient 2, whose samples were taken 5 and 6 days after injury, had markedly more ezrin than all the other patients at both time points. Similar to Patient 1, there was a slight increase at the later time point, 6 days after TBI, compared with day 5. Patient 3 had more ezrin at the earlier time point (11 days post-injury) than at 12 days after TBI. Patient 4 was only represented by one sample that contained ezrin 3 days after TBI. Patient 5's samples were taken at 12, 15, and 17 days after injury and showed reduced levels over time. Patient 6 had low levels at both time points (5 and 9 days after TBI), whereas Patient 7 had rather high levels at 12 days post-TBI. A direct comparison between the patients cannot be made, but the result suggests a delayed secretion of ezrin into human CSF compared with the rat model. As controls, we studied two ventricular CSF samples from patients with posterior fossa tumors and compared the levels to that of Patient 2 (day 6 after TBI) (Fig. 3B). The controls had very low levels of ezrin compared with the injured patient, indicating that ezrin is a possible biomarker for TBI.
Moesin levels were low at both time points in Patient 1 (2 and 5 days after trauma), but showed an inverse expression pattern over time compared with ezrin. Patient 2 had the highest levels of moesin in CSF, and the levels between day 5 and day 6 were almost identical. Patient 3, on the other hand, had no discernible moesin in CSF at either time point (Fig. 3C).
WB of pERM proteins demonstrates very low levels of activation of the extracellular ezrin and moesin, even in Patient 2, who had high levels of both ezrin and moesin (Fig. 3D). Taken together, these data demonstrate that ezrin is present in human CSF after TBI, and that the change in expression differs between individual patients and over time, supporting the notion that it may be useful as a future biomarker.
A fraction of the extracellular ERM proteins may form complexes with F-actin
During the course of the WB analyses of the individual ERM proteins, we discovered bands located at aberrant sizes. The same bands were found with all antibodies tested, and as the ERM proteins are known to interact with actin, but only when phosphorylated, these bands were of particular interest because of the possibilities of protein aggregates in medium and CSF that could have physiological properties.
The bands found in the medium samples included a band >250 kD, another at 250 kD, and a third at 150 kD. All three high-molecular bands were intense and in contrast to the bands of the predicted size, and did not change in response to injury (Fig. 4A). Rat CSF showed one clear high molecular weight band with all antibodies at 250 kD and the 150 kD band was detected at day 3, although much fainter. In contrast to the medium samples, the protein levels of the high molecular weight bands in rat CSF were increased in injured rats compared with naïve animals, and whereas no pERM of the predicted size was found in the CFS samples from the injured rats, a clear increase was observed after injury in the high molecular pERM compared with controls (Fig. 4B). Human CSF from Patient 2 also showed two bands corresponding to the 250 and 150 kD size, with antibodies against ezrin and pERM. WB for moesin and β-actin resulted in only one of the bands in human samples, ∼250 kD (Fig. 4C). In order to discern whether the high molecular bands present in the fluid phase medium were caused by nonspecific binding, two types of controls were performed. Medium 2 days post-injury was incubated with either secondary antibody only or with ezrin antibodies from another distributor (Fig. 4D). The negative controls did not show any bands, whereas the ezrin antibody confirmed the presence of high molecular complexes of ezrin in the medium (Fig. 4D).
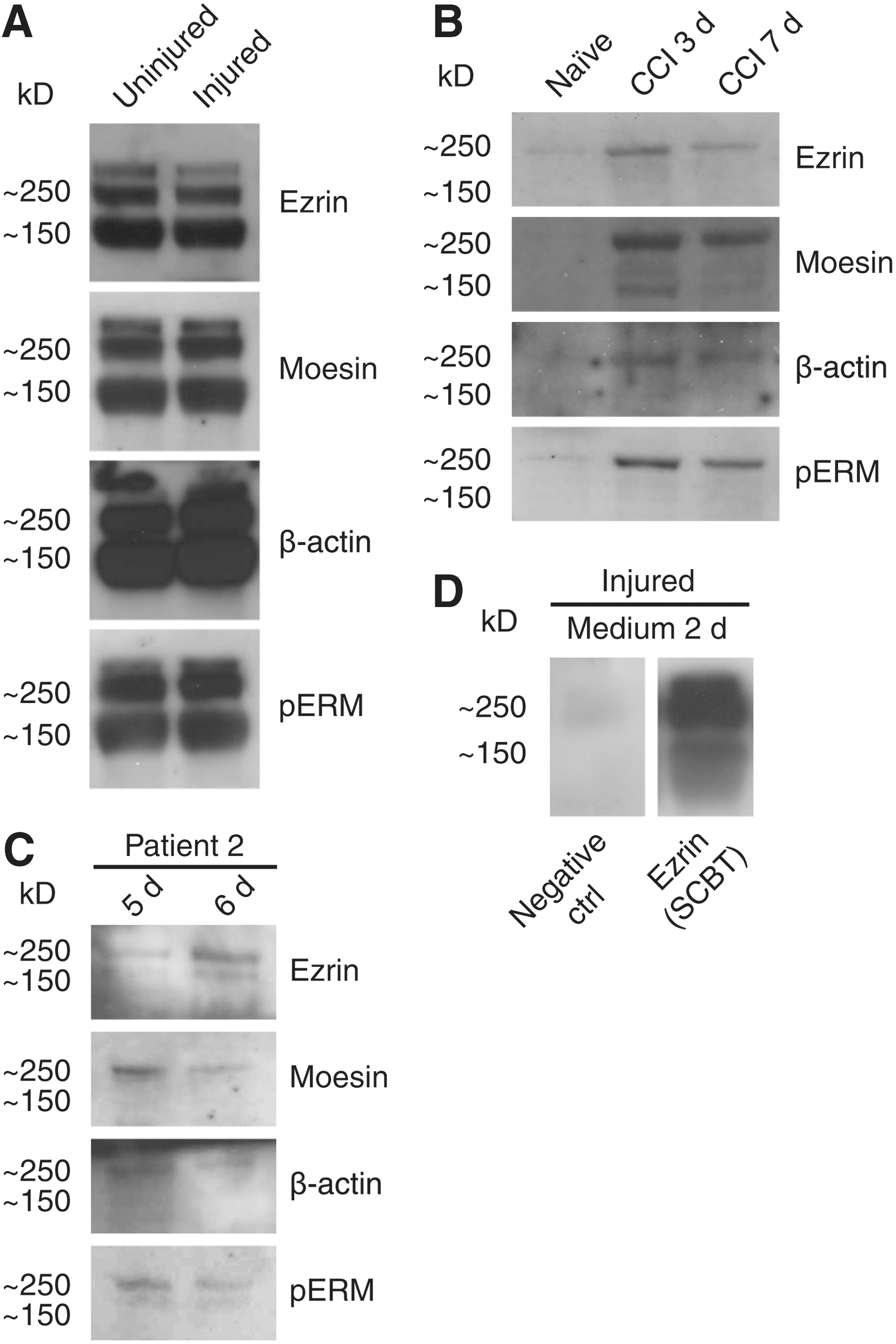
High molecular ezrin-radixin-moesin (ERM) proteins may be complexes of ERM proteins and actin. Western blot (WB) of medium samples revealed three bands at approximate sizes of >250 kD, 250 kD, and 150 kD, which appeared with antibodies against all the individual ERM proteins, β-actin and phospho-ERM (pERM). In contrast to the ezrin of the predicted size, the high molecular bands in the liquid phase did not change in response to injury
Discussion
In this study, we have shown that extracellular ezrin, in its unbound form, is a putative biomarker for TBI. We found that ezrin most likely is released actively from the cells, because moesin, pERM, and β-actin have different expression patterns in cell culture medium than ezrin after injury. Three days after TBI in rats, there is a >200% increase of ezrin in the rat cisternal CSF, and the levels remain high 7 days after trauma. Interestingly, the intracellular levels of ezrin in mice are significantly lowered 3 days after injury, suggesting that the expressed protein is secreted and leaves the immediate injury site. Although extracellular ezrin increases dramatically after TBI in both cell culture medium and in CSF from rats, the intracellular levels of this protein are the most stable over time compared with both radixin and moesin in lysates from injured mice. This validates a specified function for the respective proteins despite their homology and proposed redundancy. 8 The extracellular distribution of ezrin in human ventricular CSF showed that it was indeed present in all samples following TBI, whereas it was extremely low in ventricular CSF from patients with posterior fossa tumors. It is important to note that we have loaded the samples directly on the gel, without any purifications, which means that even a weak band in the WB analysis reflected a rather high amount of extracellular ezrin.
The inverse relationship between ezrin and moesin in patient CSF could indicate release of respective proteins by different cell types. Moesin was barely present in the cell culture medium and did not change in response to injury, whereas ezrin showed a clear upregulation. The cell culture used only contain astrocytes, neurons, and oligodendrocytes and moesin has been shown to be particularly expressed by endothelial cells. 17
Interestingly, WB of β-actin showed the same larger sized (150–250 kD) bands as the WB of ERM and pERM. We hypothesized that some of the extracellular ezrin and moesin may have remained bound to F-actin, and, therefore, did not locate to the estimated size of the respective proteins, although the nature of the interaction remains undetermined at this point. Phosphorylation of ezrin and moesin is vital for interaction with F-actin, and our hypothesis was validated by pERM WBs, showing the same high molecular bands, although there was no phosphorylated ERM of the predicted size. Moreover, antibodies against ezrin fromm another distributor also produced the high molecular bands, whereas the negative control did not. Our cell culture experiments indicated that ezrin was secreted actively, but that necrosis could lead to a passive release of actin-ezrin into the extracellular space and would also produce membrane metabolites. Inactivation of the ERM proteins through ceramide, a metabolite of membrane hydrolysis, happens within minutes. 18,19 Serum has, for example, been shown to contain the ceramide substrate sphingomyelin, 18 which could indicate that a portion of the actin-bound, phosphorylated ezrin becomes inactivated extracellularly after injury and extricates itself from F-actin. The exact mode of release is still to be determined, and could suggest possible functions of the extracellular ezrin.
F-actin has been shown to act as a damage-associated molecular pattern that binds to the C-type lectin domain family 9, member A (CLEC9A, also known as DNGR-1) receptor. 20 This receptor is required for cross-presentation of dead-cell-associated antigens by CD8α+ dendritic cells. 21 Auto-antibodies against moesin have been identified in patients with acquired aplastic anemia, 22 which indicates that the free, extracellular ERM proteins can be presented as antigens, possibly by the activation of CLEC9A receptors on dendritic cells. 21 The activation of CLEC9A and subsequent presentation of self-antigens, as for example ezrin, in TBI could prove detrimental for patient outcome.
Taken together, our data identify ezrin as a possible biomarker for brain trauma, but further investigations are needed to clarify the role of secreted ezrin in the secondary injury cascade after TBI.
Footnotes
Acknowledgments
This study was supported by the Swedish Medical Society, the Tore Nilsson Foundation, the Selander Foundation, and Uppsala University Hospital.
Author Disclosure Statement
No competing financial interests exist.