
Abstract
Microthrombus formation and bleeding worsen the outcome after traumatic brain injury (TBI). The aim of the current study was to characterize these processes in the brain parenchyma after experimental TBI and to determine the involvement of coagulation factor XI (FXI). C57BL/6 mice (n = 101) and FXI-deficient mice (n = 15) were subjected to controlled cortical impact (CCI). Wild-type mice received an inhibitory antibody against FXI (14E11) or control immunoglobulin G 24 h before or 30 or 120 min after CCI. Cerebral microcirculation was visualized in vivo by 2-photon microscopy 2-3 h post-trauma and histopathological outcome was assessed after 24 h. TBI induced hemorrhage and microthrombus formation in the brain parenchyma (p < 0.001). Inhibition of FXI activation or FXI deficiency did not reduce cerebral thrombogenesis, lesion volume, or hemispheric swelling. However, it also did not increase intracranial hemorrhage. Formation of microthrombosis in the brain parenchyma after TBI is independent of the intrinsic coagulation cascade since it was not reduced by inhibition of FXI. However, since targeting FXI has well-established antithrombotic effects in humans and experimental animals, inhibition of FXI could represent a reasonable strategy for the prevention of deep venous thrombosis in immobilized patients with TBI.
Introduction
T
A major factor of the pathogenesis of TBI is the reduction of cerebral blood flow down to ischemic levels, which result in neuronal cell death and subsequent poor functional outcome. 3 –7 Various mechanisms, such as vasoconstriction and microthrombosis, were discussed as being responsible for post-trauma ischemia. 8 –15 Since large extraparenchymal cerebral vessels seem to be neither spastic nor functionally impaired, the reason of post-trauma ischemia was increasingly believed to be located on the level of the cerebral microcirculation.
One possible explanation of how microvascular flow can be impaired is the formation of microthrombosis. The formation of microthrombi after TBI was reported in patients 11,16,17 and by histology in animal studies. 8 –10,14,17 –21 However, investigations of thrombus formation after TBI in vivo were, so far, limited to the pial microcirculation, 15,22 which is part of the cerebral microcirculation but does not provide nutritive perfusion to the brain parenchyma. Accordingly, the dynamics and mechanisms of post-trauma thrombus formation in the parenchymal microcirculation, which ultimately determines the fate of injured tissue, remain poorly understood. Therefore, one aim of the current study was to visualize the parenchymal microcirculation in vivo by 2-photon microscopy, and to investigate the spatial and temporal dynamics of microthrombus formation after TBI. The second aim of the current study was to understand better the mechanisms of this process. Microthrombi are formed by the activation of the intrinsic and/or extrinsic coagulation cascade and the adhesion of platelets. 23 –26 For the current study, we decided to investigate the hypothesis that the intrinsic coagulation cascade and one of its major components, factor XI, are involved in microthrombus formation after brain injury. Accordingly, pharmacological inhibition or genetic deletion of FXI may improve cerebral perfusion and improve histopathological outcome after TBI.
In addition to its role for microthrombus formation, FXI is also a potential pharmacological target for anti-coagulative therapies. In fact, patients suffering from brain trauma are often immobilized for significant periods of time and require anti-coagulation. 27,28 However, almost 50% of all TBI patients also have intracranial bleeding, thereby making the choice of the appropriate anticoagulant rather difficult. 29 Therefore, an additional aim of the current study was to investigate whether inhibition of FXI increases hemorrhage after TBI or if it has a favorable risk profile.
Methods
For this study, we used 6- to 8-week-old male C57Bl6 mice (n = 101 in total; purchased from Jackson Laboratories, Kent, UK) and FXI-/- mice (male, 8–9 weeks old; n = 15 in total). These mice were described previously, 30,31 and backcrossed for at least nine generations to C57Bl6J background.
All animal experiments were carried out in compliance with our institutional guidelines, and all experiments were approved both by the Research Ethics Committee of the Royal College of Surgeons in Ireland (RCSI; REC number 467) and by the Ministry for Health and Children in Dublin, Ireland (license number B100/4169). Our results are being reported in accordance with the Animal Research: Reporting of In Vivo Experiments guidelines. 32
Husbandry
The mice were housed under a 12-h light/12-h dark cycle in groups of ≤5 mice per cage with access to food and water ad libitum prior to the experiment. Once in the experiment, the animals were housed in single cages. Hygiene management checks and health screens were performed in compliance with the guidelines and recommendations established by the Federation of Laboratory Animal Science Associations. 33
Randomization
The animals were randomly assigned to the treatment groups and time-points of sacrifice. Both the researcher performing the experiments and the one carrying out the analysis were blind to the treatment/genetic background of the animals throughout the experiments.
Animal preparation for intravital microscopy
Intravital microscopy was performed as described previously. 15,34,35 Following controlled cortical impact (CCI; n = 8) or the sham procedure (n = 6; see below), the animals were anesthetized with an intraperitoneal injection of a cocktail containing medetomidine (0.5 mg/kg body weight; Domitor®; Dr. E. Graeub AG, Basel, Switzerland), fentanyl (0.05 mg/kg body weight.; Janssen-Cilag, Neuss, Germany), and midazolam (5 mg/kg body weight; Dormicum®; Roche, Basel, Switzerland). Subsequently, the animals were endotracheally intubated and ventilated in a volume-controlled mode (MiniVent 845 ventilator; Hugo Sachs Elektronik, March-Hungstetten, Germany); end-tidal pCO2 was continuously monitored and recorded using microcapnometry 36 (Capnograph 340; Hugo Sachs Elektronik). Throughout the experiment, body temperature was maintained at 37°C via a rectal probe attached to a feedback-controlled heating pad. A catheter was inserted into the femoral artery and used to monitor mean arteriolar blood pressure and administer both saline (0.9% NaCl) at 0.4 mL/h and the fluorescent dye. To maintain the anesthesia, one-third of the initial dose was administered every hour. The animal was then immobilized in a custom-made stereotaxic frame, the cranial window was prepared (see below), and the animal was transferred to the 2-photon microscope. At the end of each experiment, arteriolar blood gases and pH were measured (Rapidlab 348; Siemens, Munich, Germany).
Cranial window preparation
Following CCI (see below), a cranial window (2 mm ×2 mm) was prepared over the right fronto-parietal cortex under continuous cooling with saline as described previously. 15,34,35 Special care was applied in order to leave the dura mater intact. The location of the window was placed 1 mm laterally to the sagittal suture and 1 mm frontally to the coronal suture, thereby enabling us to view the region 1.5-3.5 mm frontal to the primary contusion (i.e., in the region where we expected maximum microthrombus formation to occur; Fig. 1). Following the craniotomy, the dura mater was removed, and the surface of the brain was rinsed with saline. Subsequently, a precise-fitting custom-made square cover glass with a thickness of 175 μm (Schott Displayglas, Jena, Germany) was carefully placed in the window and fixed to the skull using dental cement (Cyano Veneer; Hager & Werken, Duisburg, Germany).

Quantification of microthrombi following traumatic brain injury (TBI).
Two-photon microscopy
Two-photon imaging was performed as described previously 34 using a Zeiss LSM-710 upright confocal microscope equipped with a Ti:Sa laser (Coherent Chameleon Vision II; Coherent, Glasgow, UK) and two non-descanned photomultipliers for detecting red (BP 500-550 nm) and green (BP 565-610 nm) fluorescence. Visualization of the micro-vessels and microthrombi was performed by an intravenous injection of fluorescein isothiocyanate-labeled dextran (0.1 mL of a 0.5% solution, molecular weight 150,000; Sigma Chemical, St. Louis, Missouri) and Rhodamine 6G (repetitive injections of 0.05 mL of a 0.01% solution; Merck, Darmstadt, Germany), respectively. For imaging, the window was divided into five regions measuring 425 μm × 425 μm on the surface and reaching up to 500 μm into the parenchyma (cortical layers 4 and 5). The first three regions were aligned close to the primary contusion, whereas region four and five were placed more distally (Fig. 1). Beginning 1.5 h after CCI/sham, z-stacks were captured (at 3-μm increments) from all five regions within 1 h.
Analysis of data acquired by 2-photon microscopy
The images were analyzed offline as described previously
34
using the ImageJ software (
Coagulation tests
In our study, we used an antibody specifically designed to selectively inhibit prothrombotic FXI activation by factor XIIa (14E11) 37,38 and a control antibody (control immunoglobulin G [IgG]). The antibodies were injected into the femoral vein, which was subsequently ligated (4 mg/kg body weight dissolved in phosphate-buffered saline; a mouse weighing 25 g received a volume of 0.25 mL intravenously). Previously, 14E11 was shown to prolong activated partial thromboplastin time (aPTT) in mice at least for 72 h. 38
We verified the selective inhibition of the intrinsic pathway by determining the aPTT, a test for the intrinsic coagulation pathway, 39 and the prothrombin time (PT), a test for the extrinsic coagulation pathway, 40 –42 with a coagulation analyzer (ACL 200 Automated Coagulation Laboratory; Instrumentation Laboratory; Bedford, MA). APTT and PT were determined in mice that received control IgG or 14E11 via the femoral vein, and in C57Bl6 and FXI-/- mice (n = 8 per group). In this series, mice were anesthetized in an isoflurane chamber (4% in a 1:2 mix of O2:N2O), and anesthesia was maintained with a face mask using 1.2% isoflurane in a 1:2 mix of O2:N2O during surgery. A thermostatically regulated feedback-controlled heating pad was used to maintain rectal temperature at 37°C (FHC Inc., Bowdoin, ME). At 24 h after treatment, the animals were anesthetized deeply by injection of the above mentioned triple combination intraperitoneally. Following a quick thoracotomy, the right ventricle of the heart was punctured and 0.1 mL blood was withdrawn with a 1 mL syringe containing 75 μL 3.2% sodium citrate.
CCI
Traumatic brain injury was induced as described previously. 15,35,43 In brief, the mice were anesthetized with a gas mixture containing 2% isoflurane, 65% N2O, and 33% O2. A craniotomy window was drilled over the right parietal cortex under continuous cooling with saline. A cortical contusion was produced using a CCI device optimized for use in mice (cylinder diameter: 3.0 mm; velocity: 6.0 m/sec; penetration depth: 0.5 mm; contact time: 150 msec). Immediately following the impact, the bone flap was re-implanted and fixed using histoacrylic glue. The animals were then allowed to wake in a recovery chamber (33°C and 50% humidity). Sham-operated animals were treated as above, except CCI was not performed.
Histopathological analysis
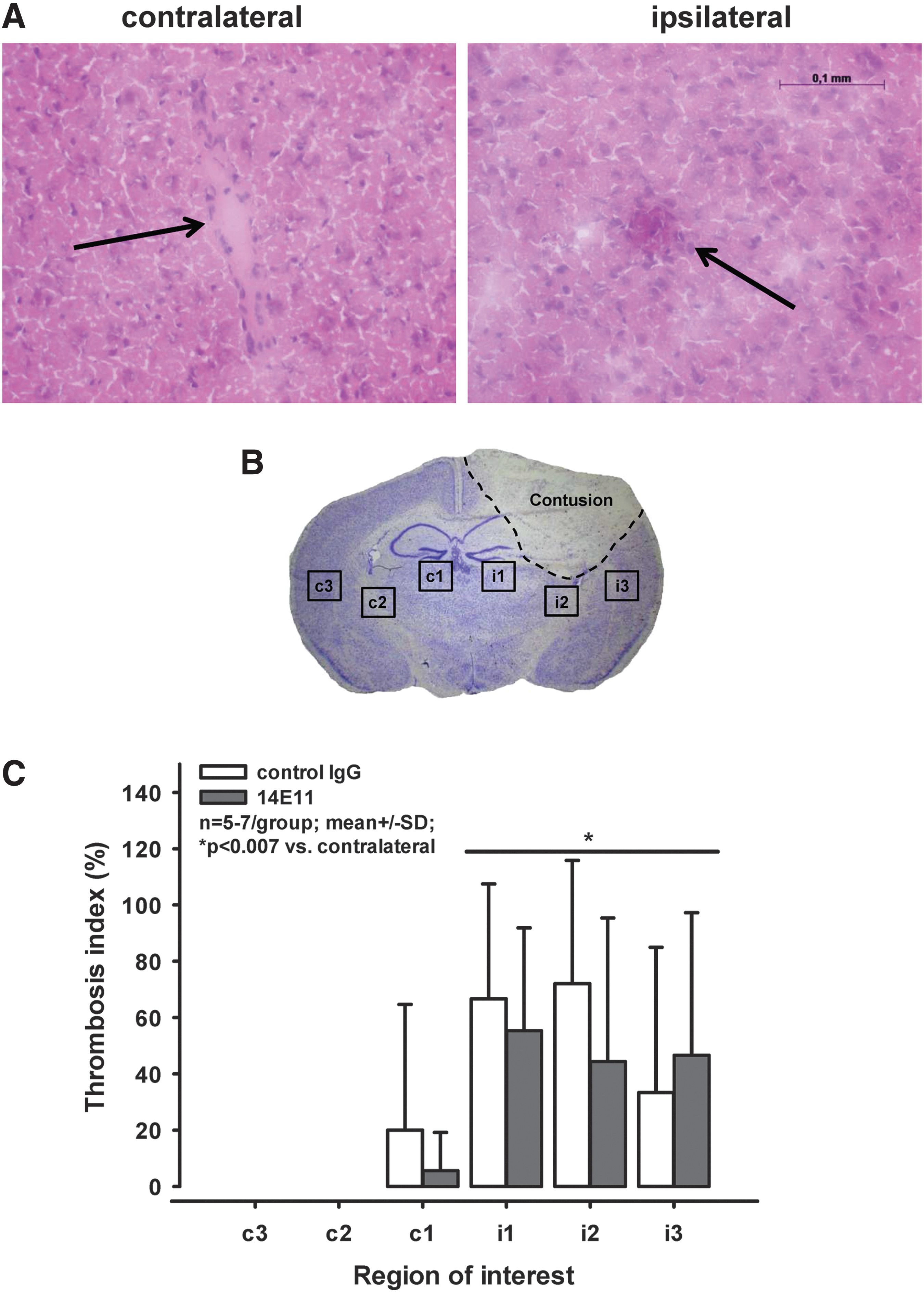
Secondary lesion expansion, brain swelling, and intracranial hemorrhage following CCI were assessed in histological sections as described before. 34,44 As a reference for the initial primary damage, these parameters also were assessed 15 min after trauma in each experimental series. At the respective end-point of the experiments, the animals were anesthetized with ketanest intraperitoneally and sacrificed by cervical dislocation. Immediately afterwards, the brains were removed carefully, frozen in powdered dry ice, and stored at −20°C. Ten-micrometer thick coronal sections were cut from 15 levels with a distance of 500 μm throughout the contusion site using a cryostat (Cryostar CM 1950; Leica, Wetzlar, Germany). Subsequently, two sections from each level were stained with cresyl violet and hematoxylin/eosine, respectively, and digitized using a camera connected to a microscope (Leica DM 3000; Wetzlar, Germany). Using an imaging analysis system (Leica), the contusion area and total volume of both hemispheres in each section were quantified in the sections stained with cresyl violet, and intracranial hemorrhage was determined in the sections stained with hematoxylin/eosin. Finally, the total volumes of contusion, each hemisphere, and intracranial hemorrhage were calculated respectively with the following formula: x = d*(A1/2 + A2 + A3…+ A15/2), where d represents the distance between neighboring sections (i.e. 0.5 mm) and A stands for the contusion area assessed on the single sections. Brain swelling was estimated by the size (area) of the ipsilateral hemisphere divided by the size of the contralateral hemisphere. The thrombosis index, defined as the number of microthrombi per total vessel number in the sample, was determined on hematoxylin/eosin–stained coronal brain sections. 45,46 In the section containing the largest contusion, three regions of interest were investigated adjacent to the contusion: a medial region (1), a region in the middle of the hemisphere (2), and a lateral region (3). Subsequently, the corresponding regions on the contralateral side were investigated.
The following treatment series were carried out: a) treatment with control IgG or 14E11 24 h before trauma and sacrifice at 15 min (no treatment) or 24 h post-trauma (n = 24); b) administration of control IgG 30 min after CCI, 14E11 30 min after CCI, 14E11 2 h after CCI after trauma and sacrifice at 15 min (no treatment) or 24 h post-trauma (n = 32); and c) CCI in wildtype C57Bl6 and FXI-/- mice and sacrifice at 15 min or 24 h post-trauma (n = 7 per group).
Statistical analysis
Results are presented as median followed by the 75th and 25th percentile in parentheses, except for the thrombosis index and the results acquired by the coagulation analyzer, which are reported as mean ± standard deviation. Differences between the groups were evaluated using the Mann-Whitney Rank Sum test with the Bonferroni correction. Differences with a p < 0.05 were considered significant. The calculations were performed with a standard statistical software package (Sigma Stat 3.0; Systat Software, Erkrath, Germany).
Results
Microthrombus formation in the parenchymal microcirculation after TBI
Following sham operation, the microcirculation was not demonstrably impaired and few leukocytes (single red dots) were visible in the three-dimensional stack (Fig. 1B, left image). By contrast, TBI lead to formation of leukocyte-platelet aggregates and microthrombi (labeled in red, Fig. 1B, right image). Overall, about 2–4% of the vessel lumen was filled by microthrombi after trauma, whereas less than 1% of the vessel lumen was filled by microthrombi following sham operation (p < 0.05; Fig. 2A, 2C). The clots were spread similarly among the different regions of interest (i.e., there was no gradient according to the distance to the contusion; Fig. 2A).
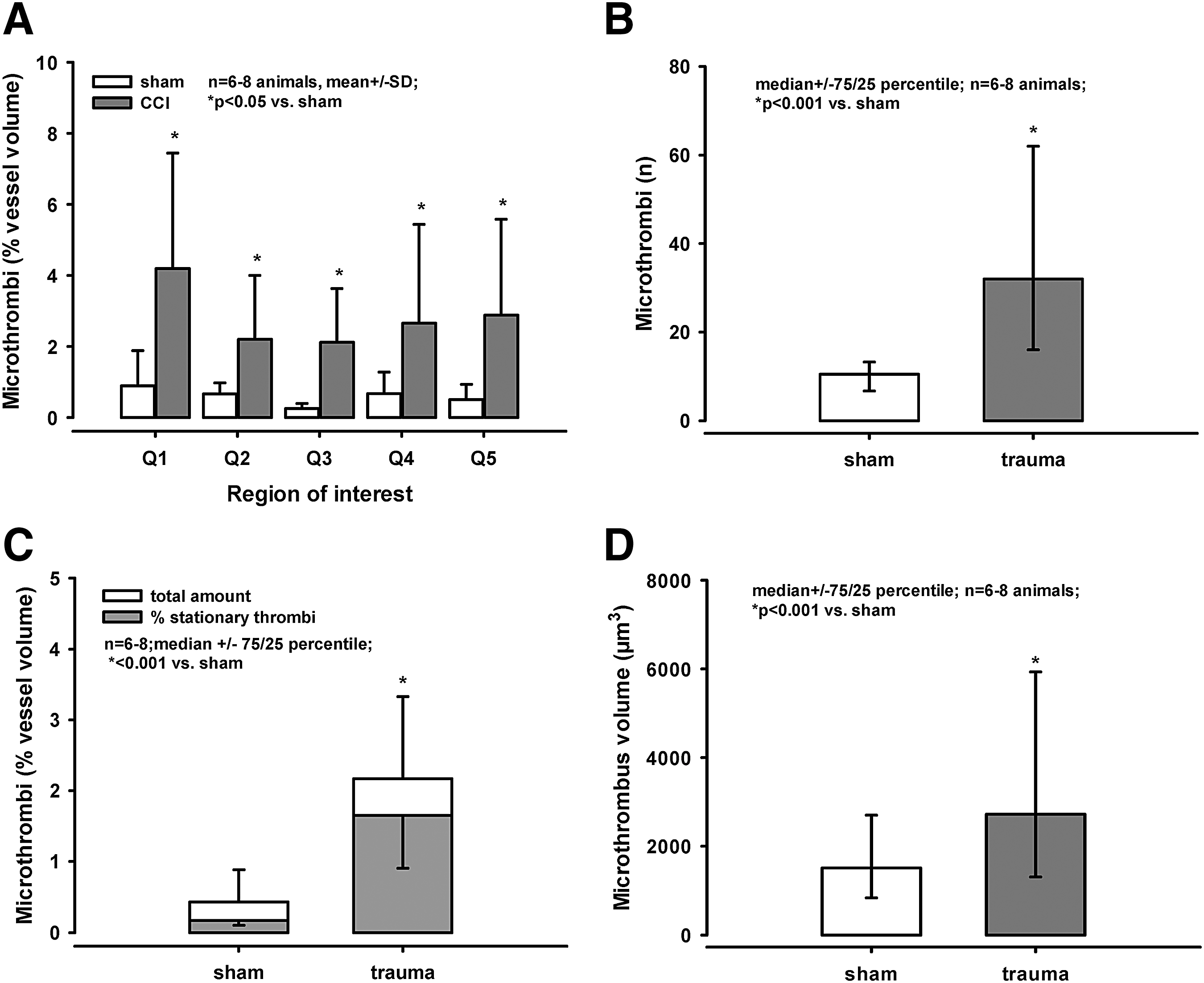
By taking two consecutive images in within 60 sec (Fig. 1C), we defined the percentage of stationary/adherent microthrombi. Following sham operation, only a small number of microthrombi was visible, but 65% of microthrombi could not be detected again after 60 sec (Fig. 1C, four images on the left; Fig. 2C). In contrast, 76% of the clots detected after trauma were stationary (Fig. 1C, two images on the right; Fig. 3C). The actual number of microthrombi was significantly higher after TBI (33[16/62]); data presented as median followed by the 75th and 25th percentile in parentheses) than after sham operation (10[6.7/13.2]; p < 0.001 vs. trauma; Fig. 2B). Also, the volume of the microthrombi formed after trauma was significantly larger, compared with the volume of microclots after sham operation (p < 0.001; Fig. 2D).
Administration of 14E11/control immunoglobulin G (IgG) before controlled cortical impact (CCI). Mice received either control IgG or 14E11 24 h before trauma.
Animals were kept in anesthesia under physiological conditions at all times during the operation and imaging (Table 1). There were no differences in blood gas parameters between groups, and no differences in mean arteriolar blood pressure or end-tidal pCO2 between groups or between different time-points within the groups. Although the values for end-tidal pCO2 are slightly lower than the physiological range, these values correspond to physiological blood gases at the end of the experiment. This can be explained by a rather large shunt volume since we do not use positive end expiratory pressure in our ventilation.
Mean ± standard deviation.
All parameters were continuously kept within the physiological range and there were no differences between groups. With a relatively low end-tidal pCO2, the animals had physiological pCO2 values in the blood gas analysis.
pCO2, partial pressure of carbon dioxide; pO2, partial pressure of oxygen; CCI, controlled cortical impact.
Inhibition of FXI
Assessment of aPTT and PT
The activated partial thromboplastin time (aPTT) is a measure for the intrinsic coagulation pathway. 39 Reference values are 20-40 sec in humans. In wild-type mice and in mice that had received control IgG 24 h before blood withdrawal, aPTT was 25.7 ± 2 and 25.1 ± 4 sec, respectively (Table 2). However, aPTT was significantly prolonged by 1.7-fold to 42.7 ± 8 sec in animals that had received 14E11 24 h before blood withdrawal (p = 0.008 vs. control IgG). The mean aPTT of FXI-/- mice was 40.6 ± 7 sec (p = 0.008 vs. wild-type). As expected, the values for PT, a measure for the extrinsic pathway, 40 –42 were not affected by administration of 14E11 or in FXI-/- mice. The PT values ranged between 11.4 ± 1 sec (after 14E11 injection) and 12.9 ± 1 sec (in wild-type mice), and there was no difference between groups.
Mean ± standard deviation; * p = 0.008 vs. aPTT following IgG treatment or in C57/Bl6 animals.
Both following treatment with 14E11 and in FXI-/- mice, aPTT was significantly prolonged, compared with aPTT following IgG treatment and in C57/Bl6 mice (* p = 0.008, respectively), while PT was not affected.
IgG, immunoglobulin G; aPTT; activated partial thromboplastin time; PT, prothrombin time.
Administration of 14E11/IgG before CCI
Cerebral hemorrhage
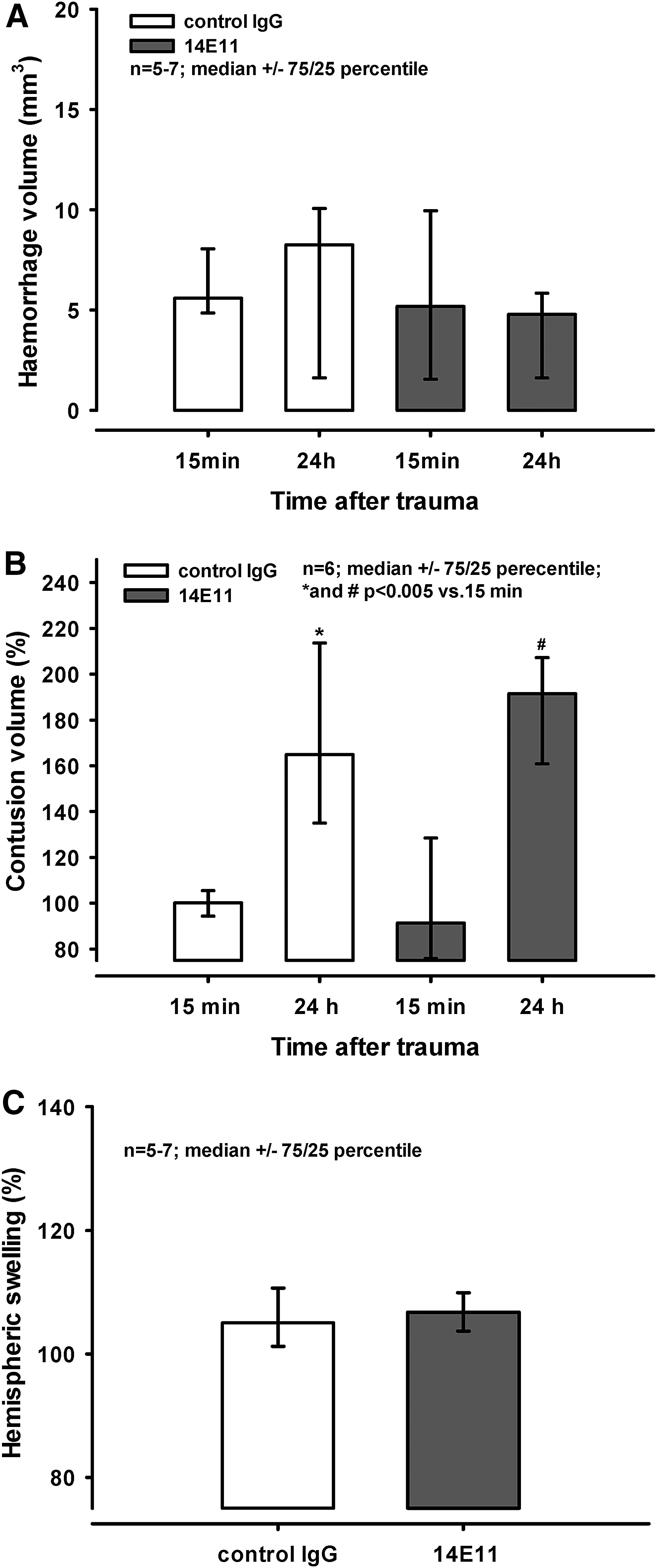
Twenty-four hours before trauma the animals received either control IgG or 14E11. The extension of cerebral hemorrhage inside the contusion and in the surrounding tissue assessed 15 min following CCI measured 5.6 (4.8/8.1) mm3 and 5.2 (1.6/9.9) mm3 after injection of control IgG and 14E11, respectively (Fig. 3A). Twenty-four hours post-trauma, intracranial hemorrhage extended to 8.3 (1.6/10.1) mm3 and 4.8 (1.6/5.8) mm3 following injection of control IgG and 14E11, respectively. There was no statistical difference between the groups or between the time-points.
Contusion volume and hemispheric swelling
Contusion size is expressed as a percentage of the size of the primary contusion determined 15 min after trauma. Contusion volume increased significantly in both groups during the 24 h post-injury to 165% (135/214%) and 192% (161/207%) after injection of control IgG and 14E11, respectively (* and #p < 0.005 vs. primary contusion; Fig. 3B). However, there was no statistically significant difference between treatment groups.
Hemispheric swelling is displayed as ipsilateral volume in percent of the contralateral volume. In animals that had received control IgG or 14E11 24 h before injury, hemispheric swelling reached values of 105% (101/111%) and 107% (104/110%) at 24 h post-trauma (Fig. 3C). No difference between groups was detected.
Thrombosis index
The number of microthrombi per total vessel number was quantified in three regions of interest in each hemisphere (Fig. 4B) in the section containing the largest contusion area 24 h after CCI. Only few microthrombi were counted contralaterally, and those were seen only in the medial region (c1; i.e., in the region closest to the contusion). By contrast, 40–60% of the vessels on the ipsilateral side were at least partly occluded by microthrombi (p < 0.007 vs. contralateral; Fig. 4A, 4C). This corresponds to values obtained at much earlier time-points (2 h after trauma) in in vivo experiments. 15
Administration of 14E11/control immunoglobulin G (IgG) before controlled cortical impact (CCI)—assessment of microclot formation.
Administration of 14E11/control IgG after CCI
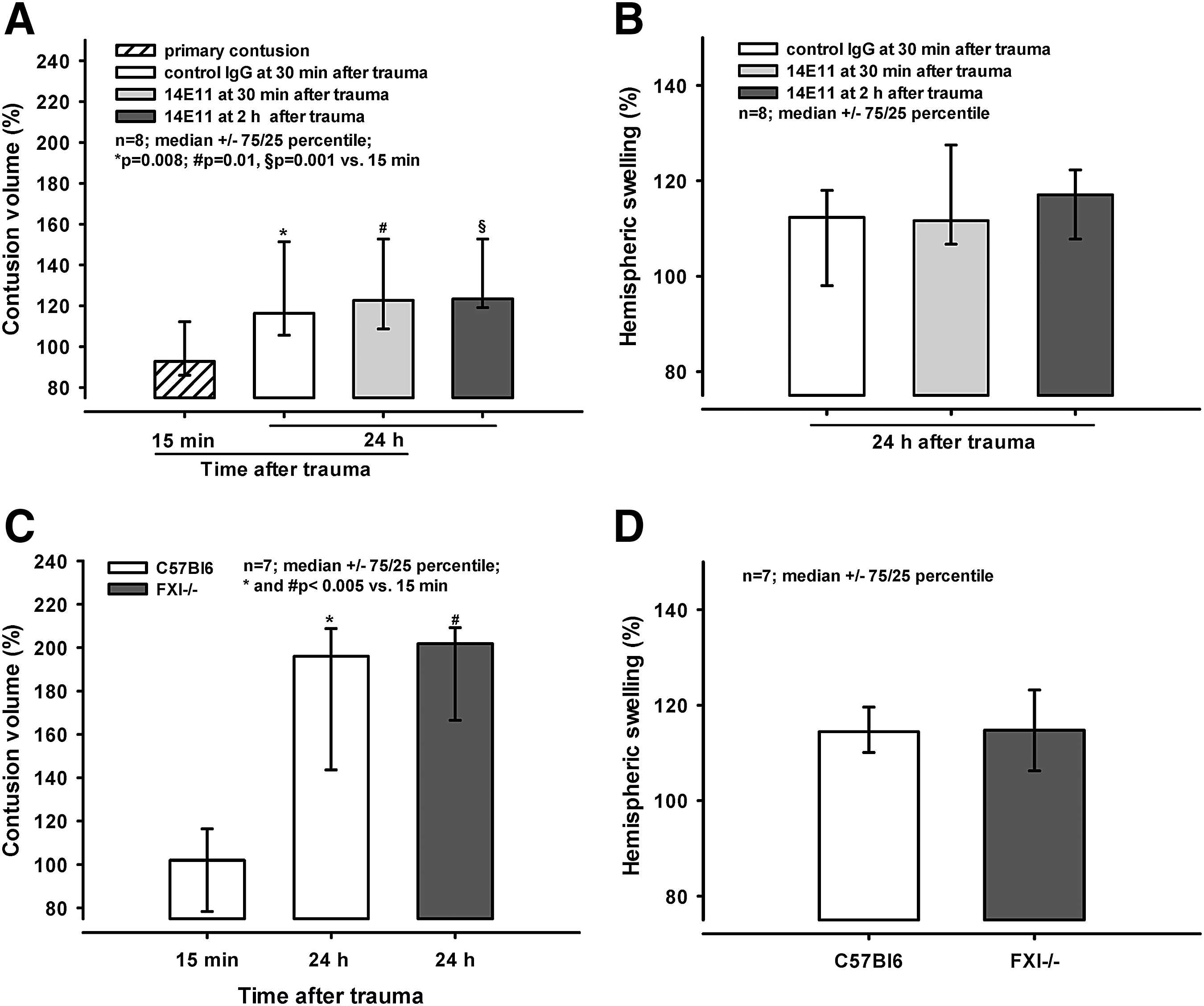
In this series, we assessed the effect of 14E11 injection 30 min and 2 h after trauma and compared it with the effect of control IgG administered 30 min after trauma. Again, we determined primary contusion volume 15 min after injury, and secondary lesion expansion and brain swelling 24 h after trauma.
Twenty-four hours following CCI, the lesions expanded to 116% (106/151%; control IgG; p = 0.008 vs. 15 min), 123% (109/153%; 14E11 30 min after trauma; p = 0.01 vs. 15 min), and 123% (119/153%; 14E11 2 h after trauma, p < 0.001 vs. 15 min; Fig. 5A). There was no difference in lesion expansion between the groups. Hemispheric swelling extended to 112% (98/118%; control IgG), 112% (107/128%; 14E11 30 min after trauma), and 117% (108/122%; 14E11 2 h after trauma; Fig. 5B).
(
Contusion volume and hemispheric swelling in C57Bl6 animals and FXI-/- mice Secondary lesion expansion 24 h after trauma expanded to 196% (144/209%) and 202 (167/209%) of the primary contusion size in wild-type animals and FXI-/- mice, respectively (p < 0.005 vs. 15 min; Fig. 5C). Hemispheric swelling also was similar 24 h after trauma, reaching 114% (110/120%) and 115% (106/123%) in wild-type and in FXI-/- animals (Fig. 5D).
Discussion
In the current study, we observed the formation of microthrombi in the brain of sham-operated and otherwise healthy mice. The total number of microthrombi, as well as their volume, were small, and most microthrombi became only transiently stuck in cerebral capillaries. After TBI, microthrombi start to form in the parenchymal microcirculation within the first few hours and occlude more than 60% of all cerebral vessels 24 h after injury. Genetic or pharmacological inhibition of FXI successfully inhibited the intrinsic coagulation cascade as evidenced by PTT and PT measurements; however, no effect was observed on post-trauma microthrombosis and histopathological outcome, suggesting that the intrinsic coagulation does not contribute to microthrombus formation. Inhibition of FXI activation by FXIIa using the 14E11 antibody or prevention of FXI activity by using FXI knockout mice, however, also did also influence post-trauma brain hemorrhage. Since knock down of FXI using antisense oligonucleotides is antithrombotic in patients undergoing major surgery,47 development and use of FXI inhibitors for systemic antithrombotic anticoagulation may be safe and also benefit TBI patients.
So far, microthrombi were observed by histology in animal experiments employing weight drop injury, 8 fluid percussion injury (FPI), 9,19,20 and CCI, 10,14,21 and by in vivo imaging following weight drop injury, 22 and CCI. 15 Additionally, there is evidence from clinical studies investigating patient samples that microthrombi form in the cerebral microcirculation after TBI. 11,16,17 While these studies provide valuable information on the occurrence of microthrombi in various settings of TBI, they do not fully elucidate information on the stability of microthrombi, the distribution in the parenchymal microcirculation, and the size of the microthrombi that are formed post-trauma (and the hemostatic systems involved (i.e., plasmatic coagulation vs. platelets). Following TBI, we observed a significant increase in the number and volume of microthrombi, and most of those were stable throughout an observation period of 60 sec. This is of particular importance because these microthrombi might well occlude capillaries in the parenchymal microcirculation, thereby impairing cerebral blood flow (CBF) in the traumatic penumbra (i.e., the region where secondary brain damage occurs). These results extend our knowledge on the spatial and temporal distribution of microthrombus formation after TBI and are well in line with our previous results, which demonstrated significant formation of microthrombi in pial vessels and in the pial microcirculation (i.e., in the vessels which have no nutritive function for the brain parenchyma). 15
Following TBI, reduced CBF is associated with poor outcome. 3,6,48 –53 In view of that, the formation of microclots after TBI, which occlude the vessels and lead to further disturbances in regional blood flow, 10,15,20 is of particular interest. So far, several therapeutic approaches targeting microthrombus formation after experimental TBI were reported in the literature: Maeda and colleagues describe an improvement of local CBF and a reduction in contusion volume after CCI in rats that had received a platelet activation factor antagonist. 10 In another study on FPI in rats, Bentzer and colleagues found an improvement of CBF but also an increase in brain water content after treatment with prostacyclin. 54 Lu and colleagues report both a reduction in thrombosis formation and improved neurological outcome after CCI in rats treated with atorvastatin, a compound that increases the production of nitric oxide (a molecule with well-known anti-adhesive effects on leukocytes and platelets). 21,55 Finally, in a study on FPI in rats, contusion volume and cell loss were significantly reduced following administration of erythrocyte-bound tissue-type plasminogen activator. 56 Despite these promising results, a safe clinical therapy directed against microclot formation following TBI is still missing. 27,28
With our approach of inhibiting FXI activation, we specifically aimed to decrease post-trauma microthrombus formation without exacerbating intracranial bleeding complications. Since the intrinsic cascade is mainly important for the amplification of coagulation 57,58 we targeted a coagulation factor from this cascade for our therapy.
Previously, 14E11 was demonstrated to be protective in a mouse model of sepsis, 38,59 and in acute ischemic stroke. 60 In the current study, we did not detect any improvement in contusion volume, hemispheric swelling, thrombus formation, and intracranial hemorrhage following FXI inhibition. The results obtained by administering 14E11 were similar to those obtained in FXI-/- mice. In both series, FXI inhibition resulted in a prolongation of aPTT but all other parameters remained unaffected. This indicates that the 14E11 antibody was successfully inhibiting FXI activation by FXIIa, but without any consequence to post-trauma microclot formation or secondary brain damage. The obtained aPTT values (around 40 sec) are practically in line with the values recommended in a prophylactic anticoagulation regime, while a therapeutic regime would require much higher aPTT levels. Interestingly, we found 60% of all cerebral microvessels to be occluded by microthrombi 24 h after trauma; these microthrombi were detected ipsilaterally outside the contusion. Accordingly, microthrombus formation is not a transient event but an ongoing and pathogenically, a highly relevant event after trauma, as has also been reported by others. 13
So far, the mechanisms of post-trauma microclot formation are poorly understood. It has been hypothesized that the process could be driven by traumatic exposure of the subendothelium that contains collagen and laminin, which support hemostatic platelet activation, as well as intravascular thrombin generation. This thrombin generation would be driven by both hemostatic tissue factor activation and prothrombotic contact activation. Inhibiting the activation of FXI did not affect post-trauma microthrombus formation, implying that the intrinsic coagulation cascade is only of minor importance for the formation of microthrombi after TBI. In turn, this suggests that TBI-induced microthrombus formation is driven by the extrinsic coagulation pathway. This is supported by the fact that the brain expresses significant amounts of tissue factor and that TBI-induced small vessel damage presumably exposes this subendothelial factor rapidly. In that regard, investigating the potential effect of inhibition of the extrinsic pathway might be worth pursuing. However, while targeting extrinsic coagulation factors or platelet function might prevent microvascular thrombus formation, it would at the same time impair hemostasis and significantly increase the risk of detrimental intracranial hemorrhage. Hence, the therapeutic potential of such an approach would be limited or even dangerous.
Inhibition of FXI activation by FXIIa 24 h before trauma using 14E11, which is active for at least 72 h, 38 did not aggravate intracranial hemorrhage; this was consistent with the lack of relevance for FXI in post-trauma hemostasis. The values assessed 15 min and 24 h after injury were similar in the groups receiving 14E11 or control IgG; additionally, there was no increase in hemorrhage in either group over time. Since 14E11 has been shown to be antithrombotic in rodent and primate models, 37,38 this is of particular importance regarding a second therapeutic approach: the safe prophylactic anticoagulation, which is required, for example, in patients who are immobilized in an intensive care unit; this is usually the case in the first days or weeks following TBI. 27,28
Until now, prophylactic anticoagulation in patients suffering from TBI remains a double edged sword 27,28,61 –67 : there is the risk of secondary thrombosis due to immobilization and altered coagulation after TBI versus the risk of exacerbating intracranial hemorrhage. After inhibiting FXI activation by FXIIa with 14E11 in our CCI model, we specifically determined intracranial hemorrhage and could not detect any detrimental effect 15 min or 24 h after trauma. Since the compound increased aPTT without exacerbating intracranial hemorrhage, further tests regarding a clinical implication for patients suffering from brain trauma and requiring anticoagulative prophylaxis might be promising. To clarify the safety and efficacy of FXI treatment following TBI, the effect on both behavioral outcomes and formation of deep venous thrombosis needs to be determined in future studies.
In summary, we showed for the first time the post-trauma accumulation of microthrombi in the parenchymal microcirculation in vivo (i.e., the microvessels that are directly responsible for the nutritive perfusion of the brain tissue). Reducing FXI-mediated thrombin generation by targeting FXI (knockout) or FXII (14E11) did not attenuate microthrombus formation, secondary lesion expansion, or brain swelling after trauma; hence, it did not work as a specific therapy for TBI in this model of a focal cortical contusion. However, post-trauma intracranial hemorrhage was not aggravated either, and investigation of targeting FXI as a prophylactic antithrombotic therapy for TBI patients is warranted.
Footnotes
Acknowledgments
AG is supported by National Institute of Health grants AI088937, HL106919 and HL101972. CK was supported by the Deutsche Forschungsgemeinschaft, SFB 688 and the Interdisziplinäres Zentrum für Klinische Forschung (IZKF) Würzburg. We would like to thank Joe Guerin for editing the manuscript.
Author Disclosure Statement
Andras Gruber and Oregon Health and Science University may have financial interest in the results of this study. For the other authors, no competing financial interests exist.