
Abstract
Oligodendrocyte cell death and axon demyelination after spinal cord injury (SCI) are known to be important secondary injuries contributing to permanent neurological disability. Thus, blocking oligodendrocyte cell death should be considered for therapeutic intervention after SCI. Here, we demonstrated that fluoxetine, an antidepressant drug, alleviates oligodendrocyte cell death by inhibiting microglia activation after SCI. After injury at the T9 level with a Precision Systems and Instrumentation (Lexington, KY) device, fluoxetine (10 mg/kg, intraperitoneal) was administered once a day for the indicated time points. Immunostaining with CD11b (OX-42) antibody and quantification analysis showed that microglia activation was significantly inhibited by fluoxetine at 5 days after injury. Fluoxetine also significantly inhibited activation of p38 mitogen-activated protein kinase (p38-MAPK) and expression of pro-nerve growth factor (pro-NGF), which is known to mediate oligodendrocyte cell death through the p75 neurotrophin receptor after SCI. In addition, fluoxetine attenuated activation of Ras homolog gene family member A and decreased the level of phosphorylated c-Jun and, ultimately, alleviated caspase-3 activation and significantly reduced cell death of oligodendrocytes at 5 days after SCI. Further, the decrease of myelin basic protein, myelin loss, and axon loss in white matter was also significantly blocked by fluoxetine, as compared to vehicle control. These results suggest that fluoxetine inhibits oligodendrocyte cell death by inhibiting microglia activation and p38-MAPK activation, followed by pro-NGF production after SCI, and provide a potential usage of fluoxetine for a therapeutic agent after acute SCI in humans.
Introduction
O
Resident microglia are activated and transformed into macrophages after SCI and contribute to secondary damage by producing proinflammatory cytokines, reactive oxygen species (ROS), nitric oxide, and proteases, which result in tissue destruction, lesion enlargement, and apoptotic cell death of oligodendrocytes as well as neurons. 3,4 Several studies showed that inhibition of microglia activation by minocycline alleviates oligodendrocyte cell death after SCI, thereby improving functional recovery. 5,6 We also reported that p38 mitogen-activated protein kinase (p38-MAPK) is activated in microglia after SCI and thereby mediates pro-nerve growth factor (pro-NGF) production, which induces oligodendrocyte cell death through the p75 neurotrophin receptor (p75NTR)-signaling cascade. 7 In addition, activation of c-Jun N-terminal kinase 3 (JNK3) has been known to be involved in oligodendrocyte cell death as a downstream molecule of Ras homolog gene family member A (RhoA) activation after SCI. 8 Thus, blocking microglia activation appears to prevent oligodendrocyte cell death after SCI. Although, preservation of the WM from secondary injury by inhibiting microglia activation followed by inflammation, thereby attenuation of oligodendrocyte cell death, is critical for limiting functional impairment, there are no effective therapies available clinically to block microglia activation after SCI.
The antidepressant drug, fluoxetine, as a selective serotonin reuptake inhibitor, is known to exert neuroprotective effects by inhibiting neuroinflammation in neurodegenerative animal models. For example, fluoxetine prevents lipopolysaccharide (LPS)-induced degeneration of substantia nigral dopaminergic neurons by inhibiting microglia activation and oxidative stress. 9 It was also reported that fluoxetine provides the neuroprotective effect by inhibiting microglia activation in a cerebral ischemia model and a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinson's disease (PD) model. 10,11 Our recent studies also showed that fluoxetine exhibits the neuroprotective effect by preventing blood–brain barrier (BBB) and blood–spinal cord barrier (BSCB) disruption after transient global ischemia and SCI. 12,13
Here, we examined whether fluoxetine may inhibit oligodendrocyte cell death by attenuating microglia activation after SCI. Our results showed that fluoxetine inhibits apoptotic cell death of oligodendrocytes, axon loss, and myelin loss after SCI by inhibiting microglia activation and p38-MAPK activation followed by pro-NGF production in activated microglia.
Methods
Spinal cord injury
Adult male C57BL/6 (18–22 g; Samtako, Osan, Korea) mice were anesthetized with chloral hydrate (500 mg/kg), and a laminectomy was performed at the T9 level, exposing the cord beneath without disrupting the dura, as previously described. 12 The spinous processes of T8 and T11 were then clamped to stabilize the spine, and the exposed dorsal surface of the cord was subjected to moderate contusion injury (50 kdyn force per 500- to 600-μm displacement) using an Infinite Horizons impactor (Infinite Horizons Inc., Lexington, KY). For sham-operated controls, animals underwent a T9 laminectomy without contusion injury. Surgical interventions and postoperative animal care were performed in accord with the Guidelines and Policies for Rodent Survival Surgery provided by the Animal Care Committee of Kyung Hee University (Seoul, Korea).
Drug treatment
Fluoxetine (Sigma-Aldrich, St. Louis, MO) dissolved in sterile phosphate-buffered saline (PBS) was immediately administered into injured mice by intraperitoneal (i.p.) injection (10 mg/kg) after SCI and then further treated once a day for 14 days for analysis of loss of myelin and axons or for the indicated time points, as previously described. 12 To determine the therapeutic time window, fluoxetine (10 mg/kg) was also administered at 2, 6, and 12 h after SCI and further treated with the same dose once a day for 14 days. PBS was administered for vehicle control. All data presented here are from experiments with fluoxetine administration starting immediately after injury, except in Figure 8. For sham-operated controls, animals underwent a T9 laminectomy without contusion injury and received no pharmacological treatment. Significant side effects, such as changes in body weight or an increase in mortality, resulting from fluoxetine treatment were not observed during our experiments.
Tissue preparation
At the indicated time points after SCI, animals were anesthetized with chloral hydrate and perfused by cardiac puncture initially with 0.1 M of PBS and subsequently with 4% paraformaldehyde in 0.1 M of PBS. A 20-mm section of spinal cord, centered at the lesion site, was dissected out, postfixed by immersion in the same fixative for 4 h, and placed in 30% sucrose in 0.1 M of PBS (pH 7.4). The segment was embedded in optimal cutting temperature compound for frozen sections, and transverse sections were then cut at 10 or 20 μm on a cryostat (CM1850; Leica Microsystems Nussloch GmbH, Nussloch, Germany). For molecular work, animals were perfused with 0.1 M of PBS and segments of spinal cord (8 mm), including the lesion site, were isolated and frozen at −80°C.
Immunohistochemistry
At 5 and 38 days after SCI, frozen sections were processed for immunofluorescence (IF) staining or double labeling (n=18) with antibodies (Abs) against phosphorylated (p)-p38MAPK (1:100; Cell signaling Technology, Danvers, MA), p-c-Jun (1:100; Cell Signaling Technology), cleaved caspase-3 (1:100; Millipore, Billerica, MA), CC1 (1:100; Millipore), CD11b (OX-42; 1:200; Invitrogen, Carlsbad, CA), myelin basic protein (MBP; 1:500; Millipore), and 200-kDa neurofilament protein (NF200; Sigma-Aldrich). Fluorescein isothiocyanate– or cyanin 3–conjugated secondary Abs were used (Jackson Laboratories, Danvers, MA). Also, nuclei were labeled with 4′,6-diamidino-2-phenylindole (DAPI), according to the protocol of the manufacturer's (Invitrogen). For NF200 staining, the ABC method was used to detect labeled cells using a Vectastain kit (Vector Laboratories, Inc,, Burlingame, CA). 3,3′-Diaminobenzidine (DAB) served as the substrate for peroxidase. In all immunohistochemistry controls, reaction to the substrate was absent if the primary Ab was omitted or if the primary Ab was replaced with a nonimmune, control Ab. Some serial sections were also stained with Cresyl violet acetate for histological analysis. For quantification of p-c-Jun and cleaved caspase-3-positive oligodendrocytes, serial transverse sections (10-μm thickness) were collected every 100 μm from 8 mm rostral to 8 mm caudal to the lesion epicenter (total, 160 sections), and p-c-Jun/CC1 or cleaved caspase-3/CC1 double-positive oligodendrocytes in the lateral and ventral funiculus of the WM were manually counted, as previously described. 8
Quantitation of the proportion of resting and activated microglia
Percentage of field analysis was used to provide a quantitative estimate (proportional) of changes in the activation state of microglia, as previously described. 3,14 After IF staining with CD11b Ab (Millipore), images were taken with an Olympus BX-52 microscope connected by a CoolSNAP digital camera (Roper Scientific, Tucson, AZ). Quantitative analysis was performed by blinded observers (S.R.K. and T.Y.Y.) by using MetaMorph software (Molecular Devices, Sunnyvale, CA). For cell-density determination, the number of positively labeled cells was counted for a predefined area of ventral horn. Resting and activated microglia were classified and counted based on a previous report. 3,14,15 Resting microglia displayed small compact somata bearing long, thin, ramified processes. Activated microglia exhibited marked cellular hypertrophy and retraction of processes, such that the process length was less than the diameter of the soma compartment. Cells were sampled only if the nucleus was visible within the plane of section and if cell profiles exhibited distinctly delineated borders. Background levels of signal were subtracted, and control and experimental conditions were evaluated in identical manners.
RNA isolation and real-time reverse-transcription polymerase chain reaction
Total RNA was isolated from spinal cords at 5 days (n=9) after injury using TRIzol Reagent (Invitrogen), according to the manufacturer's instructions. Complementary DNA was synthesized from 5 μg of the total RNA using Moloney murine leukemia virus reverse transcriptase (Invitrogen), and real-time polymerase chain reation (PCR) was performed using SYBR Green PCR master mix (Invitrogen), as previous described. 16 Primers used for NGF and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were synthesized by Genotech (Daejeon, Korea), and the sequences of the primers are as follows (5′-3′): NGF forward, 5′-CAG GCA GAA CCG TAC ACA GA-3′, reverse, 5′-GTC CGA AGA CCT GGG TGG AG-3′; GAPDH forward, 5′- AAC TTT GGC ATT GTG GAA GG-3′; reverse, 5′- GGA GAC AAC CTG GTC CTC AG-3′.
Western blot
For Western blot analysis, segments of spinal cord (8 mm) containing the lesion site were isolated and the tissue homogenates were prepared (n=24), as previously described. 12 Total protein was prepared with a lysis buffer containing 50 mM of Tris-HCl (pH 8.0), 150 mM of NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 10 mM of Na2P2O7, 10 mM of NaF, 10 μg/mL of aprotinin, 10 μg/mL of leupeptin, 1 mM of sodium vanadate, and 1 mM of phenylmethylsulfonyl fluoride (PMSF). Tissue homogenates were incubated for 20 min at 4°C and centrifuged at 25,000g for 30 min at 4°C. Protein sample (50 μg) was separated on SDS/polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane (Millipore). Membranes were blocked with 5% nonfat skim milk or 5% bovine serum albumin in Tris-buffered saline solution with 0.1% Tween 20 for 1 h at room temperature and then incubated with primary Abs against p-p38-MAPK (1:1000; Cell Signaling Technology), p38-MAPK (1:1000; Cell Signaling Technology), pro-NGF (1:1000; Alomone Labs, Jerusalem, Israel), p-c-Jun (1:1000; Cell signaling Technology), c-Jun (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), cleaved caspase-3 (1:1000; Cell Signaling Technology), MBP (1:500; Millipore), and RhoA (1:1000; Santa Cruz Biotechnology) overnight at 4°C. Membranes were then processed with horseradish-peroxidase–conjugated secondary Ab (Jackson ImmunoResearch, West Grove, PA). Immunoreactive bands were visualized by chemiluminescence using Supersignal™ (Thermo Scientific, Pittsburgh, PA). Experiments were repeated three times to ensure reproducibility. Densitometric values of the bands on Western blots obtained by AlphaImager software (Alpha Innotech Corporation, San Leandro, CA) were subjected to statistical analysis. Background in films was subtracted from optical density measurements.
Pull-down assay for Ras homolog gene family member A activity
Purification of glutathione S-transferase (GST)/Rho-binding protein domain (RBD) was performed as described previously. 7 At 5 days after injury, frozen spinal cord tissue was homogenized in a modified radioimmunoprecipitation assay buffer containing 50 mM of Tris (pH 7.2), 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 500 mM of NaCl, 10 mM of MgCl2, 1 μg/mL of aprotinin, 10 μg/mL of leupeptin, and 1 mM of PMSF (n=9). Homogenates were clarified by centrifugation twice for 10 min at 13,000g at 4°C. The supernatant was incubated for 50 min at 4°C with GST-RBD–coupled beads (30 μg/sample). Beads were washed four times with lysis buffer and eluted in sample buffer. Trimeric G-protein (GTP)-bound RhoA and total RhoA present in tissue homogenates were detected by Western blot.
Terminal deoxynucleotidyltransferase-mediated deoxyuridine triphosphate-biotin nick end labeling staining
Five days after injury, serial spinal cord sections (20-μm thickness) were collected every 200 μm and processed for terminal deoxynucleotidyltransferase-mediated deoxyuridine triphosphate-biotin nick end labeling (TUNEL) staining by using the Apoptag in situ kit (Millipore), according to the manufacturer's instructions (n=10). A DAB substrate kit (Vector Laboratories) was used for peroxidase staining, and sections were then counterstained with methyl green. Negative control sections were treated similarly, but incubated in the absence of TdT enzyme, dUTP-digoxigenin, or antidigoxigenin Ab, and positive control sections were incubated in DNase1. Only those cells showing morphological features of nuclear condensation and/or compartmentalization in the WM were counted as TUNEL positive. The investigators who were blind as to the experimental conditions carried out TUNEL analysis. Quantification was assessed by blinded observers (T.Y.Y. and T.H.O.). TUNEL-positive cells in both lateral and ventral funiculus of the WM from 8 mm rostral to 8 mm caudal to the lesion epicenter (total, 80 sections) were manually counted.
Myelin staining
Myelin staining was performed as described previously. 7 Mice treated with vehicle or fluoxetine were anesthetized at 38 days after injury, and frozen sections were prepared as described above. For Luxol fast blue (LFB) staining for myelin, serial transverse cryosections (16-μm thickness) were incubated in 0.1% LFB (Solvent Blue 38; Sigma-Aldrich) in acidified 95% ethanol overnight at 60°C. Differentiation was performed with 0.05% lithium carbonate and 70% ethyl alcohol.
Quantitation of the spared axons
At 38 days after injury, spinal sections were prepared as described above. Axons were stained using an Ab specific for NF200. For quantitative analysis of axonal densities, serial transverse cryosections (10-μm thickness) were collected every millimeter section rostral and caudal 3 mm to the lesion site. Axonal densities were determined within preselected fields (40×40 μm, 1600 μm2) in the ventral funiculus, as previously described. 7 The location of these sites was carefully conserved from group to group using anatomical landmarks, and neurofilament-stained axons were manually counted from each field. The number of axons in vehicle- or fluoxetine-treated spinal cord is expressed as a percentage relative to that in sham control (100%). Quantification was assessed by blinded observers (T.Y.Y. and T.H.O.).
Behavioral tests
Locomotor outcome after spinal cord contusion injury was assessed using the Basso Mouse Scale (BMS; n=50). 17 Mice were scored in an open-field environment by blinded observers (S.R.K. and T.Y.Y.). Consensus scores for each animal were averaged at each time point for a maximum of 9 points for the BMS score and 11 points for the subscore, which assess finer aspects of locomotion.
Statistical analysis
Data are presented as the mean±standard deviation values. Comparison in between vehicle- and fluoxetine-treated groups were made by an unpaired Student's t-test. Multiple comparisons between groups were performed by one-way analysis of variance (ANOVA). Quantitative analysis of axonal densities was analyzed by repeated-measures ANOVA (distance vs. treatment). Behavioral scores from BMS score and BMS subscore were analyzed by repeated-measures ANOVA (time vs. treatment). Tukey's multiple comparison was used as post-hoc analysis. Statistical significance was accepted with p<0.05. All statistical analyses were performed by SPSS software (15.0; SPSS Science, Chicago, IL).
Results
Fluoxetine inhibits microglia activation after spinal cord injury
Microglia are activated after SCI and produce proinflammatory cytokines and mediators, which lead to neuronal and oligodendrocyte cell death. 18 –20 Because an anti-inflammatory effect of fluoxetine was reported in cerebral ischemia and a MPTP-induced PD model, 10,11 we expected that fluoxetine would inhibit microglia activation after SCI. To determine the effect of fluoxetine on microglia activation, IF staining with CD11b Ab, the marker of microglia, was done with cross-sectioned spinal tissues at 5 days after injury. As a result, most microglia in vehicle-treated spinal cord exhibited an activated phenotype at the penumbra region (1 mm) rostrocaudal from the lesion site, with marked cellular hypertrophy and retraction of cytoplasmic processs. 14 However, fluoxetine treatment resulted in a significant reduction (p<0.05) in the proportion of microglia, demonstrating an activated phenotype (Fig. 1A,B), indicating that fluoxetine significantly inhibits microglia activation after SCI.
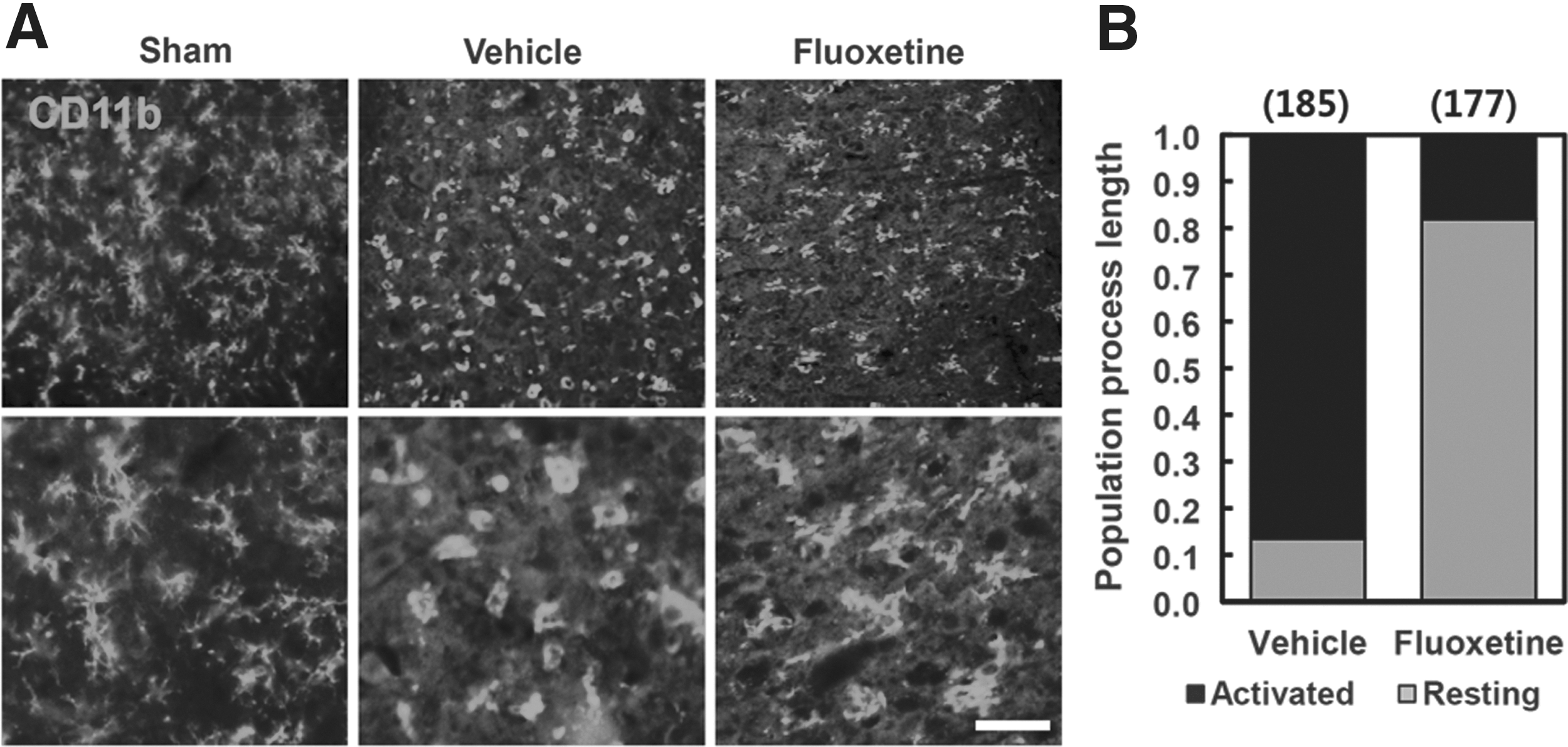
Fluoxetine inhibits microglial activation after spinal cord injury (SCI). After SCI, fluoxetine (10 mg/kg) was administrated immediately by intraperitoneal injection and further treated once a day for 5 days (n=3). (
Fluoxetine inhibits p38 mitogen-activated protein kinase activation and pro-nerve growth factor expression after spinal cord injury
It is known that p38-MAPK mediates inflammatory responses in microglia. 21 Our previous report also showed that oligodendrocyte cell death is mediated by pro-NGF, which is produced by p38-MAPK activation in activated microglia after SCI. 7 Given that fluoxetine reduced the proportion of activated microglia (Fig. 1), we determined whether fluoxetine would inhibit p38-MAPK activation and pro-NGF expression in microglia after SCI. IF staining using an Ab against p-p38-MAPK revealed that immunoreactivity of p-p38-MAPK in uninjured spinal cord was very weak (Fig. 2A, sham). After SCI, there was a marked increase in p-p38-MAPK immunoreactivity (Fig. 2A, vehicle). However, immunoreactivity of p-p38-MAPK was markedly reduced in fluoxetine-treated spinal cord (Fig. 2A, fluoxetine). Western blot analysis also revealed that the level of p-p38-MAPK was increased and peaked at 3 and 5 days after injury, but the level of total p38-MAPK was not changed, as reported previously (Fig. 2B,C). 7 Further, fluoxetine treatment significantly decreased the level of p-p38-MAPK, when compared to vehicle-treated control (vehicle, 6.7±0.61 vs. fluoxetine, 2.2±0.33 at 5 days; n=3; p<0.05; Fig. 2B,C). In addition, levels of NGF messenger RNA (mRNA) and pro-NGF (26 kDa) were significantly inhibited by fluoxetine treatment, when compared to vehicle-treated mice (NGF mRNA: vehicle, 11.4±0.6 vs. fluoxetine, 5.9±0.59 at 3 days; pro-NGF: vehicle, 3.7±0.45 vs. fluoxetine, 2.1±0.33 at 5 days; n=3; p<0.05; Fig. 2D–F). These data indicate that fluoxetine treatment inhibits p38-MAPK activation and pro-NGF expression in microglia after SCI.
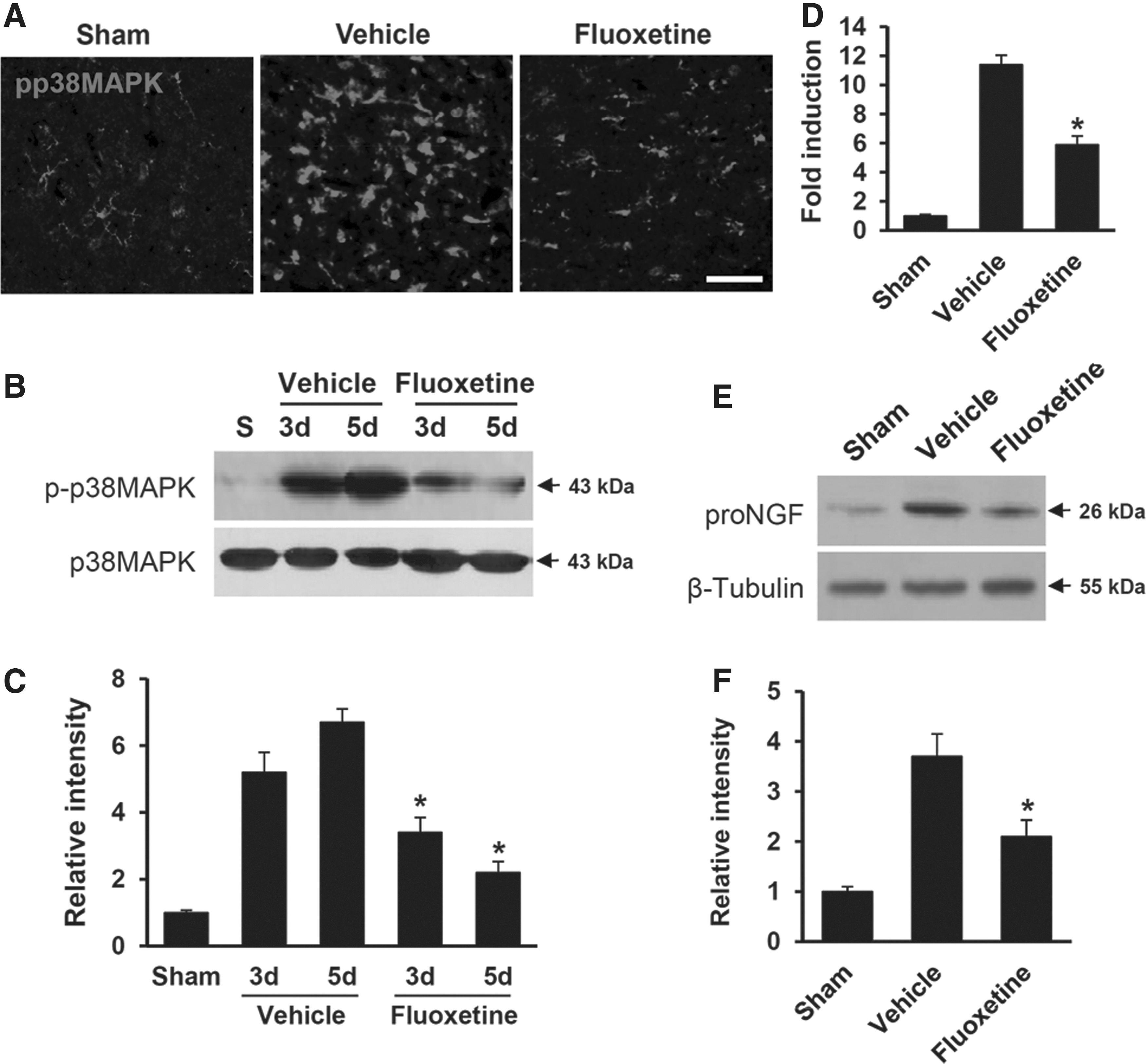
Fluoxetine inhibits p38-MAPK activation and pro-NGF production in microglia after spinal cord injury (SCI). Spinal cord from mice treated with fluoxetine at 3 or 5 days after injury was prepared as described in the
Methods
section (n=3). (
Fluoxetine inhibits Ras homolog gene family member A activation after spinal cord injury
It has been known that RhoA is activated after SCI and involved in oligodendrocyte cell death. 7,8,22 To investigate whether fluoxetine inhibits RhoA activation after SCI, we measured the level of GTP-bound RhoA in spinal cord homogenate from vehicle- and fluoxetine-treated spinal cord by pull-down assays using immobilized GST fusion constructs of the RBD of Rhotekin, as previously reported. 7 The level of GTP-bound RhoA was increased at 5 days after SCI, but the level of total RhoA, as detected by Western blots from tissue extracts used for isolation of GTP-RhoA, was not changed (Fig. 3A), as previously reported. 7 Further, fluoxetine treatment significantly inhibited RhoA activation, when compared to vehicle-treated control (vehicle, 4.6±0.59 vs. fluoxetine, 2.1±0.27; n=3; p<0.05; Fig. 3B). These results suggest that fluoxetine treatment significantly inhibits RhoA activation after SCI.
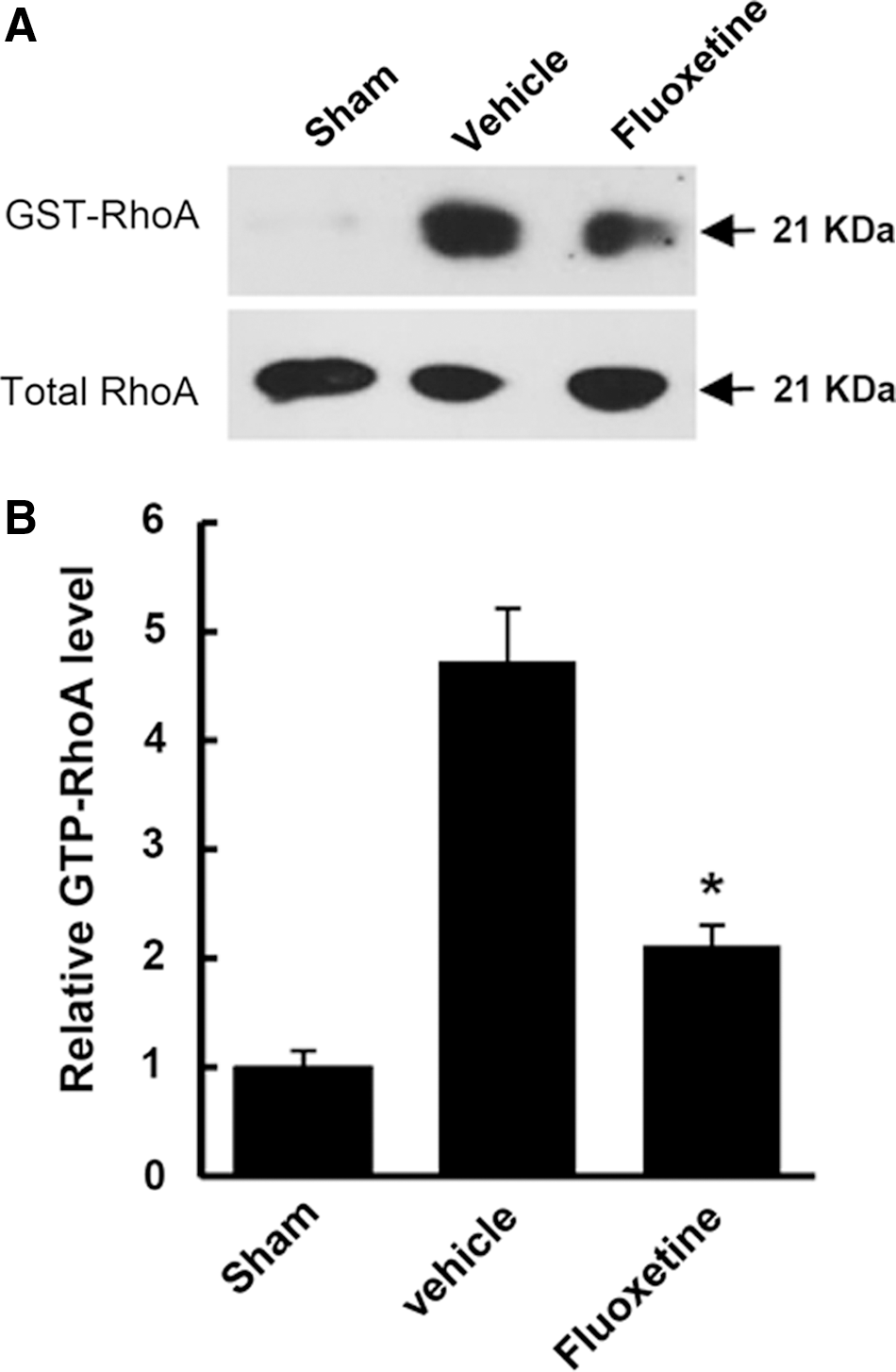
Fluoxetine inhibits RhoA activation after spinal cord injury (SCI). Spinal cord extracts at 5 days after injury were prepared, and GTP-bound RhoA was isolated by pull-down assay and detected by Western blot using RhoA antibody (n=3). (
Fluoxetine inhibits c-Jun phosphorylation in oligodendrocytesafter spinal cord injury
It is also known that JNK3 activation after SCI is implicated in predominantly oligodendrocyte cell death, but not in cell death of neurons. 23 Moreover, we recently demonstrated that RhoA activation mediates JNK3 activation and c-Jun phosphorylation during oligodendrocyte cell death after SCI. 8 Thus, we next examined the effect of fluoxetine on c-Jun phosphorylation, a downstream activator of JNK3, at 5 days after SCI. As shown in Figure 4A, there was little expression of p-c-Jun in sham-operated control. However, the level of p-c-Jun was markedly increased at 5 days after injury, as previously reported. 8 Further, fluoxetine significantly reduced the level of p-c-Jun, compared to vehicle-treated control (vehicle, 4.3±0.5 vs. fluoxetine, 2.7±0.25; n=3; p<0.05; Fig. 4B). These results were also confirmed with double staining and counting the number of p-c-Jun/CC1–positive cells in the WM after SCI. Double staining with CC1, an oligodendrocyte-specific cell marker, showed that p-c-Jun was colocalized in oligodendrocytes (Fig. 4C), as previously reported. 8 However, p-c-Jun was not detected in uninjured normal or sham spinal cord (data not shown). When we counted double-positive cells of p-c-Jun/CC1 at 5 days after SCI, fluoxetine treatment significantly reduced the number of p-c-Jun-positive oligodendrocytes in the WM, as compared to vehicle control (vehicle, 125±11 vs. fluoxetine, 78±15; n=3; p<0.05; Fig. 4C,D), indicating that fluoxetine treatment inhibits c-Jun phosphorylation in oligodendrocytes after SCI.

Fluoxetine reduces the level of c-Jun phosphorylation after spinal cord injury (SCI). Spinal cord extracts and sections were prepared at 5 days after injury for Western blot and immunofluorescence staining using anti-c-Jun and p-c-Jun antibodies (n=3). (
Fluoxetine inhibits caspase-3 activation and cell death of oligodendrocytes after spinal cord injury
It is known that caspase-3 is activated after SCI and mediates apoptotic cell death of neurons and oligodendrocytes. 7,24,25 To investigate the effect of fluoxetine on apoptotic cell death of oligodendrocytes after SCI, we first examined the effect of fluoxetine on caspase-3 activation after SCI. Double labeling with Abs against activated (cleaved) caspase-3 and CC1 revealed that fluoxetine treatment significantly reduced the number of activated caspase-3-positive oligodendrocytes in the WM at 5 days after injury, compared to vehicle control (vehicle, 243±15 vs. fluoxetine, 155±12; n=3; p<0.05; Fig. 5A,B). However, caspase-3-positive oligodendrocytes were not observed in sham-operated tissues (data not shown). In addition, Western blot and quantification analysis showed that the level of cleaved caspase-3 was increased after SCI and its level was significantly decreased in the fluoxetine-treated group, as compared to vehicle control (vehicle, 4.2±0.25 vs. fluoxetine, 2.8±0.37; n=3; p<0.05; Fig. 5C,D), indicating that fluoxetine inhibits caspase-3 activation in oligodendrocytes after SCI.
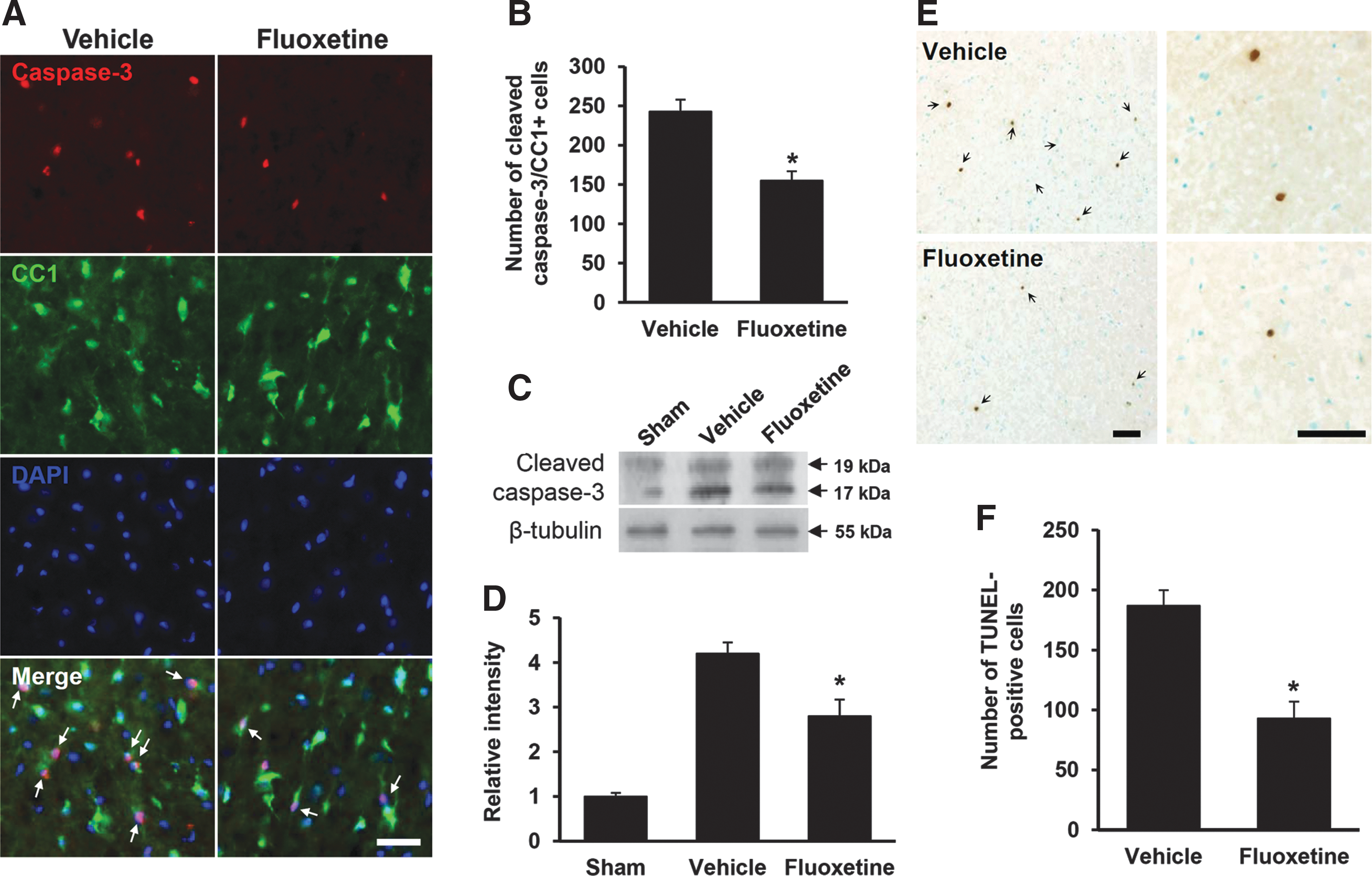
Fluoxetine inhibits oligodendrocyte cell death after spinal cord injury (SCI). At 5 days after injury, spinal cord tissues from sham, vehicle-treated, and fluoxetine-treated mice were prepared and performed with Western blot, immunohistochemistry, and TUNEL staining, as described in the
Methods
section. (
Next, we examined the effect of fluoxetine on apoptotic cell death of oligodendrocytes by TUNEL staining at 5 days after SCI. Serial transverse sections (20-μm thickness) were collected every 200 μm from 8 mm rostral to 8 mm caudal to the lesion epicenter (total, 80 sections), and TUNEL staining was performed. As in our previous reports, most TUNEL-positive cells were observed in the outside of the lesion area, extending the entire length of the section (20 mm) in the WM, which are identified as most oligodendrocytes 24,26 (Fig. 5E). Counting analysis showed that fluoxetine treatment significantly decreased the numbers of TUNEL-positive cells in the WM at 5 days, when compared to vehicle-treated control (vehicle, 187±13 vs. fluoxetine, 93±14; n=3; p<0.05; Fig. 5F). Thus, our results indicate that fluoxetine inhibits apoptotic cell death of oligodendrocytes after SCI.
Fluoxetine inhibits axon and myelin loss after spinal cord injury
Given that loss of oligodendrocytes results in demyelination and secondary axon damage, 27 –29 we examined the effect of fluoxetine on loss of myelin and axons after SCI. The extent of myelin loss at 38 days after injury was assessed by LFB staining. Myelin loss was observed rostrally and caudally from the lesion site after injury. As shown in Figure 6A, representative images indicated that extensive myelin loss was observed in the lateral column of the WM at 2000 μm rostral from the lesion after injury, as compared to sham control (Fig. 6A, vehicle), whereas fluoxetine treatment markedly attenuated myelin loss (Fig. 6A, fluoxetine). Myelin integrity was also evaluated by immunostaining and Western blot with an anti-MBP Ab. Immunostaining revealed that the intensity of MBP immunoreactivity was higher in fluoxetine-treated mice than that in vehicle-treated mice at 38 days after injury (Fig. 6B). Further, Western blot analysis showed that the level of MBP was markedly decreased at 14 days after SCI, as compared to sham control, as previously reported. 24 However, the level of MBP was significantly higher in the fluoxetine-treated group, when compared to the vehicle-treated group (vehicle, 0.27±0.1 vs. fluoxetine, 0.76±0.08; n=3; p<0.05; Fig. 6C,D).
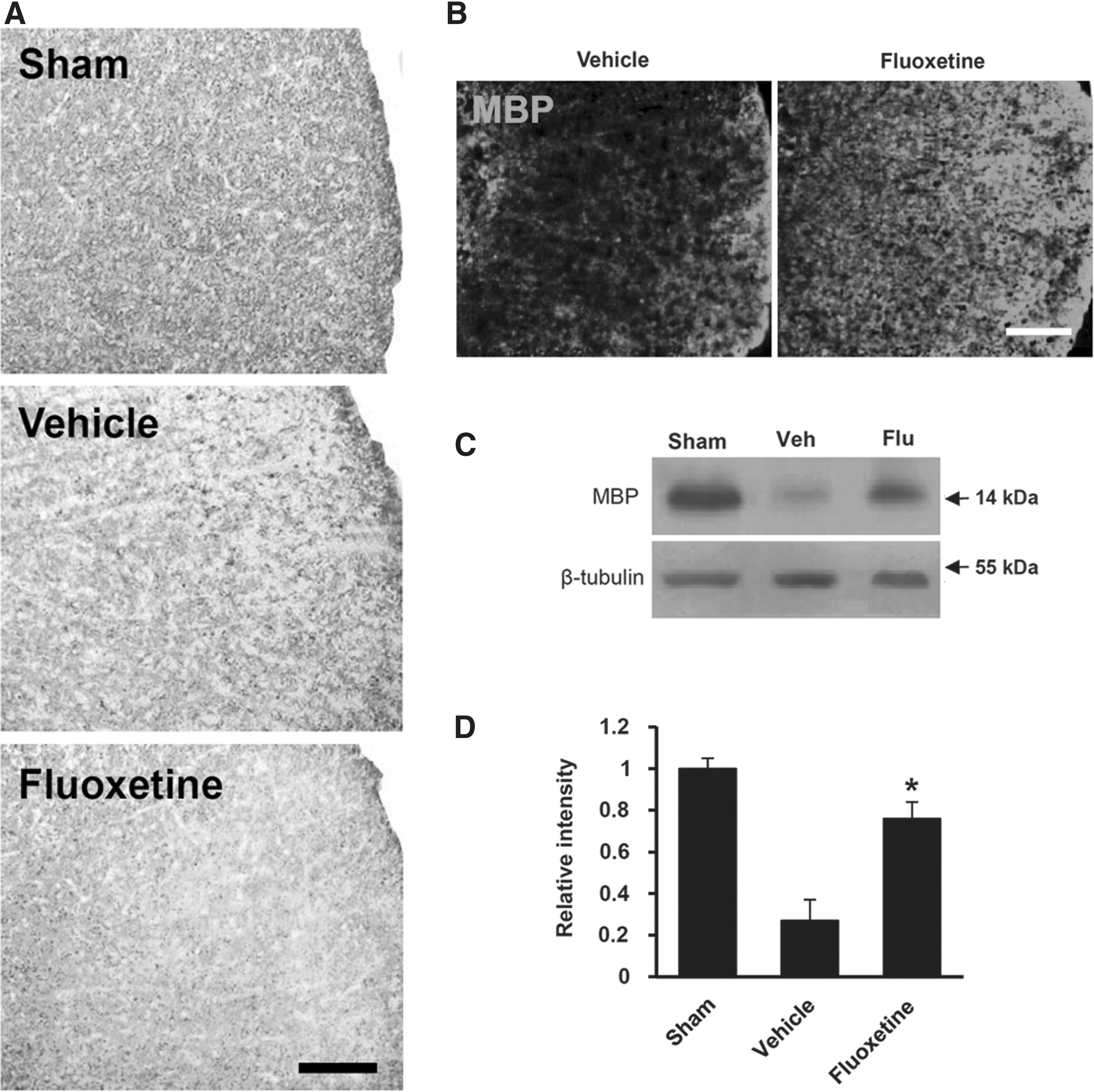
Fluoxetine alleviates myelin loss after spinal cord injury. Spinal cords at 38 days after injury were processed for Luxol fast blue (LFB) and MBP immunostaining (n=3). (
To determine whether fluoxetine preserves axons after SCI, immunstaining with NF200 Ab was performed to detect remaining axons at 38 days after injury. In sham controls, NF200-positive axons in the WM were dense and axonal packing was uniform (Fig. 7B, sham). By contrast, density of axons in injured tissues was markedly decreased and exhibited a patchy distribution (Fig. 7B, vehicle). Quantification analysis showed that the number of NF200-positive axons in the ventral funiculus at 3 mm rostrally and caudally was significantly higher in the fluoxetine-treated group, as compared to those in the vehicle-treated control group (rostral, 2 mm; vehicle, 39±8% vs. fluoxetine, 60±6%; n=3; p<0.05; Fig. 7B,C). These results indicate that fluoxetine treatment reduces loss of myelin and axons after SCI.
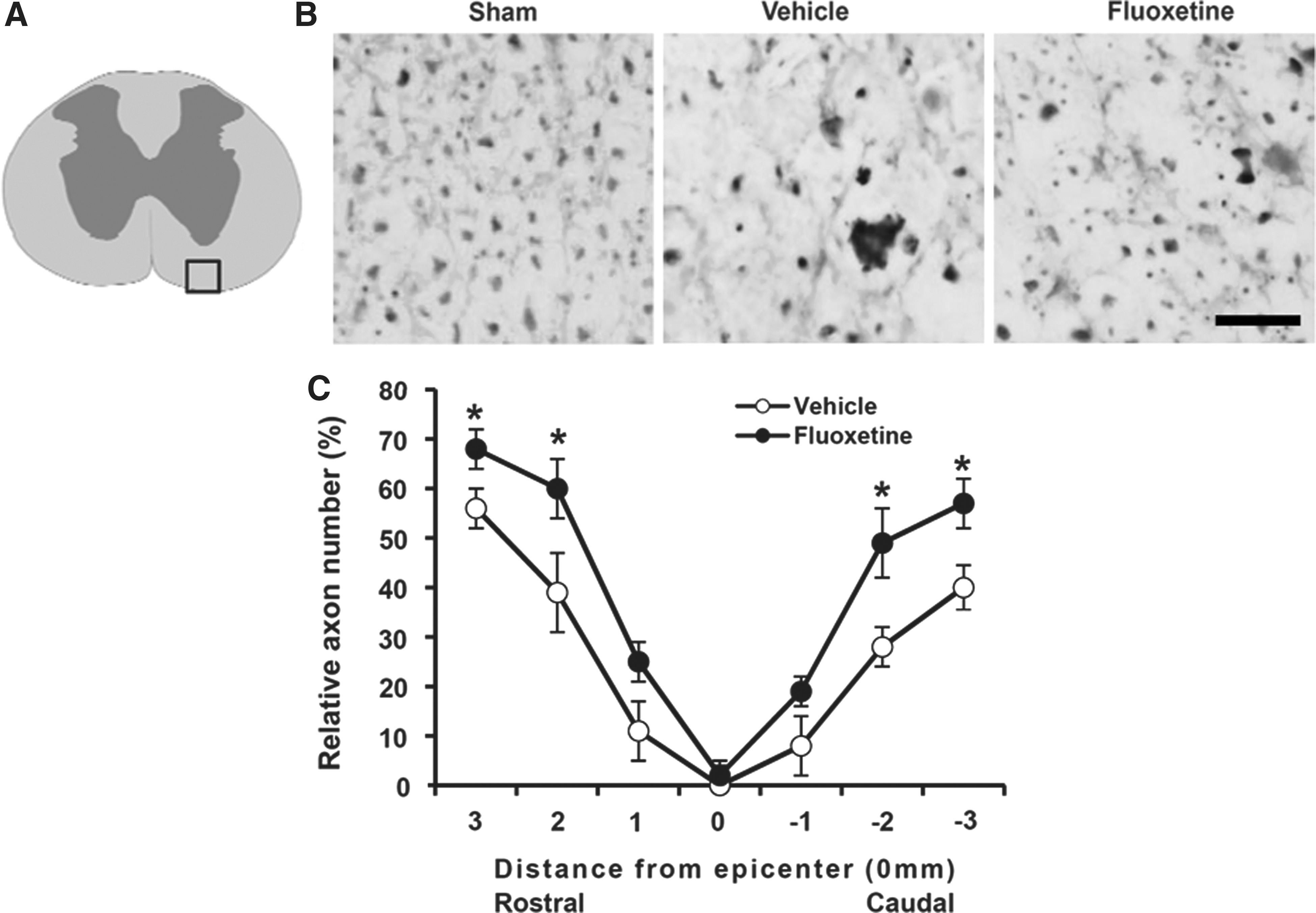
Fluoxetine attenuates axon loss after spinal cord injury (SCI). At 38 days after SCI, spinal cord transverse cryosections were selected 3 mm rostrocaudal to the lesion site for neurofilament staining with 200-kDa neurofilament protein (NF200) antibody (n=3). (
Delayed administration of fluoxetine also improves functional recovery after spinal cord injury
The time window for administration of neuroprotective compounds in acute SCI is a critical factor with respect to its possible therapeutic use. To determine the therapeutic time window, fluoxetine (10 mg/kg) was also injected at 2, 6, and 12 h after injury and further treated with the same dose once a day for 14 days; locomotor functional recovery was then evaluated using the 9-point BMS score and 11-point BMS subscore. As shown in Figure 8, fluoxetine treatment at 2 and 6 h after SCI significantly improved both BMS score and BMS subscore, as compared to those of the vehicle-treated control. However, when fluoxetine was treated at 12 h after injury, the BMS score and BMS subscore were not different from those of the vehicle-treated control.
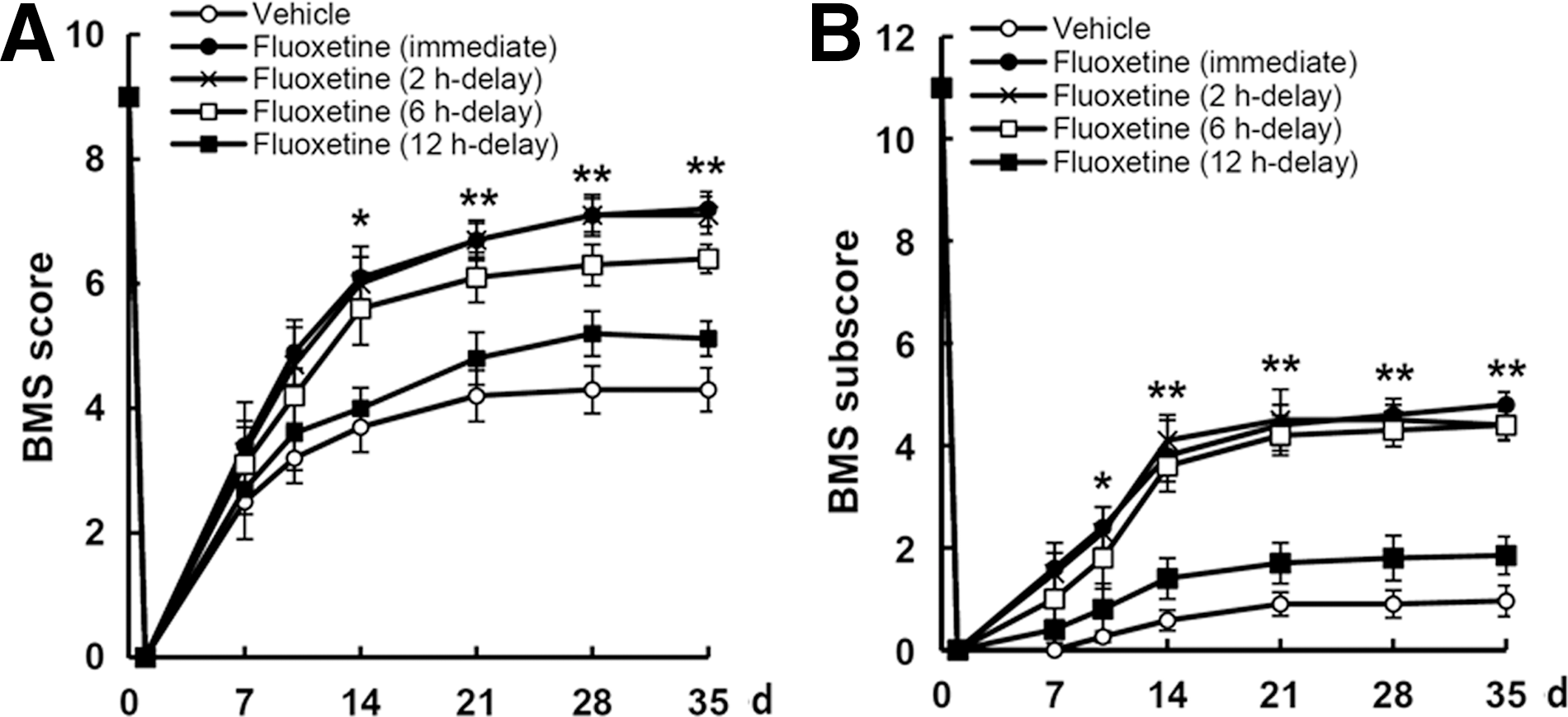
Delayed administration of fluoxetine improves functional recovery after spinal cord injury (SCI). After SCI, fluoxetine (10 mg/kg) was administered at 2, 6, and 12 h after injury and then treated with the same dose of fluoxetine once a day for 14 day, and functional recovery was assessed by BMS score and BMS subscore (n=10). Fluoxetine treatment (given at 2 and 6 h after injury) significantly improved locomotor function, when compared to vehicle. Values are presented as means±standard deviation. *p<0.05; **p<0.01 versus vehicle control. BMS, Basso Mouse Scale; d, days.
Discussion
Here, we demonstrated that post-treatment with fluoxetine after SCI inhibits microglia activation and p38-MAPK activation, followed by pro-NGF expression, in microglia. Fluoxetine treatment also inhibited RhoA activation and c-Jun phosphorylation in oligodendrocytes after SCI. In addition, we showed that fluoxetine treatment inhibited caspase-3 activation in oligodendrocytes and attenuated apoptotic cell death of oligodendrocytes at 5 days after injury. Finally, our data showed that both myelin and axon loss in the WM after SCI are significantly inhibited by fluoxetine treatment.
Fluoxetine, used as an antidepressant, has been known to exhibit anti-inflammatory effects. 30,31 Fluoxetine is also known to improve motor recovery in stroke patients. 32,33 In addition, the neuroprotective effect of fluoxetine is known to be mediated by its anti-inflammatory effect after middle cerebral artery occlusion, 11 as well as MPTP- and LPS-induced degeneration of nigral dopaminergic neurons by inhibiting microglia activation. 9 In this study, we found that fluoxetine also inhibits activation of microglia after SCI and attenuates p38-MAPK activation, followed by pro-NGF expression (see Figs. 1 and 2), which is elucidated by our group to occur in activated microglia after SCI. 7 Further, we recently reported that fluoxetine prevents disruption of the BSCB after SCI by inhibiting activation of matrix metalloprotease 12 and inhibits transient global ischemia-induced hippocampal neuronal death and memory impairment by preventing BBB disruption. 13 These reports also suggest that fluoxetine attenuates infiltration of inflammatory blood cells, such as neutrophils and macrophages, thereby inhibiting inflammatory reaction after SCI. Based on our studies, including this study, this indicates that fluoxetine exhibits a neuroprotective effect by inhibiting inflammation, which might be mediated by activated microglia and/or BSCB disruption, followed by blood cell infiltration, after SCI. However, it should be pointed out that the precise action mechanism of fluoxetine is still unknown.
Activated microglia after SCI produce proinflammatory cytokines and ROS, which contribute to secondary damage. 34,35 It has been shown that attenuation of microglia activation inhibits oligodendrocytes cell death and demyelination after SCI. 6,7,36 Although NGF can be considered as one of the therapeutic options for neurodegeneration, 37 both NGF and pro-NGF expression are increased by a variety of pathological conditions, including SCI, and lead to p75NTR-mediated apoptosis of oligodendrocytes after SCI. 38 In addition, we previously reported that pro-NGF is produced in activated microglia by p38-MAPK-mediated signaling after SCI, indicating that activated microglia are a major cell type producing pro-NGF and contributing to cell death of oligodendrocytes after SCI. 7 The present study also shows that fluoxetine treatment after SCI inhibited p38-MAPK activation and pro-NGF expression in microglia (see Figs. 1 and 2). Given that the present study was focused on the protective effect of fluoxetine on apoptosis of oligodendrocytes after SCI, we determined the effect of fluoxetine on production of pro-NGF, a death-inducing ligand for p75NTR. However, the level of mature NGF that was increased after SCI was not examined in this study. In addition, fluoxetine treatment significantly inhibited caspase-3 activation and apoptotic cell death of oligodendrocytes after SCI (see Fig. 5). Thus, our data suggest that the antiapoptotic effect of fluoxetine on oligodendrocyte cell death may be mediated, in part, by decreasing pro-NGF production through inhibition of p38-MAPK activation in microglia after SCI. However, the mechanism for the inhibition of microglia activation, p38-MAPK activation, and pro-NGF production by fluoxetine requires further study.
A recent report indicated that microglia activation is not always associated with oligodendrocyte apoptosis after SCI. The report by Sun and colleagues 39 showed that the extent of oligodendrocyte cell death was different between rhizotomy and contusion injury, although similar amounts of axonal degeneration and microglia activation were observed in both injury models. These results indicate that different stimuli may induce the different activated status of microglia and thereby differentially affect neighboring cells. It has been suggested that microglia are highly heterogeneous, and cells exhibiting the same morphological features of activation can apparently be detrimental, protective, or can induce differentiation, depending upon the presence of different environmental stimuli. 40 –42 Thus, we cannot rule out a possibility that fluoxetine may influence the status of activated microglia, and further study is needed to examine the effect of fluoxetine on the status of activated microglia.
It is known that RhoA activation contributes to apoptotic cell death and axonal degeneration after SCI. 22,43 Some reports showed that inhibition of Rho or Rho kinase, an effector of Rho, improves axonal regeneration and functional recovery after SCI. 44,45 In addition, we reported that 17β-estradiol inhibits apoptotic cell death of oligodendrocytes by inhibiting RhoA and JNK3 activation after SCI. 8 In this study, we also found that fluoxetine treatment significantly inhibited RhoA activation and decreased the level of p-c-Jun after SCI (see Figs. 3 and 4). Further, loss of myelin and axons after SCI were significantly attenuated by fluoxetine treatment, when compared to vehicle control (see Figs. 6 and 7). A significant sparing of axons within the ventral and dorsolateral funiculus is known to contribute to locomotor control. 46 Our data demonstrated that density of spared axons within the ventral funiculus was higher in the fluoxetine-treated group than the vehicle-treated control group (see Fig. 7). In mice, ventral funiculus of the spinal cord contains the vestibulospinal tract, which is known to be important for initiation of stepping. 46,47 Therefore, these results indicate that the effect of fluoxetine on locomotor behavior after SCI may be mediated, in part, by preventing axonal loss in the ventral funiculus. Taken together, our results suggest that inhibition of oligodendrocyte cell death, as well as myelin and axon loss by fluoxetine, might, in part, be mediated by inhibition of RhoA activation after SCI. However, the specific type of axonal tract preserved by fluoxetine after injury was not examined in the present study. Thus, further study examining the mechanism underlying fluoxetine-mediated inhibition of RhoA activation and axon degeneration after SCI is needed.
Although methylprednisolone, as an anti-inflammatory agent, has been used for treatment of acute SCI in humans, no beneficial effect can be guaranteed in SCI patients if it is not administered early after injury. 48 Therefore, the determination of the therapeutic time windows for candidate agents is pivotal to judge the possibility for clinical use after SCI. Here, our data showed that fluoxetine significantly improved functional recovery when administered at 2 and 6 h after injury (see Fig. 8). These results provide a possibility that fluoxetine, showing the anti-inflammatory effect as well as the blocking effect of BSCB disruption after SCI, can be potentially useful as a therapeutic agent for acute SCI.
Footnotes
Acknowledgments
The authors thank Dr. Tae H. Oh at Kyung Hee University for assistance with data analysis. This research was supported by the Pioneer Research Center Program through the National Research Foundation of Korea funded by the Ministry of Science, Information and Communications Technology & Future Planning (grant no.: 2010-0019349) and a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare (grant no.: HI13C14600000), Republic of Korea.
Author Disclosure Statement
No competing financial interests exist.