
Abstract
Focal neocortical brain injuries lead to functional alterations, which can spread beyond lesion-neighboring brain areas. The undamaged hemisphere and its associated disturbances after a unilateral lesion, so-called transhemispheric diaschisis, have been progressively disclosed over the last decades; they are strongly involved in the pathophysiology and, potentially, recovery of brain injuries. Understanding the temporal dynamics of these transhemispheric functional changes is crucial to decipher the role of the undamaged cortex in the processes of functional reorganization at different stages post-lesion. In this regard, little is known about the acute-subacute processes after 24–48 h in the brain hemisphere contralateral to injury. In the present study, we performed a controlled cortical impact to produce a unilateral traumatic brain injury (TBI) in the motor and somatosensory cortex of mice. In vitro extracellular multi-unit recordings from large neuronal populations, together with single-cell patch-clamp recordings in the cortical network contralateral to the lesion, revealed a strong, but transient, neuronal hyperactivity as early as 24–48 h post-TBI. This abnormal excitable state in the intact hemisphere was not accompanied by alterations in neuronal intrinsic properties, but it was associated with an impairment of the phasic gamma aminobutyric acid (GABA)ergic transmission and an increased expression of GABAA receptor subunits related to tonic inhibition exclusively in the contralateral hemisphere. These data unravel a series of early transhemispheric functional alterations after diffuse unilateral cortical injury, which may compensate and stabilize the disrupted brain functions. Therefore, our findings support the hypothesis that the undamaged hemisphere could play a significant role in early functional reorganization processes after a TBI.
Introduction
T
A growing number of studies suggest that processes of functional reorganization are not limited to the ipsilateral hemisphere, but can also be observed in the primarily undamaged contralateral cortical network, thereby indicating its potential contribution in the recovery post-TBI. First coined by Von Monakow 17 and later refined by Andrews, 18 the concept of transhemispheric diaschisis gathers observations of altered brain functions in areas contralateral to a cortical lesion. In accordance, data from brain injury animal models reported an enhanced neuronal excitability in the uninjured hemisphere, often accompanied by a transient increase in metabolism. 7,19,20 An impairment of gamma aminobutyric acid (GABA)ergic synaptic transmission is thought to play a major role in the development of such abnormal neuronal activity in the contralesional cortex. 21 –24 In this context, several studies suggested that the observed altered GABAergic transmission may be even beneficial given that the reduced inhibition can modulate cortical map plasticity and thus promote recovery of function. 24 In support of this, imaging studies in humans reported an enhanced recruitment of the contralateral hemisphere, progressively raising the hypothesis that the functional alterations in the undamaged hemisphere are critical for functional recovery after a unilateral lesion. 6,25 –30 Although the majority of the mentioned studies revealed functional and structural alterations in subacute or chronic phases post-TBI, little is known about the physiological changes occurring in the contralateral hemisphere in the first 24–48 h post-TBI.
With respect to the injury model, stroke-like brain lesion models (with vascular-ischemic etiology) were relatively often investigated, 19,31,32 whereas brain injuries induced by a mechanical trauma received substantially less attention despite its high clinical relevance, especially considering its prevalence in young adults. 33 Therefore, a detailed characterization of the functional alterations at early stages after a mechanical brain injury is still missing. In the present study, we used a clinically relevant TBI model of unilateral controlled cortical impact (CCI) in the motor and somatosensory cortex of mice to investigate populations of neurons as well as single cells in the contralateral cortical network at 24–48 h post-TBI with an ex vivo/in vitro approach. This way, we unraveled a neuronal hyperactivity specifically in the contralateral hemisphere that was not associated to changes in neurons intrinsic properties, but rather to an impaired function of GABAergic inhibition as early as 24 h post-TBI. These findings suggest that, already in the acute phase after lesion, the intact hemisphere may play a role in processes of functional reorganization. The observed acute functional changes post-TBI might represent an interhemispheric homeostatic mechanism to stabilize the overall brain function post-trauma.
Methods
Animals and ethical statement
C57BL/6 wild-type mice at the age of 19–22 days (n = 80) were housed at a constant room temperature of 23 ± 2°C with a standard 12-h light/dark cycle and free access to food and water. All animal experiments were performed in strict accord with the European regulations and with the institutional guidelines of the Johannes Gutenberg-University (Mainz, Germany). The protocol was approved by the Animal Ethics Committee of the Landesuntersuchungsamt Rheinland-Pfalz (23 177-07/G 14-1-037). The number of animals was kept to a minimum, and all efforts were made to minimize the suffering of the mice.
Induction of traumatic brain injury
Experimental TBI was induced, as previously described, in our lab by CCI. 34 In brief, mice were initially anesthetized with 4% isoflurane (AbbVie, Wiesbaden, Germany) and maintained under anesthesia during the surgery by face mask application of 1.5% isoflurane in a mixture of 40% O2 and 60% N2. Mice were placed in a stereotactic frame, the body temperature was kept constant at 37°C using a heating pad and a rectal temperature probe (Physitemp Instrument Inc., Clifton, NJ). After midline incision, a 4-mm2 craniotomy was made over the right parietal sensorimotor cortex, between the bregma and the lambda suture, using a high-speed drill. The craniotomy corresponded to a square area of 2 mm width starting at bregma 0, extending first following the coronal suture, then parallel to the sagittal suture, and, finally, extending back to the midline. Brain trauma was induced mechanically with a stereotaxic impactor for CCI (Impact One™; Leica Mikrosysteme, Wetzlar, Germany) by positioning the impactor tip (diameter = 1.5 mm) directly on the brain surface, perpendicular to the intact dura. The impact was always performed at a speed of 6 m/s, with a dwell time of 200 ms and to a depth of 0.8 mm. Immediately after cortical impact, the craniotomy was closed by repositioning the previously removed bone flap and sealing it with Histoacryl surgical glue (B. Braun-Melsungen, Melsungen, Germany). The skin was sewed up with polypropylene sutures (Ethicon, Somerville, NJ), and the isoflurane flux was interrupted to terminate anesthesia. Animals were then placed back in their cage and recovered under an infrared lamp for at least 2 h. Sham-operated age-matched littermates were used as controls to compare the different experimental groups in order to minimize variability attributed to developmental changes.
Tissue preparation
Animals were deeply anaesthetized with 4% isoflurane and decapitated at 1–4 days after lesion induction. Brains were quickly removed and placed in 4°C standard artificial cerebrospinal fluid (aCSF) containing (in mM): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.5 MgCl2, 2 CaCl2, 1.25 NaH2PO4, and 25
Electrophysiology
Multi-electrode array recordings
The extracellular spontaneous neuronal activity in slices was recorded in both cortical hemispheres with perforated multi-electrode array chips (pMEA; pMEA32S12 Layout 4; Multi Channel Systems GmbH, Reutlingen, Germany) equipped with titanium nitride electrodes consisting of 32 recording electrodes (four rows of eight electrodes), 12 stimulating electrodes (two rows of six electrodes), and one internal reference electrode. The recording electrodes had a diameter of 30 μm and an impedance ranging from 30 to 50 kΩ. The interelectrode distance (center to center) was 100 μm. The pMEA chips were mounted underneath a small circular perfusion chamber. Acute slices were transferred into this chamber and carefully positioned in a way that the upper border of cortical layer 1 overlaid the first row of the chip's recording electrodes. As a consequence, the majority of recording electrodes was located in the supragranular layers 2 and 3. The center of the pMEA chip was placed at around a 1-mm distance from the border of the lesion. Recordings in the contralateral hemisphere of injured animals and sham-operated mice were performed by placing the pMEA in the homotopic region of the brain slice. To improve contact between slices and electrodes, a constant negative pressure (15–30 mbar) was applied through the perforation using a constant vacuum pump (Multi Channel Systems). The quality of the pMEA chips was controlled before each experiment. On average, each chip could be used for 1 month. One and the same chip was always used for a set of experiments including measurements of CCI-treated animals and the respective sham control.
All recordings were performed at 35°C. To induce spontaneous activity, slices were perfused with a modified artificial cerebrospinal fluid (mACSF) containing (in mM): 126 NaCl, 25 NaHCO3, 4.3 KCl, 1 MgCl2, 1 CaCl2, 1.25 NaH2PO4, 25
Patch-clamp recordings
Single slices were transferred into a submerged recording chamber of an upright Olympus BX51 microscope (Olympus Corporation, Tokyo, Japan) with a continuous perfusion (3 mL/min) of oxygenated aCSF at 35°C. Whole-cell patch-clamp recordings were performed using borosilicate glass electrodes (3–5 MΩ resistance, GB 150F-8P; Science Products, Hofheim am Taunus, Germany). For the extracellular stimulation of ascending fibers projecting onto superficial layers, a glass pipette electrode was placed in layer 4 underneath the recorded cell.
Access resistance was monitored throughout the recordings and neurons were discarded if access resistance was >20 MΩ or changed more than 20% during the recordings. Data were recorded with an Axopatch-200B amplifier (Molecular Devices, Sunnyvale, CA), digitized at 10 KHz with a Digidata 1440 (Molecular Devices). Data acquisition and offline analysis were performed with pCLAMP 10 Software (Molecular Devices).
Neuronal spontaneous activity and membrane/firing properties were acquired in current-clamp mode with the brain slices bathed in mACSF. The intracellular solution contained (in mM): 130 K-gluconate, 10 Hepes, 11 ethylene glycol tetraacetic acid (EGTA), 2 MgCl2, 2 CaCl2, 4 Na-ATP (adenosine triphosphate), and 0.2 Na-GTP (guanosine triphosphate). Spontaneous neuronal activity of pyramidal cells was assessed throughout 5 min of recordings. If one spike or several spikes occurred during this period, cells were considered as active. If no spikes were recorded, cells were considered as silent. Firing frequency of active cells was calculated by counting all spikes detected during the recording period. Resting membrane potential (Vm rest) was measured immediately after the whole-cell patch-clamp configuration was achieved. All membrane and firing properties of neurons (input resistance, rheobase, adaptation coefficient, spike half-width, spike threshold Vm, spike amplitude, and maximal firing rate) were analyzed after applying 1-sec lasting square pulses of current through the patch-clamp electrode (at 0.1 Hz). The magnitude of the first current pulse was −50 pA. The injected current was then increased by 10 pA in each step. During this protocol, the Vm of neurons was kept at −70 mV by constant current injection, and, when not possible, those cells were not used for further analysis. Input resistance was measured using the voltage shift obtained in response to the smallest negative square current pulse (−50 pA). The rheobase was calculated as the smallest current injected capable to generate an action potential. Spike threshold, spike half-width, and spike amplitude were assessed at the rheobase of each neuron. To calculate the spike threshold, we first obtained the derivative of the membrane potential. Spike threshold was then defined as the Vm at which the derivative of the membrane potential reached 3% of its maximum value. 35 Spike amplitude was calculated as the difference in membrane potential between the spike threshold and spike peak. Spike half-width was defined as the duration of an action potential at 50% of the spike amplitude. The coefficient of adaptation and maximum firing rate were calculated from the square current pulse where the firing rate reached its maximum. The coefficient of adaptation was calculated by dividing the interspike intervals between the two first and two last spikes in the train. All current-clamp recordings were compensated for electrode resistance by applying the bridge balance correction.
Spontaneous and evoked postsynaptic currents (PSCs) were recorded in voltage-clamp mode at (or very close to) the reversal potential of either alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors (AMPARs; +10 mV) or GABA receptors (GABARs; −60 mV) in the presence of the N-methyl-D-aspartate receptor blocker, D(–)-2-amino-5-phosphonopetanoic acid (D-APV; 25 μM). This procedure allowed us to acquire spontaneous as well as evoked inhibitory and excitatory signals within the same cell. The intracellular solution used for this set of experiments contained (in mM): 125 Cs-gluconate, 5 CsCl, 10 EGTA, 2 MgCl2, 2 Na2-ATP, 0.4 Na2-GTP, and 10 HEPES. QX-314 (5 mM) was also added to block all activity from voltage-sensitive Na+-channels. Spontaneous excitatory post-synaptic current (sEPSC) and spontaneous inhibitory postsynaptic currents (sIPSCs) were semiautomatically identified and subsequently validated by careful visual inspection. Frequency, amplitude, and kinetics of sEPSC and sIPSCs were calculated as the mean of 300 events for each cell.
Evoked signals were recorded by first adjusting the intensity of the extracellular stimulation to obtain eEPSC with an amplitude of 200 pA (±11.9 pA) while the cell was voltage-clamped to −60 mV. Then, the holding potential was shifted to +10 mV and the same stimulation strength was applied to evoke a corresponding eIPSC. Six to eight consecutive responses were repeated every 15 sec and finally averaged to calculate the ratio of AMPAR/GABAR-mediated signal amplitudes and the kinetics.
Histology and immunohistochemistry
Some of the acute brain slices were fixated immediately after the preparation with 4% paraformaldehyde (Carl Roth, Karlsruhe, Germany) for 2 h. After overnight cryoprotection in 30% sucrose in phosphate-buffered saline (PBS), fixed brain slices were sectioned at 50 μm using a microtome (Leica CM1325; Leica Mikrosysteme, Wetzlar, Germany). Brain sections were subsequently used for Nissl staining or immunohistochemistry (IHC). For IHC, cortical sections were first treated with Block A buffer containing 10% goat serum, 20% Avidin D, in PBS 0.2% Triton-X 100 (Blocking kit; Vector Laboratories, Burlingame, CA) for 1.5 h at room temperature. Brain sections were then incubated overnight at 4°C with Parvalbumin (1:1000, PV27; Swant, Marly, Switzerland), glial fibrillary acidic protein (GFAP; 1:250, Z0334; Dako, Glostrup, Denmark) and CD68 antibody (ED-1; 1:500, MAB1435; Millipore, Billerica, MA) antibodies diluted in Block B buffer containing 1% goat serum, 20% biotin, in PBS 0.2% Triton-X 100 (Blocking kit; Vector Laboratories). The following day, secondary antibodies (1:100 goat antirabbit Cy2, A120-201C2; Bethyl Laboratories, Montgomery, TX; 1:100 goat antimouse Cy3, 115-165-166; Jackson Immuno Research, West Grove, PA) were diluted in 1% goat serum in PBS 0.1% Triton-X 100 and added to cortical sections for 1.5 h at room temperature. Slices were subsequently mounted using immunoselect antifading mounting medium 4′,6-diamidino-2-phenylindole (DAPI) (SCR-38448; Dianova, Hamburg, Germany) and imaged with a confocal microscope (Leica TCS SP5; Leica Mikrosysteme).
Reverse-transcriptase quantitative polymerase chain reaction
Following preparation of acute coronal slices of 400-μm thickness, a stamp of tissue (volume, 0.3 mm3) was harvested with a biopsy punch (Standard ø 1 mm; pfm medical, Köln, Germany) in the area where the electrophysiological studies were performed (around 1 mm away from the border of the lesion). Total RNA was phenol-chloroform extracted and ethanol precipitated (miRNeasy Mini Kit; Qiagen, Hilden, Germany) according to the kit's handbook protocol (miRNeasy Mini Handbook, p. 22), endogenous DNA being additionally cleared away (RNase-Free DNase Set; Qiagen). RNA purity and concentration were subsequently measured with a spectrophotometer (NanoDrop 2000; Thermo Fisher Scientific, Waltham, MA), and complementary DNA (cDNA) was produced (Transcriptor First Strand cDNA Synthesis Kit; Roche, Basel, Switzerland) from 0.15 μg of RNA, following the manufacturer's protocol (Handbook Version 6.0, Procedure A, p. 8). RNA expression was assessed through template cDNA amplification using the TaqMan® Fast Advanced Master Mix (Thermo Fisher Scientific) and a combination of specific primers and dual-labeled hydrolysis probes (Universal Probe Library technology; Roche, Basel, Switzerland), according to the manufacturer's protocol (TaqMan Fast Advanced Master Mix Guide p21). Polymerase chain reaction (PCR) plates (MicroAmp® Fast Optical 96-Well Reaction Plate; Applied Biosystems, Foster City, CA) were then transferred to a StepOnePlus™ real-time PCR system (Applied Biosystems, Foster City, CA), monitored through the associated software (StepOne™ and StepOnePlus Software v2.3; Applied Biosystems). Designed according to the provider, the thermal-cycling program contained one holding cycle (2 min, 50°C; 20 sec, 95°C), followed by 45 amplification cycles (1 sec, 95°C; 20 sec, 60°C).
Drugs
The following blockers were used: D-APV (25 μM; Tocris Bioscience, Wiesbaden-Nordenstadt, Germany); 6,7-dinitroquinoxaline-2,3-dione (dNQX; 20 μM; Tocris Bioscience); picrotoxin (PTX; 50 μM; Tocris Bioscience); and QX-314 (5 mM; Tocris Bioscience).
Statistical analysis
Results are presented as mean ± standard error of the mean. The statistical evaluation of the data was performed with SPSS software (Statistics V22.0; IBM Corp., Armonk, NY). To test for normal distribution of the data, Kolmogorov-Smirnov's test in association with Shapiro-Wilk's test were performed. If the data were normally distributed, one-way analysis of variance and post-hoc least significant difference were used to compare sham, lesion ipsilateral, and lesion contralateral. In case data were not normally distributed, a one-way Kruskal-Wallis test and pair-wise Mann–Whitney U test were used. Chi-square test was performed for the statistical evaluation of active and silent cells. Significantly different values are indicated by asterisks in the figures and table (*p < 0.05; **p < = 0.01; ***p < = 0.001).
Results
Histology of the lesion induced by the controlled cortical impact
CCI led to a reproducible lesion affecting all cortical layers at all time points of investigation, as shown by Nissl-stained coronal section of a CCI-treated brain. The contusion area covered both part of motor (M1 and M2) and somatosensory (S1) cortices, measured 1.5–2.0 mm in mediolateral extent, and extended along the rostrocaudal axis between 0 and −2 mm relative to bregma (Fig. 1A and inset).

Histochemistry of the controlled cortical impact (CCI) lesion. (
To evaluate a potential neuroinflammatory reaction of cortical tissue in the vicinity of the lesion, 36,37 we performed IHC stainings for GFAP revealing reactive hypertrophic astrocytes, and for the the single-chain glycoprotein, ED-1, a known marker of activated microglia/macrophage. At 1 and 2 days post-lesion, an astro- and microgliosis was visible in the vicinity of the injured area spreading in both medial and lateral directions of the ipsilateral hemisphere. Reactive astrocytes could be detected up to a distance of 700–1000 μm from the border of the lesion whereas activated microglia/macrophages were limited up to 300–500 μm from the lesion border (Fig. 1C). Importantly, no astro- or microgliosis reaction as well as no overt structural damage could be observed in the cortex of sham-operated mice and in the contralateral hemisphere of CCI-treated mice (Fig. 1B,D).
Acute contralateral hyperactivity in the somatosensory cortex post-lesion
Acute cortical slices from the injured as well as from the contralateral hemisphere were selected between bregma −0.5 and −1.5 mm to measure spontaneous neuronal activity of the surviving cortical microcircuits in the acute (1 and 2 days post-lesion) and subacute (3 and 4 days post-lesion) phase post-lesion.
We recorded spontaneous neuronal activity from 32 channels with the pMEA chip positioned in the superficial cortical layers of CCI- and sham-treated mice (for details, see Methods). Electrodes were positioned 1 mm away from the border of the lesion or in homotopic regions both contralaterally and in sham animals (Fig. 2A). Interestingly, at 1 day post-lesion, the contralateral hemisphere showed a significantly increased number of channels in which we detected spontaneously occurring spikes (active channels; for details, see Methods; 19.6 ± 4.5 active channels; n = 5 from 5 mice; 190.6 ± 43.5%) compared to sham (10.3 ± 2.2 active channels; 100 ± 23.3%; n = 7 from 7 mice; p = 0.042; Fig. 2B,C). Neuronal activity contralateral to the lesion was not different anymore at 3 days post-TBI (19.3 ± 7.7 active channels; 130.6 ± 51.9%; n = 3 from 3 mice; sham, 14.8 ± 3.7 active channels; 100 ± 28.3%; n = 5 from 5 mice). Surprisingly, the ipsilateral hemisphere showed no change in the number of active channels compared to sham in neither the acute phase (sham, 10.3 ± 2.2 active channels; 100 ± 23.3%; n = 7 from 7 mice; ipsilateral, 8.9 ± 2.1 active channels; 86.3 ± 20.4%; n = 8 from 8 mice) nor the subacute phase post-lesion (sham, 14.8 ± 3.7 active channels; 100 ± 28.3%; n = 5 from 5 mice; ipsilateral, 18.2 ± 5 active channels; 123 ± 33.7%; n = 5 from 5 mice). We also analyzed the overall firing rate (for details, see Methods), but no differences were observed between CCI- and sham-treated animals, either at 1 day post-lesion (ipsilateral, 0.07 ± 0.2 Hz; 106.7 ± 41.7%; n = 8 from 8 mice; contralateral, 0.1 ± 0.2 Hz; 175.2 ± 39.5%; n = 5 from 5 mice; sham, 0.1 ± 0.1 Hz; 100 ± 29.3%; n = 7 from 7 mice) nor at 3 days post-lesion (ipsilateral, 0.09 ± 0.2 Hz; 104.9 ± 27.9%; n = 5 from 5 mice; contralateral, 0.2 ± 0.2 Hz; 128.6 ± 34.3%; n = 3 from 3 mice; sham, 0.1 ± 0.2 Hz; 100 ± 39.3%; n = 5 from 5 mice). These results strongly indicate that the acute phase post-TBI is characterized by a rapid and transient increase in the percentage of spontaneously active cells located exclusively in the contralateral hemisphere.
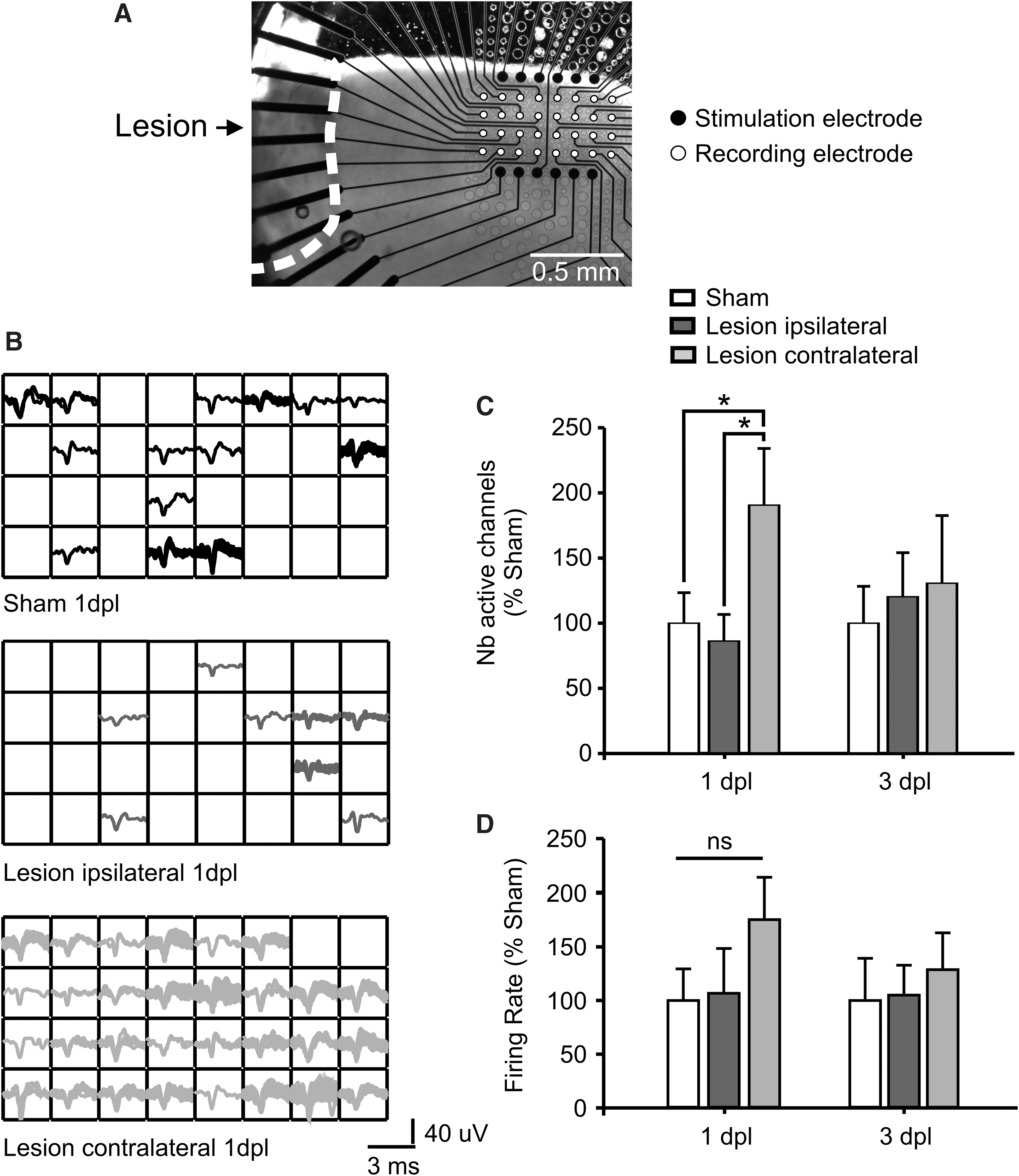
Extracellular pMEA recordings showing a hemispheric-specific increased spontaneous neuronal activity post-lesion. (
After exploring neuronal activity changes at the cortical network level, we investigated lesion-induced changes in spontaneous neuronal activity at the level of single cells. To this end, we performed whole-cell patch-clamp on pyramidal neurons from cortical layers 2 and 3 in current-clamp mode. We defined an acute and a subacute phase corresponding to 1–2 and 3–4 days post-lesion. Cells were considered active or silent based on the all-or-none presence of spontaneous action potentials during a 5-min recording period (for details, see Methods; Fig. 3A). In line with the extracellular recordings, we observed an increase in the number of spontaneously active cells only in the noninjured hemisphere at 1–2 days post-lesion (sham, 26.7%; 8 of 30 cells, from 10 mice; ipsilateral, 40%; 10 of 25 cells, from 10 mice; p > 0.05; contralateral, 51.5%; 17 of 33 cells, from 10 mice; p = 0.044), which fell back to control level in the subacute period post-CCI (sham, 28%; 7 of 25 cells, from 7 mice; ipsilateral, 40.4%; 11 of 27 cells, from 12 mice; contralateral, 28.6%; 8 of 28 cells, from 12 mice; Fig. 3B). The increased number of active cells did not correlate with a significant increase in the frequency of spontaneous firing at 1–2 days post-lesion (ipsilateral, 4.9 ± 2.8 Hz; 44.7 ± 25.9%; n = 10 from 7 mice; contralateral, 11.3 ± 2.5 Hz; 103.8 ± 24.4%; n = 17 from 9 mice; sham, 10.9 ± 3.4 Hz; 100 ± 31.2%, n = 8 from 7 mice; Fig. 3C). However, at 3–4 days post-lesion, a significant difference in frequency of spontaneous firing was revealed contralaterally compared to sham (ipsilateral, 9.6 ± 4.2 Hz; 400.6 ± 176.0%; n = 11 from 7 mice; contralateral, 15.1 ± 2.7 Hz; 632.1 ± 113.9%; n = 7 from 6 mice; p = 0.02; sham, 2.9 ± 1.8 Hz; 100 ± 64.9%; n = 6 from 4 mice; Fig. 3C). These results thus corroborate our extracellular data by confirming the contralateral-specific raise of neuronal activity at 1–2 days post-lesion.
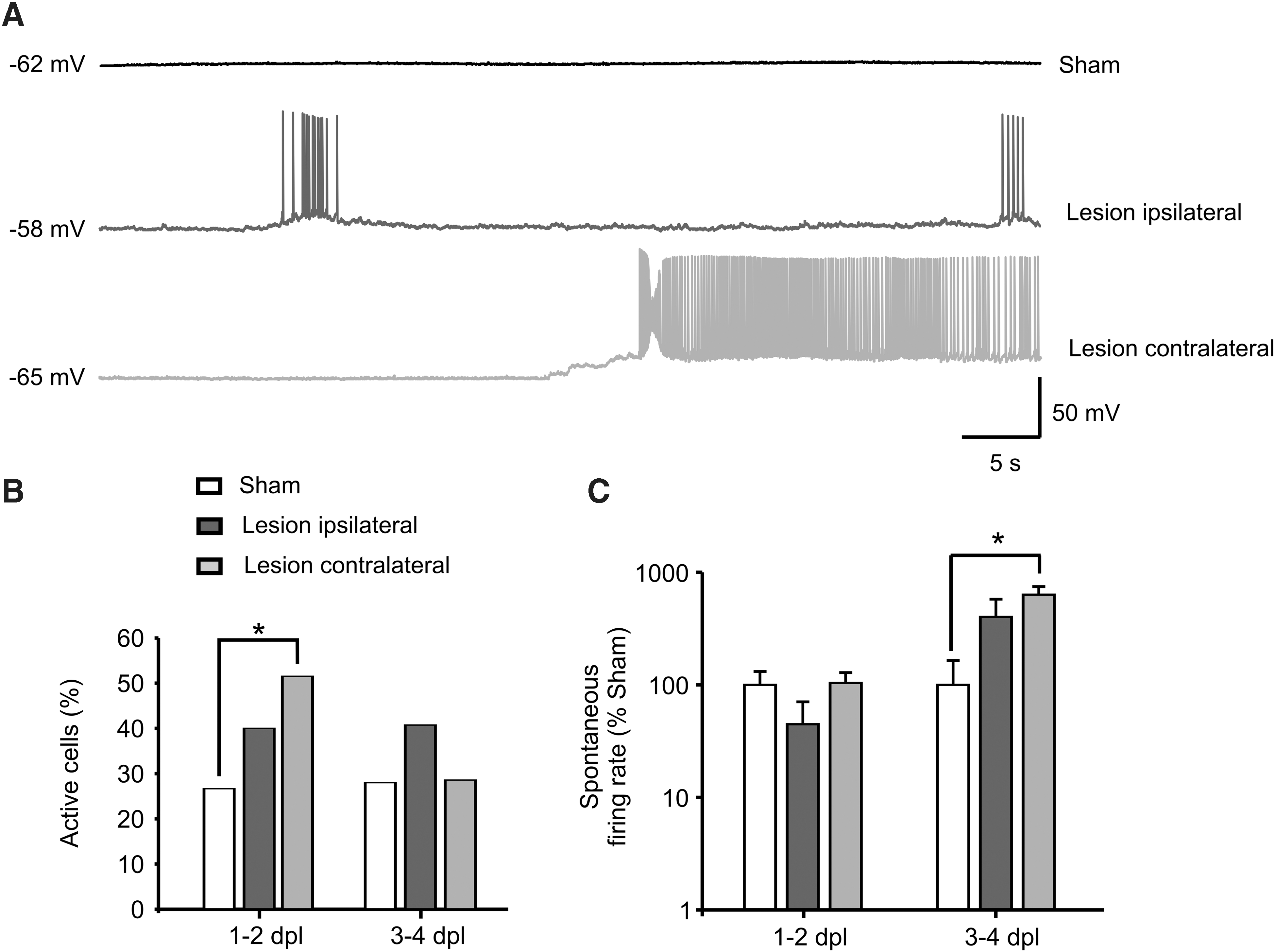
Whole-cell patch-clamp recordings confirming a hemispheric-specific increase in spontaneous neuronal activity of layer 2 and 3 pyramidal cells post-lesion. (
No lesion-induced changes in membrane and firing properties of contralateral pyramidal neurons
To further determine whether this acute raise of neuronal activity was linked to changes in the intrinsic excitability of neurons, we current-clamped pyramidal neurons in layers 2 and 3 and studied the intrinsic membrane properties in the injured and noninjured hemispheres and in sham-operated animals at 1–2 days post-lesion. Resting membrane potential was not altered after the lesion (Table 1), revealing that the increased number of active cells contralaterally could not be attributed to a general depolarization of neurons.
The numbers in brackets indicate the number of recorded neurons and the number of mice, respectively.
Input resistance (sham, 70.4 ± 5.1 MΩ; n = 28 from 9 mice; ipsilateral, 103.7 ± 8.9 MΩ; n = 22 from 10 mice; p < 0.001; contralateral, 73.8 ± 5.9 MΩ; n = 27 from 10 mice) and the rheobase (sham, 207.3 ± 12.1 pA; n = 28 from 9 mice; ipsilateral, 147 ± 13.4 pA; n = 22 from 10 mice; p = 0.002; contralateral, 190 ± 17.7 pA; n = 27 from 10 mice) of neurons recorded at −70 mV were significantly altered in the ipsilateral hemisphere post-TBI (Fig. 4A,B). The adaptation coefficient, spike half-width, threshold, and amplitude were also not altered by the lesion in neither the ipsilateral nor the contralateral cortex (Table 1). The action potential frequency–injected current relationship revealed significant differences for both contra- and ipsilateral hemispheres for relatively lower currents amplitude, slightly above the rheobase (Fig. 4A–C). This indicates a modulation in neuronal gain, which may be important to maintain a specific neuronal output in the face of a lesion-induced reduction of synaptic input. The maximal number of spikes was also not altered in between the different experimental conditions (Table 1).
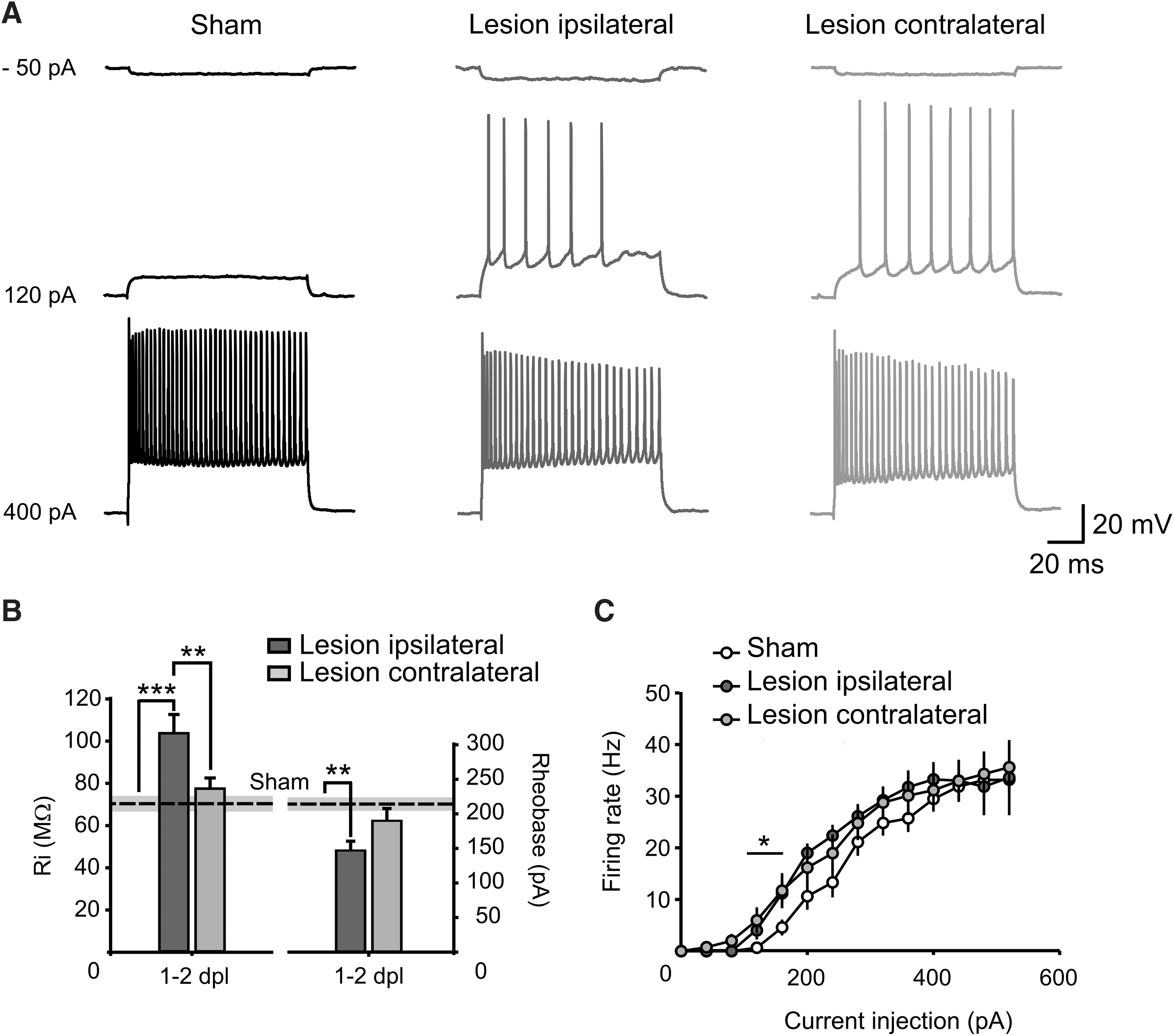
Lesion effects on input resistance and firing properties of layer 2 and 3 neurons at 1–2 days post-CCI. (
All together, these data suggest TBI-induced alterations in the intrinsic membrane properties of neurons in the injured hemisphere, but not in the undamaged cortex. From these findings, we conclude that an enhanced intrinsic excitability of neurons is very unlikely to explain the hyperactivity observed in the acute phase post-lesion in the contralateral hemisphere.
Contralateral specific lesion-induced changes in inhibitory inputs onto pyramidal neurons
Next, we investigated the impact of the lesion on synaptic efficacy. To do so, we recorded both spontaneous and evoked IPSCs and EPSCs in layer 2 and 3 pyramidal neurons at the reversal potential of AMPARs and GABARs, both in presence of D-APV (25 μM). The recordings were performed in the acute phase post-lesion (days 1 and 2 post-lesion), where we observed changes in neuronal activity.
sEPSCs were recorded at a holding potential of −60 mV (Fig. 5A). Neither the amplitude nor the frequency of these spontaneous events were altered post-TBI (Fig. 5B). In line with this, also sEPSC rise time and decay time remained unaltered post-CCI (Fig. 5B). These results suggest that basal glutamatergic transmission was not altered in the acute period post-TBI and did not contribute to the observed contralateral neuronal hyperactivity.
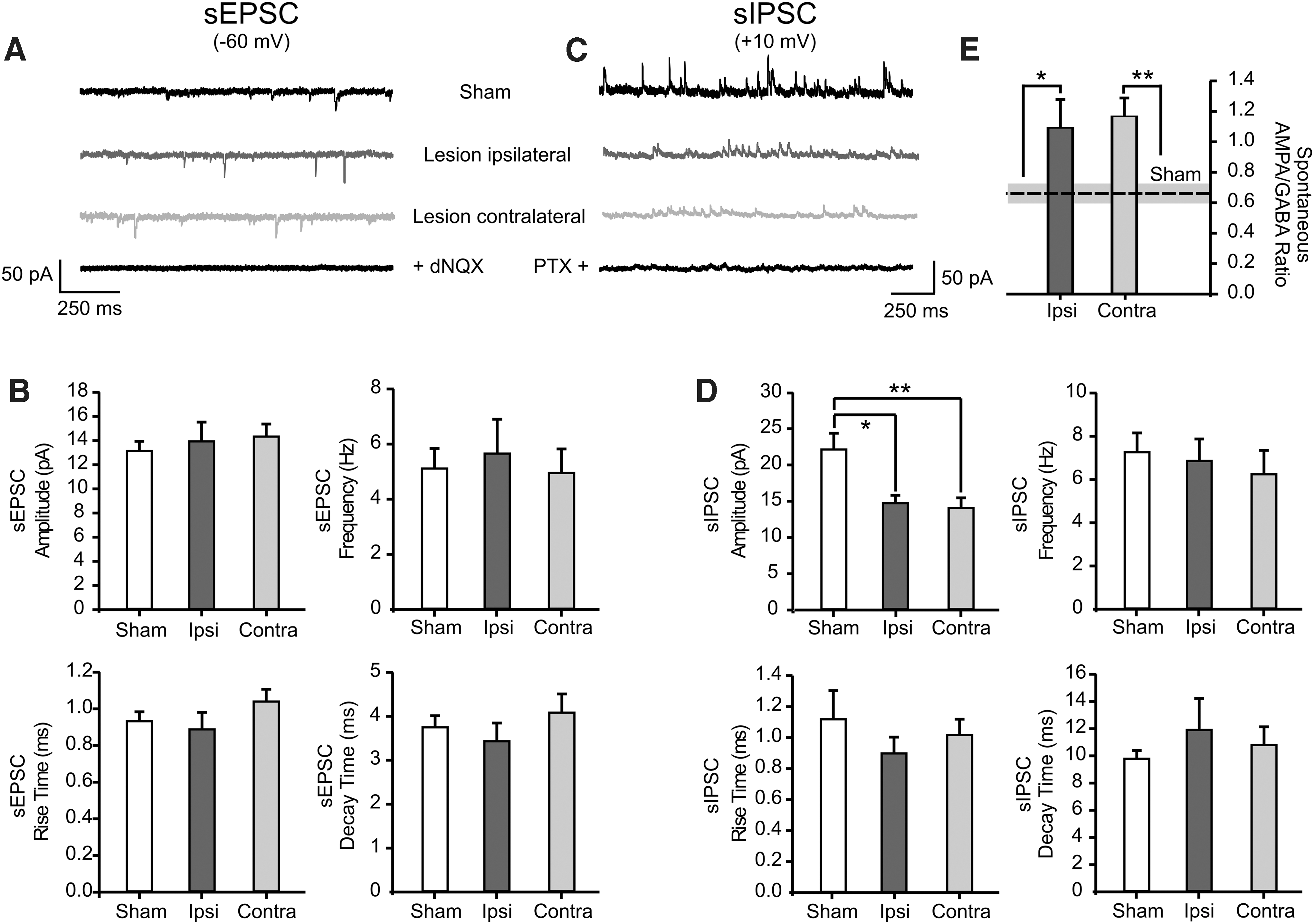
Lesion-induced changes in the basal synaptic transmission in layer 2 and 3 pyramidal neurons. (
sIPSCs were recorded in the same neurons by voltage clamping to +10 mV (Fig. 5C). Similarly to sEPSCs, frequency, rise time, and decay time were unaltered after the lesion (Fig. 5D). However, sIPSC amplitude was significantly reduced in both ipsi- and contralateral hemispheres post-lesion (sham, 22.1 ± 2.2 pA; n = 16 from 5 mice; ipsilateral, 14.7 ± 1.1 pA; n = 13 from 7 mice; p = 0.042; contralateral, 14.0 ± 1.4 pA; n = 17 from 7 mice; p = 0.002; Fig. 5D).
To maintain neuronal activity within a physiological range, pyramidal cells receive a balanced excitatory and inhibitory synaptic input. The analysis of the sIPSCs revealed an impaired GABAergic basal synaptic transmission post-lesion. This suggests that a shift toward excitability attributed to an imbalance between excitatory/inhibitory synaptic inputs may occur in CCI-treated mice. In order to further investigate this hypothesis, we monitored the excitatory/inhibitory (E/I) balance in layer 2 and 3 neurons either in spontaneous or evoked conditions. We used the recorded sPSCs to calculate the ratio of the amplitude of spontaneous AMPAR- versus GABAR-mediated currents, both recorded in the same neuron (Fig. 5A,B). The E/I balance for spontaneous events was significantly altered in both hemispheres post-lesion (sham, 0.7 ± 0.06; n = 16 from 5 mice; ipsilateral, 1.1 ± 0.2; n = 13 from 7 mice; p = 0.023; contralateral, 1.2 ± 0.1; n = 17 from 7 mice; p = 0.001; Fig. 5E). Evoked EPSCs (eEPSCs) and IPSCs (eIPSCs) induced by extracellular activation of afferent fibers were also recorded in the same neuron by clamping the cell either to the reversal potential of GABARs- or of AMPARs-mediated currents while keeping the intensity of the extracellular stimulus constant throughout the experiment (Fig. 6A). This allowed us to directly measure the strength of the excitatory (E) versus the inhibitory (I) synaptic currents received by layer 2 and 3 neurons. Consequently, we plotted the ratio between the amplitude of evoked AMPAR- and GABAR-mediated currents. The resulting E/I balance was significantly increased exclusively in the contralesional cortex (sham, 0.4 ± 0.1; n = 15 from 5 mice; ipsilateral, 0.7 ± 0.2; n = 13 from 5 mice; contralateral, 1.8 ± 0.4; n = 16 from 7 mice; p = 0.001; Fig. 6B). The kinetics of eIPSCs were also altered in the noninjured hemisphere. The decay time of the GABAR-mediated response was significantly increased compared to both sham and ipsilateral hemisphere (sham, 16.8 ± 1.3 ms; n = 15 from 5 mice; ipsilateral, 19.8 ± 2.0 ms; n = 13 from 5 mice; contralateral, 27.7 ± 3.1 ms; n = 16 from 7 mice; p = 0.001; Fig. 6C). These alterations of the eIPSCs in the uninjured hemisphere, together with the observed decreased amplitude of sIPSCs (Fig. 5D), strongly suggest an acute lesion-induced effect on the GABAergic post-synaptic site in the contralateral hemisphere.
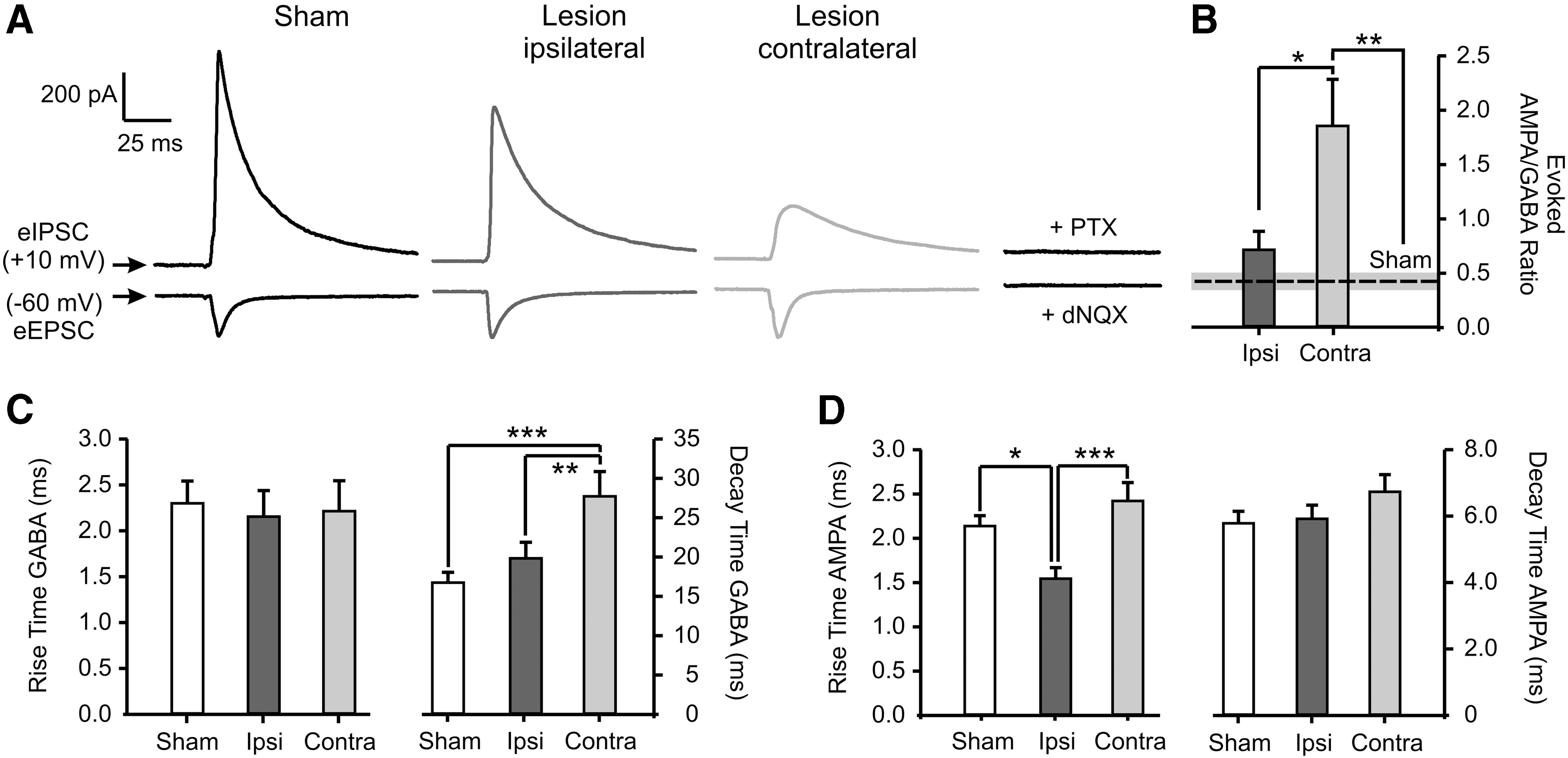
Hemispheric-specific alteration of the excitatory/inhibitory balance at 1–2 days post-CCI. (
The kinetics of evoked AMPAR-mediated currents were largely unaltered. However, the rise time of eEPSCs showed a decrease in the ipsilateral hemisphere (sham, 2.1 ± 0.1 ms; n = 15 from 5 mice; ipsilateral, 1.5 ± 0.1 ms; n = 13 from 5 mice; p = 0.02; contralateral, 2.4 ± 0.2 ms; n = 16 from 7 mice; Fig. 6D).
Changes in the messenger RNA expression of gamma aminobutyric acid type A receptor subunits
Our results on basal and evoked inhibitory transmission post-TBI point toward changes in the functional properties of post-synaptic GABA type A receptors (GABAARs). Differences in synaptic receptors function are very often related to changes in the combination of the subunits that the receptors are made of. Indeed, the subunit composition of the GABAARs is important to determine the channel intrinsic properties as well as the synaptic versus extrasynaptic localization. More specifically, receptors containing α1/γ2 GABAA subunits are mainly located synaptically and they are associated with a faster kinetics therefore being involved in phasic inhibition, whereas receptors containing α4/δ subunits are located extrasynaptically and have a much slower desensitization and they are thus associated to tonic inhibition. 38 –40 We investigated the specific messenger RNA (mRNA) expressions of the α1, γ2, α4, and δ subunits of GABAARs at 1–2 days post-injury in the same areas where we collected the electrophysiological data. In this time window, the expression of α1 and γ2 mRNA was not significantly altered in CCI-treated animals as compared to sham-operated mice (Fig. 7). This indicates that post-synaptic GABAARs mediating primarily the phasic GABAergic transmission are not altered (Figs. 5 and 6B). However, the expression of both the α4 and δ subunits was significantly enhanced in the contralateral hemisphere (α4, 1.4 ± 0.1; n = 6; p = 0.025; δ, 1.6 ± 0.2; n = 6; p = 0.028), potentially indicating a lesion induced increase of tonic inhibition in the contralateral hemisphere (Fig. 7).
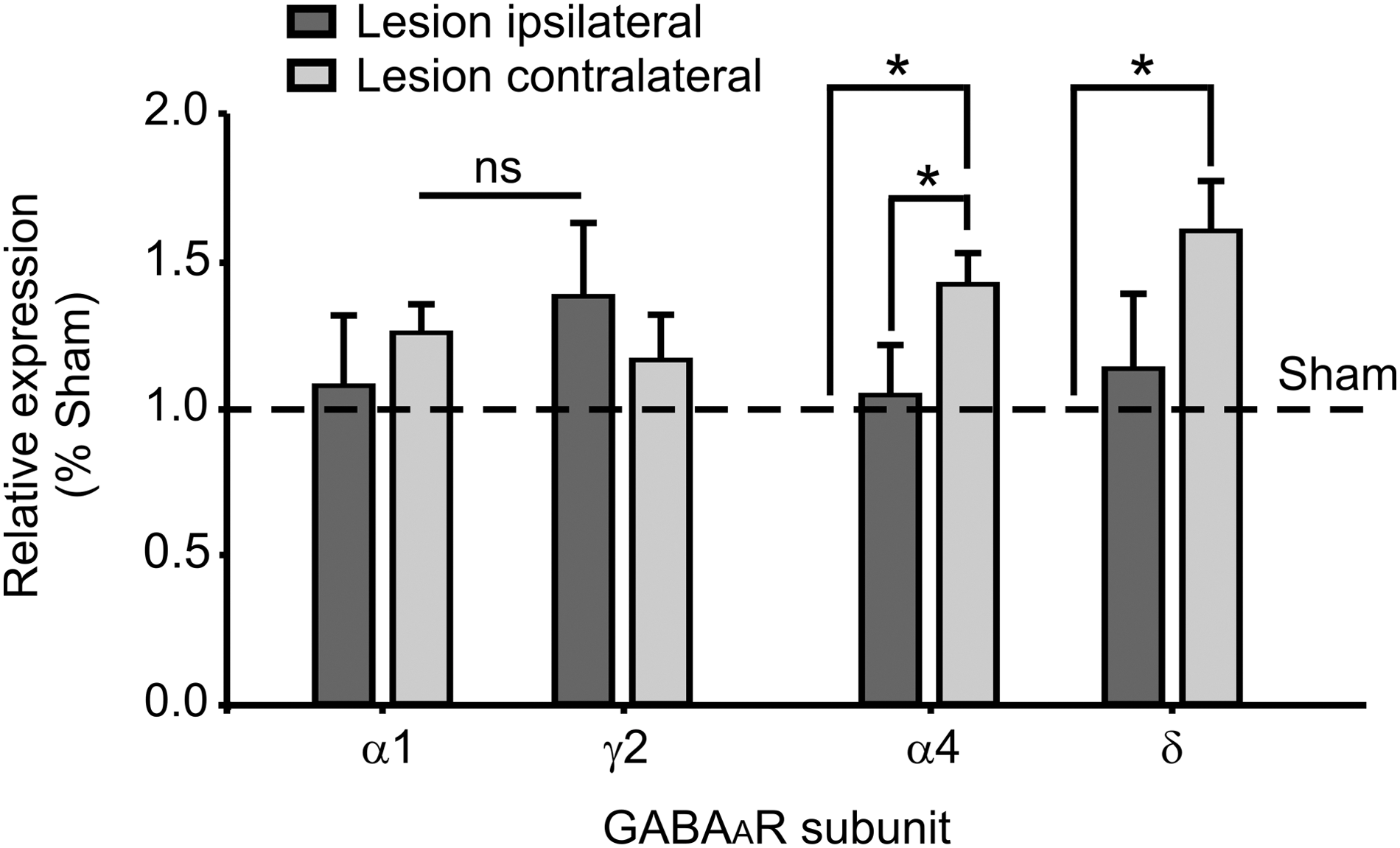
Lesion-induced increase in mRNA expression for the α4 and δ subunits of GABAA receptors. Bar graphs representing the relative expression of the mRNA for the GABAAR subunits α1, γ2, α4, and δ in the area of electrophysiological recordings at 1–2 days post-CCI. The dashed line shows the mRNA expression in sham-operated animals. CCI, controlled cortical impact; GABA, gamma aminobutyric acid; GABAAR, GABA type A receptor; mRNA, messenger RNA; ns, not significant.
In summary, we observed an acute hyperexcitable state in the noninjured cortex accompanied by an exclusive alteration in the GABAergic synaptic transmission in the same hemisphere.
Discussion
The brain is organized in a large number of highly specialized areas dedicated to specific functions. These functional brain areas are not separated entities, but are anatomically and functionally interconnected with other brain regions in a specialized fashion. It has been reported that the complex functional connectivity across distant brain regions is crucial for several tasks, from the integration of sensory inputs 41 and coordination of bilateral movements 42 to the elaboration of high cognitive processes. 43 As a logical consequence, an injury to one particular brain region will most likely affect directly and/or indirectly the other connected brain areas, thus modifying global functions of the brain. In this context, a phenomenon called transhemispheric diaschisis was intensively discussed since the early last century. This term was used to describe substantial disturbances of electrical activity, metabolism, and connectivity across the two cortical hemispheres after a unilateral cortical lesion. 17 Notably, a recent study from our lab started to consider transhemispheric diaschisis not as a merely functional disturbance, but rather an adapting mechanism to compensate for the functional loss caused by the lesion. 23 In line with this, some studies showed abnormal recruitment of the undamaged cortex for stimuli normally represented in the injured hemisphere at early time points after lesion induction. 7,32 To provide further insights into the role of the spared cortex in early processes of functional reorganization post-lesion, we functionally characterized both the injured and undamaged sensory-motor cortices in a well-established mouse model of mechanical cortical injury.
Morphology of the traumatic brain injury–treated neocortex
It is well known that a focal brain injury can trigger a gliosis reaction in the vicinity of the injury. Importantly, activated astrocytes as well as microglia can influence the responses of other neighboring cell types, notably neurons. 36,44 –46 Our IHC stainings showed that glial cells quickly reacted to the TBI, but stayed spatially restricted to the lesion region, most likely attributed to release of cellular debris, and disruption of the blood–brain barrier. Although resident inflammatory cells possess the ability to migrate post-injury, remote areas such as the hemisphere contralateral to the lesion were not affected according to our stainings. These results are in line with other studies after trauma in rodents, 47 –49 thus pointing out that, in our lesion model, glial reaction at early stages post-lesion is unlikely to influence remote cortical networks activity.
Acute hyperactivity in the contralateral hemisphere of somatosensory cortex post–traumatic brain injury
In the last decades, the study of brain hyperexcitability after cortical trauma has gained increasing attention because it often evolves into epilepsy months to years after the injury. 50 First experiencing a period of functional suppression, the neural network surrounding the lesion rapidly develops abnormal activity, which can spread across distal brain areas ultimately affecting global brain function. 35,51,52 Based on these findings, studies on unilateral cortical lesions have started not to focus on the damaged cortex alone, but also to investigate potential functional alterations in remote, connected brain areas such as the contralateral, homologous cortex. However, most of the studies were performed in subacute and chronic periods post-injury (days to weeks post-trauma), 18 given that structural plasticity and development of recurrent connectivity 53 were considered the most prominent cause for the development of hyperexcitable neuronal networks. 7,8 Despite these findings, recent developments in the field of functional neuroimaging have highlighted that the functional connectivity between distant brain areas can dynamically change within short time scales and have a great impact on global neuronal excitability. 54 Thus, interhemispheric alterations affecting the function of the undamaged cortex may occur in a time window shorter than previously thought. In light of these new findings, we started to investigate functional changes as early as 1 day after lesion induction. By using a modified aCSF in combination with extra- (multi-electrodes array) and intracellular (patch-clamp) electrophysiological recordings, we disclosed a biphasic change in neuronal activity throughout the contralateral, primarily undamaged brain area both at the cortical network level as well as at the single-cell resolution. The use of modified aCSF supplemented with low concentrations of kainate and carbachol was required to induce any spontaneous activity. The inevitable loss of afferent fibers through the process of acute slicing was most likely responsible for the absence of spontaneous spikes under normal aCSF perfusion. This technical caveat could here be compensated by using an aCSF with higher potassium concentration and with specific pharmacological agents that recreate inputs normally present in vivo. 11,55 Remarkably, in our injury model, we could distinguish an acute (1–2 days post-lesion) hyperactive contralateral cortical network from a subacute (3–4 days post-lesion) rather control-like active state (Figs. 2C and 3B), underlying the transient component of this over-responsiveness. In parallel, the ipsilateral hemisphere stayed unaffected. The firing frequency of both damaged and intact hemispheres was also unchanged (Figs. 2D and 3C). Therefore, the observed hyperexcitable state in the contralateral cortical microcircuits, as observed under our experimental conditions, seems to be mainly attributed to an increased number of spontaneously active cells and not to an increase in the firing frequency of neurons.
These results strongly indicate that the very acute period after a TBI is characterized by a rapid and transient neuronal hyperactivity in the contralateral hemisphere. To the best of our knowledge, these findings reveal, for the first time, an early hyperactivation of the intact hemisphere in a clinically relevant rodent model of TBI both at the cortical network level as well as at the single-cell resolution.
No lesion-induced changes in membrane and firing properties of contralateral pyramidal neurons
Previous studies from our lab using a different lesion model reported alterations of intrinsic properties in both pyramidal neurons from layer 2 and 335 and in interneurons 11 during the first week post-lesion in the damaged cortex. In the present study, we also investigated membrane and firing properties of neurons post-TBI given that alterations at these levels may promote neuronal firing and explain the observed increased neuronal activity. Our current-clamp recordings indeed revealed lesion-dependent alterations in input resistance and rheobase. However, these changes did not expand into the contralateral cortical microcircuits at 24–48 h post-TBI. Therefore, under our experimental conditions, changes in the intrinsic properties of neurons cannot explain the increased neuronal activity that we observed contralaterally to the lesion (Table 1; Fig. 4). Membrane potential remained unaltered, implying no general depolarization of neurons. Input resistance, rheobase, and spike threshold were as well not affected, suggesting an unchanged sensitivity to synaptic inputs in the contralateral hemisphere. Finally, firing and spikes properties investigated were similar to sham condition.
A significant increase in input resistance and a lower rheobase were found only in the cortex ipsilateral to the lesion at 24 and 48 h post-TBI. Possibly, the loss of a large brain area ipsilaterally to the lesion may temporally suppress neuronal function and the observed proexcitatory changes in membrane properties may be sufficient to homeostatically adjust the otherwise impaired cortical activity to, but not above, the control-like level. This may explain why we did not observe an abnormal neuronal activity ipsilateral to the lesion despite the proexcitatory alterations in membrane properties. Notably, the observed intrinsic alterations may not be sufficient to cause hyperexcitability at such an early stage post-lesion, but may contribute to its later development. In line with this, our lab 11,35 and others 51,52 have reported that hyperexcitability in the perilesional area is slowly arising in the subacute period (first week post-lesion).
Although both hemispheres are partially commonly affected on certain parameters, all the above findings suggest that different mechanisms with different timing affect the two cortices shortly after lesion, most likely producing different outcome on the pathophysiology and recovery.
Contralateral specific lesion-induced changes in inhibitory inputs onto pyramidal neurons
Because no significant alterations could be disclosed in the intrinsic excitability of layer 2 and 3 neurons in the contralateral hemisphere, other mechanisms should underlie the observed hyperactivity post-lesion. Alternatively, the rapid recruitment of a higher number of neurons in the active network of the undamaged cortex may be achieved by unmasking previously unused subthreshold neuronal connections through changes in synaptic strength or in the balance between excitatory and inhibitory synaptic transmission. Therefore, we investigated single-cell synaptic efficacy by voltage-clamp recordings of spontaneous and evoked PSCs to directly measure inhibitory and excitatory synaptic signals at the reversal potentials of AMPARs and GABARs, respectively. Amplitude of sIPSCs was significantly reduced in both cortices, whereas no alterations were observed in amplitude of sEPSCs. Generally, a decrease in amplitude of spontaneous synaptic events suggests changes in the expression, localization, or activation of post-synaptic receptors, even though alterations in neurotransmitter release cannot be ruled out. 56 An impaired phasic inhibition has been already put forward after focal lesion, however, at later time points and ipsilaterally. 57 In addition, the significant increase of the AMPA/GABA ratio in CCI-treated animals suggested an altered spontaneous E/I balance post-lesion. Given that such spontaneous events are considered as relying on the stochastic release of neurotransmitters, computing the E/I ratio of evoked signals would bring more insight into the neuron capacity of processing incoming stimuli. Contrary to spontaneous synaptic transmission, when AMPAR- and GABAR-mediated synaptic currents were evoked with an extracellular stimulation electrode, the ipsi- and contralateral cortices disclosed a different outcome. Contrary to the E/I ratio of spontaneous signals, the ratio between the amplitude of evoked EPSCs and IPSCs in the lesioned hemisphere remained unaltered. It is possible that attributed to the different recording conditions (spontaneous currents vs. evoked currents) a different subset of receptors was activated. For instance, extrasynaptic GABA receptors are more likely to be recruited during evoked responses. 58 Therefore, this discrepancy might be linked to a different expression or localization of synaptic and extrasynaptic GABA receptors. Instead, the E/I ratio showed a drastic increase in the contralesional cortex, and the GABAR-mediated currents displayed a slower kinetics. This strongly suggests a shift in the E/I balance in direction of excitability, potentially contributing to the hyperexcitability observed in the acute time window post-lesion.
Changes in the messenger RNA expression of gamma aminobutyric acid type A receptor subunits
The observed alterations in the inhibitory synaptic transmission suggest possible changes in the expression level of different GABAARs subunits. Special focus was given at the α1/γ2 and α4/δ subunits, considering their respective affiliation to either phasic (synaptic GABAARs) or tonic inhibition (extrasynaptic GABAARs). These two forms of GABAergic neurotransmission and the GABAARs have been showed to have a distinct role in controlling neuronal excitability and plasticity. 58,59 Additionally, it was recently shown that these two forms of inhibition were differentially affected subsequent to cortical injuries, epilepsy, and stroke. 57,60,61
Under our experimental condition, no alteration could be detected for any of the investigated GABAARs subunits in the ipsilateral hemisphere at 1–2 days post-lesion. Previous work by Redecker and colleagues 62 as well revealed no changes in α1/γ2 subunits expression ipsilaterally at 1 day post-lesion. In contrast, in the cortex contralateral to the lesion, we found a significantly higher expression of the extrasynaptic-related GABAARs subunits α4 and δ, strongly suggesting an enhanced tonic inhibition. This result is in line with a recent study by Drexel and colleagues, 63 where the same subunits where upregulated in the contralateral hippocampus at 6 h post-lesion. Possibly, a shift from phasic to tonic GABAergic transmission may partially explain the hemisphere-specific impairment in evoked GABAergic responses and the slower kinetics of evoked IPSCs. As a matter of fact, tonic GABAAR-mediated currents have been shown by others as largely preserved in epileptic state, 61 even in the case of impaired phasic inhibition. As well, our lab has put forward the hypothesis that an enhanced tonic inhibition may compensate for an impaired synaptic GABAergic transmission, and this may be important to prevent runaway excitation in an already hyperexcitable network. 57
Despite the increasing interest from the scientific community in the last years, the involvement of diaschisis in processes of functional recovery post-TBI is still under debate. Here, we add a new piece to the puzzle of transhemispheric post-lesional contribution to functional reorganization processes after traumatic injury. Considering ours and the work of others, 7,32 the very early enhanced recruitment of the contralateral hemisphere with a parallel unaltered ipsilateral neuronal activity might be a beneficial attempt of the brain to compensate for the functional loss post-lesion. Future studies should disclose the functional relevance of this acute and transient contralateral hyperactivity, for example, by silencing the contralateral hemisphere through the use of local anesthesia 64 or optogenetic, 65 combined with relevant behavioral tasks or in vivo electrophysiological recordings.
Footnotes
Acknowledgments
We thank Tobias Hirnet for performing part of the CCI, Simone Dahms-Prätorius for the help with molecular biology, and the support of the Microscopy Core Facility of the IMB, Mainz. This work is supported by the German Research Foundation (DFG, CRC 1080, TP A7 CO2 to K.E. and T.M.).
Author Disclosure Statement
No competing financial interests exist.