
Abstract
Although sleep-wake disturbances are prevalent and well described after traumatic brain injury, their pathophysiology remains unclear, most likely because human traumatic brain injury is a highly heterogeneous entity that makes the systematic study of sleep-wake disturbances in relation to trauma-induced histological changes a challenging task. Despite increasing interest, specific and effective treatment strategies for post-traumatic sleep-wake disturbances are still missing. With the present work, therefore, we aimed at studying acute and chronic sleep-wake disturbances by electrophysiological means, and at assessing their histological correlates after closed diffuse traumatic brain injury in rats with the ultimate goal of generating a model of post-traumatic sleep-wake disturbances and associated histopathological findings that accurately represents the human condition. We assessed sleep-wake behavior by means of standard electrophysiological recordings before and 1, 7, and 28 days after sham or traumatic brain injury procedures. Sleep-wake findings were then correlated to immunohistochemically labeled and stereologically quantified neuronal arousal systems. Compared with control animals, we found that closed diffuse traumatic brain injury caused increased sleep need one month after trauma, and sleep was more consolidated. As histological correlate, we found a reduced number of histamine immunoreactive cells in the tuberomammillary nucleus, potentially related to increased neuroinflammation. Monoaminergic and hypocretinergic neurotransmitter systems in the hypothalamus and rostral brainstem were not affected, however. These results suggest that our rat traumatic brain injury model reflects human post-traumatic sleep-wake disturbances and associated histopathological findings very accurately, thus providing a study platform for novel treatment strategies for affected patients.
Introduction
A
Among the most commonly reported post-traumatic SWD are excessive daytime sleepiness (EDS) and excessive sleep need per 24 h. 7,8 For the sake of clarity—the term “hypersomnia” has been used for both EDS and excessive sleep need—we recently introduced the term “post-traumatic pleiosomnia” for excessive sleep need per 24 h after TBI. 7 In autopsy samples of deceased TBI victims, we found a >40% loss of wake-maintaining histamine (HA) neurons in the tuberomammillary nucleus (TMN; i.e., in the posterior hypothalamus), and more moderate damage to other sleep-wake regulating hypothalamic and brainstem nuclei. 9,10 Because human TBI is heterogeneous with reference to type, localization, severity of trauma, and to the brain it is affecting, it is difficult to systematically study specific alterations in sleep electroencephalography acutely after TBI and to compare findings to neuropathological outcomes.
In rodents, on the other hand, the effects of reproducible brain lesions on sleep-wake behavior and histological outcomes can be studied more systematically. Although decades of animal studies on TBI have thoroughly investigated cognitive performance, neuropsychiatric outcome, and motor skills after trauma, 11 –14 there are only a few systematic reports on post-traumatic SWD in rodents. 15 –22 Most of these studies examined sleep-wake behavior only within the first few days after TBI, 15,16,19,20 while human post-traumatic SWD persist up to months and years. 4,5,23,24
Sleep-wake outcomes up to five weeks after mouse or rat fluid percussion or closed cortical impact TBI have been analyzed recently, but electroencephalographic sleep assessments were not performed 18,21 or baseline (BL) sleep-wake levels were not analyzed. 17 Willie and colleagues 15 observed by intracerebral microdialysis abnormal dynamics of hypocretin (orexin) signaling in association with reduced wakefulness three days after murine TBI. Similarly, Lim and associates 16 reported that mild TBI in mice causes a persistent inability to maintain wakefulness and decreases arousal-promoting hypothalamic hypocretin neuron activation during wakefulness. Skopin and coworkers 22 also observed the appearance of chronic increased sleep in relation to a significant loss of hypocretin cells after lateral fluid percussion, while post-traumatic changes in other neurotransmitter systems, such as the highly affected histaminergic one, 9 were not explored. 22
To date, thus, there is no study to explore changes in short- and long-term post-traumatic sleep-wake behavior compared with BL recordings and in relation to the integrity of multiple sleep-wake modulating neurotransmitter systems. Such knowledge should add validity to a rodent TBI model as representative of the human condition and, thus, offer novel windows of opportunity to develop treatments for affected patients with TBI.
With this study, we aimed to investigate sleep-wake behavior after TBI in rats and to examine whether the integrity of sleep-wake modulating neurotransmitter systems in the hypothalamus and brainstem is disturbed in association with sleep-wake findings.
Methods
Animals
We used adult male Sprague-Dawley rats (Harlan Laboratories Inc, NL) weighing 230–270 g (∼8–9 weeks of age) at arrival for all experiments. The rats were housed in groups for 2–3 days before electroencephalography/electromyography (EEG/EMG) electrode implantations and afterward single-caged with ad libitum access to food and water. The animal room temperature was constantly maintained at 21–23°C, and animals were kept under a 12 h light-dark cycle starting at 8 or 9
Surgeries
We performed all surgical procedures under deep anesthesia with isoflurane (4.5% for induction, 2.5% for maintenance). We administered buprenorphine subcutaneously (0.05 mg/kg) for analgesia. Over the first week after surgical procedures, we monitored all animals on a daily basis and weekly thereafter by assessing weight progression and wound healing.
EEG/EMG electrodes implantation
We implanted rats with EEG/EMG electrodes for recording of vigilance states as described earlier. 25 Briefly, we inserted four gold-plated miniature screws, one pair for each hemisphere, bilaterally into the rat's skull following specific stereotaxic coordinates: the anterior electrodes were implanted 3 mm posterior to bregma and 2 mm lateral to the midline, and the posterior electrodes 6 mm posterior to bregma and 2 mm lateral to the midline. We left the skull above the frontal cortex unimplanted for TBI impact. For the monitoring of muscle tone, a pair of gold wires inserted into the rat's neck muscle served as EMG electrodes. All electrodes were connected to a head connector (Farnell AG, Switzerland) and fixed to the skull with dental cement. A recovery of 8–10 days was granted to all animals before further interventions.
TBI
This novel TBI model was introduced earlier. 25 Briefly, after 8–10 days of recovery, the EEG/EMG implanted rats weighed 300–350 g (∼10–11 weeks old) and underwent closed skull TBI in the pre-frontocortical area of the brain using a closed diffuse TBI-inducing device (Fig. 1a). Rats under deep anesthesia received a 0.5–0.7 cm scalp incision over the midline in the pre-frontocortical area of the brain, thus anterior to the implanted electrodes. For TBI, using a system of sliding rings, we released a 2500 g stainless steel rod provided with a flat silicon tip adjusted to an angle of 70 degrees and falling from a height of 25 cm over the exposed skull. The latter was covered by a 1 mm thick metal plate to prevent bone fractures. Thereafter, we closed and disinfected the skin, and animals returned to their home cage and were monitored continuously for at least 1 h or until full recovery from sedation and normal home cage behavior were observed. Analgesia was provided using buprenorphine. The SHAM procedures were identical to TBI ones, except the rod was not released to its intended fall.
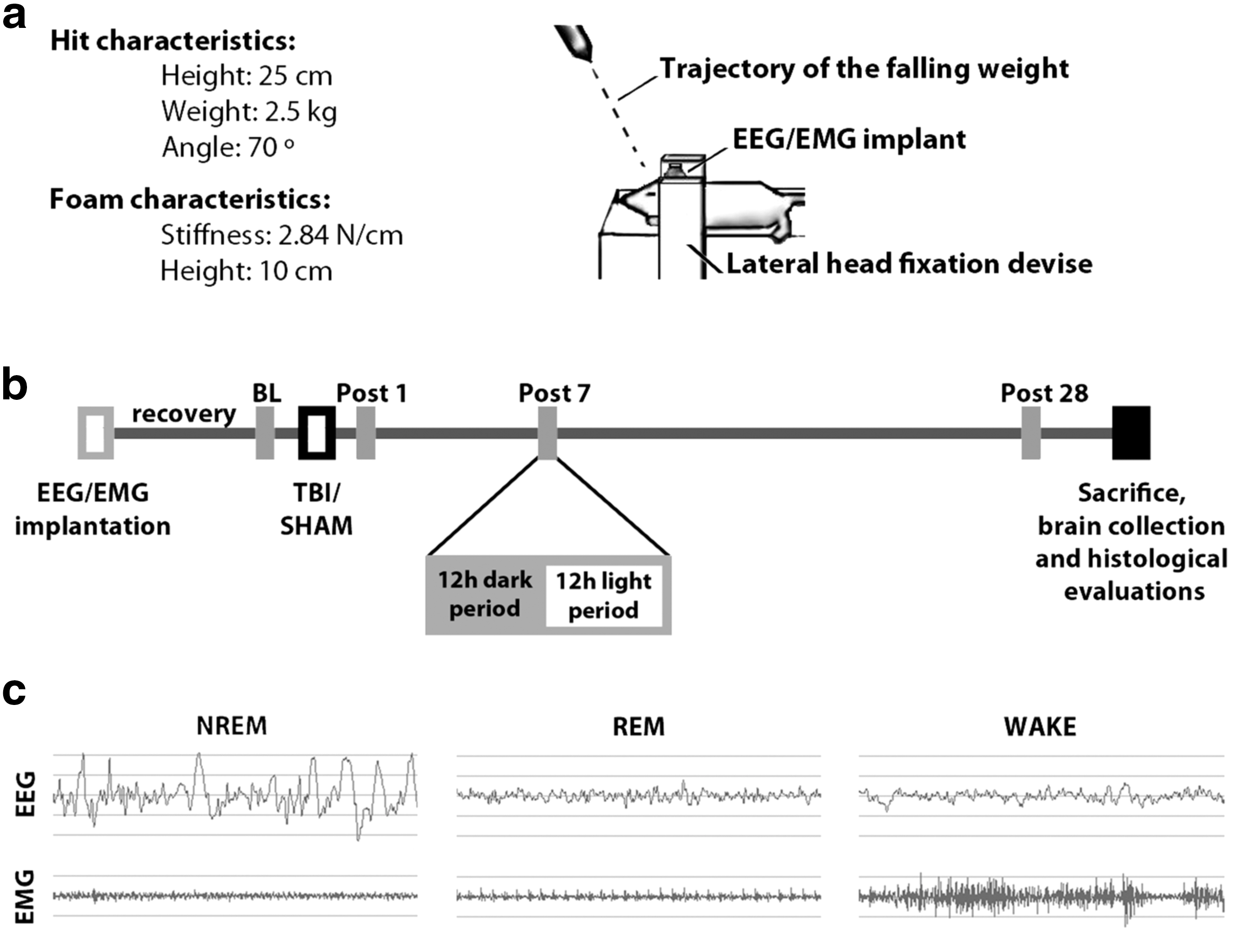
Experimental design.
As reported earlier, 25 despite memory function being impaired, rats survived TBI without major neurological or motor deficits. The EEG/EMG implants proved stable for at least one month and enabled qualitative and quantitative sleep analyses. Histologically, we observed no major bleeding or necrosis but intense diffuse axonal damage after TBI.
EEG/EMG recording and scoring
We conducted bilateral 24 h EEG/EMG recordings one day before injury to serve as BL, as well as 1, 7, and 28 days after trauma (Fig. 1b). For this purpose, we transferred animals to special recording cages with food and water available ad libitum, where they had an adaptation period of two days before recordings took place. During every recording session, we recorded SHAM and TBI rats simultaneously. For analysis purposes, we divided the 24 h scoring sessions into 12 h light and dark periods (Fig. 1b), and assessed the proportion of each vigilance state separately for each period. As before, based on EEG/EMG patterns, we assessed three behavioral states: non-rapid eye movement (NREM) sleep, rapid eye movement (REM) sleep, and wakefulness (WAKE) (Fig. 1c). 25 Sleep-wake proportion results from the light period were reported previously. 25
To further explore the characteristics of sleep in the dark period 28 days after TBI, we calculated the index of sleep fragmentation as introduced before. 4 Briefly, sleep fragmentation is defined as the number of sleep bouts divided by the total number of epochs in the same sleep stage. The sleep fragmentation index reflects, therefore, the relative number of behavioral state bouts per time (e.g., highly fragmented sleep with many changes in sleep-wake behavioral states will result in a high sleep fragmentation index). Finally, we determined the delta power (1–4 Hz) of NREM sleep at BL, and 1, 7, and 28 days after both TBI and SHAM procedures to assess whether TBI caused changes in sleep depth compared with SHAM controls. The TBI effect 28 days after trauma was calculated as ΔTBI-SHAM% per 24 h in 2 h bins during both dark and light periods.
Immunohistochemistry
Rats were sacrificed 30 days after trauma by classic cardiac perfusion under pentobarbital anesthesia (75 mg/kg). We dissected brains, post-fixed and dehydrated them in 4% paraformaldehyde/15% sucrose (Sigma-Aldrich) in phosphate buffered saline (PBS) at 4°C overnight. A further dehydration step in 30% sucrose in PBS followed. After complete dehydration, we froze brains in dry ice and stored them at −80°C until further use.
To assess post-traumatic histological outcomes in relation to sleep-wake behavior, we explored four sleep-wake regulating neurotransmitter nuclei in the hypothalamus and brainstem. We incubated free-floating 40 μm coronal brain cryostat sections at 4°C with agitation with primary antibodies against: (a) HA, (rabbit anti-HA, 1:100, Millipore, cat#AB5885, seven days incubation); (b) melanin-concentrating hormone (MCH, rabbit anti-MCH, 1:10,000, Phoenix Europe GmbH, H-070-47, 16 h incubation); (c) hypocretin/orexin (Hcrt-1, goat anti-hcrt1 [C19], 1:5,000, Santa Cruz; sc-8070, 16 h incubation); and (d) tyrosine hydroxylase (TH, mouse anti-TH, 1:8,000, Millipore, MAB318, 16 h incubation), a marker of catecholaminergic cells.
After primary antibody incubation, the sections were washed and then incubated in the corresponding secondary antibodies: biotinylated goat anti-rabbit secondary antiserum (1:250; VectorLabs), biotinylated horse anti-goat secondary antiserum (1:600, VectorLabs) or biotinylated goat anti-mouse secondary antiserum (1:500; Santa Cruz Biotechnology) for 2 h at room temperature. Then, the sections were incubated with avidin-biotin complex (1:100, VectorLabs) and exposed to 0.025% 3,3' diaminobenzidine in Tris buffered saline in the presence of 0.05–0.1% H2O2 to produce dark brown precipitates and finally mounted. All results were compared with SHAM controls.
In the TMN, the only nucleus in which we observed significant loss of neurotransmitter immunoreactivity, we examined by double immunofluorescence the association between such loss and other histological sequelae of TBI. To this end, we co-incubated TMN hypothalamic sections with anti-histidine decarboxylase, HDC—a marker of histaminergic cells—(rabbit anti-HDC, 1:5000, American Research Products, CAT#03-16045) and one of the following antibodies: anti-amyloid precursor protein-1, APP-1—a marker of diffuse axonal injury—(mouse anti-APP-1, 1:300, Millipore, MAB348); anti-Ox42—a neuroinflammation marker—(mouse anti-Ox42/anti- CD11b/c, 1:500, Abcam, ab1211); anti-glial fibrillary acidic protein, GFAP—a marker of astrogliosis—(mouse anti-GFAP, coupled with Cy3, 1:400, Sigma-Aldrich, C9205).
After primary antibody incubation overnight, the sections were washed and then incubated in the corresponding secondary antibodies: goat anti-rabbit Alexa 488 (Molecular Probes, 1:500) and donkey anti-mouse Alexa 568 (Molecular Probes, 1:500). After washings, the fluorescent sections were mounted (Aquamount, Fischer sci), coverslipped, and stored in the dark at 4°C until microscopy for qualitative evaluation.
Stereological counting
We quantified hypothalamic and brainstem neuronal populations by blinded semi-automatic stereological countings using the optical fractionator method on a Zeiss Axio Imager M2 microscope (Karl Zeiss, Jena, Germany) equipped with a Ludl MAC 6000 stage (Ludl Electronic Products, Hawthorne, NY) and MicroBrightField Stereo Investigator 10.5 software (MBF Bioscience, Williston, VT). We used a 2.5 × objective for outlining brain areas with immunopositive cells, and a 40 × oil immersion objective (1.3 numerical aperture) for stereological counting. We defined the borders of the reference space by staining pattern. Only animals with a complete set of undamaged sections were included in the final analysis. We optimized counting frames and sampling grid sizes for each antigen to achieve a Gundersen coefficient of error (CE) <0.08. A guard distance of 2.0 μm was used during cell counting to avoid introduction of errors because of sectioning artifacts.
Statistical analyses
Two-tailed, paired and unpaired t tests with or without Bonferroni corrections, repeated measures analysis of variance (ANOVA), post hoc analysis of Fisher (least significant difference), analysis of covariance (ANCOVA), and Pearson correlations were performed as indicated in the Results section and figure legends. All bars in the graphs represent the groups' mean, and the error bars indicate standard error of mean (SEM). The sample size is indicated in each figure legend as well as the statistics p value. Significant differences were acknowledged only to p values lower than 0.05. Thus, the performed analyses provide a margin of confidence of 95% to our results.
Results
Increased sleep need 4 weeks after closed diffuse TBI in rodents
As reported previously, 25 sleep-wake proportions in the light period (rats' resting phase) were unchanged in TBI compared with SHAM animals. Analysis of dark period (rats' active phase) EEG signals, however, revealed an increase of NREM sleep in injured rats 28 days after TBI, as evidenced by the significant decrease of WAKE (Fig. 2a, SHAM [n = 6] vs. TBI [n = 7], *p < 0.05, two-tailed unpaired t test with Bonferroni correction) and increase of NREM sleep (Fig. 2b, SHAM [n = 6] vs. TBI [n = 7], *p < 0.05, two-tailed unpaired t test with Bonferroni correction) compared with SHAM rats. Differences of WAKE and NREM sleep proportions between SHAM and TBI groups were not observed at any other time point analyzed in the dark period (BL, P1, P7; Fig. 2a,b). The REM sleep was unaltered after TBI for all time points (Fig. 2c).
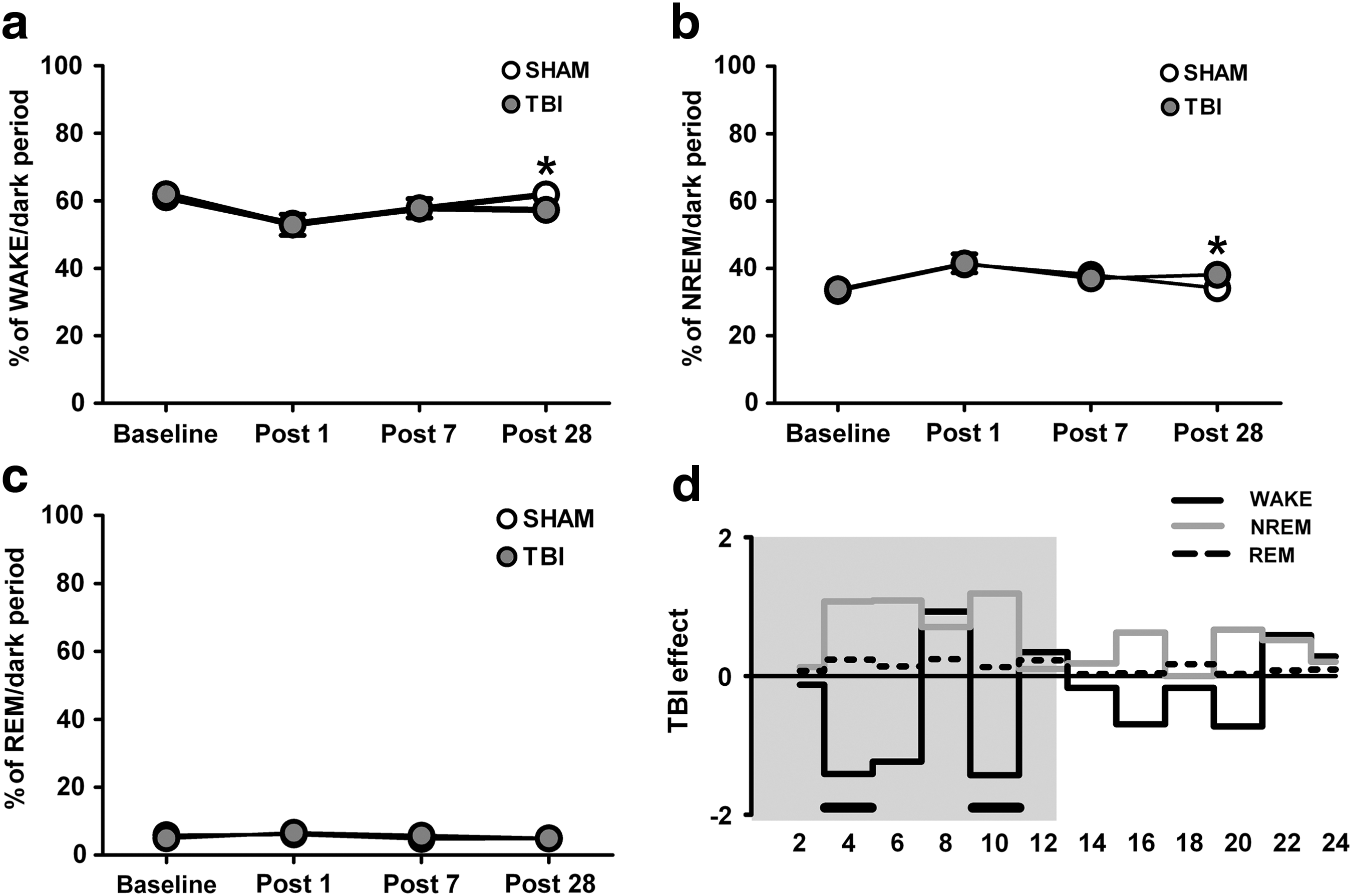
Proportion of behavioral states after traumatic brain injury (TBI). Decrease in WAKE (
To gain more detailed insights into the 24 h time course of vigilance state proportions 28 days after TBI, we analyzed the recordings of both SHAM (n = 6) and TBI (n = 7) animals in 2 h time bins. This analysis revealed that TBI animals had intermittent periods of increased sleep need at the cost of wakefulness between the third and fifth hour (WAKE: *p < 0.05; NREM: # p = 0.06, repeated measures ANOVA, post hoc analysis of Fisher) and the ninth and eleventh hour (WAKE and NREM: *p < 0.05, repeated measures ANOVA, post hoc analysis of Fisher) during the dark period (Fig. 2d). Again, the distribution of REM sleep throughout the 24 h period was similar between both groups (Fig. 2d).
To further determine the existence of post-traumatic pleiosomnia, we calculated the cumulated total sleep (TS, NREM+REM) per 24 h and in separate dark and light periods (Fig. 3) in TBI animals compared with SHAM controls. The TS was increased both in the dark period (Fig. 3a; SHAM [n = 6] vs. TBI [n = 7], **p < 0.005, one-tailed unpaired t test) and per 24 h (Fig. 3c; *p < 0.05, SHAM [n = 6] vs. TBI [n = 7], one-tailed unpaired t test), but unaltered in the light phase (Fig. 3b).
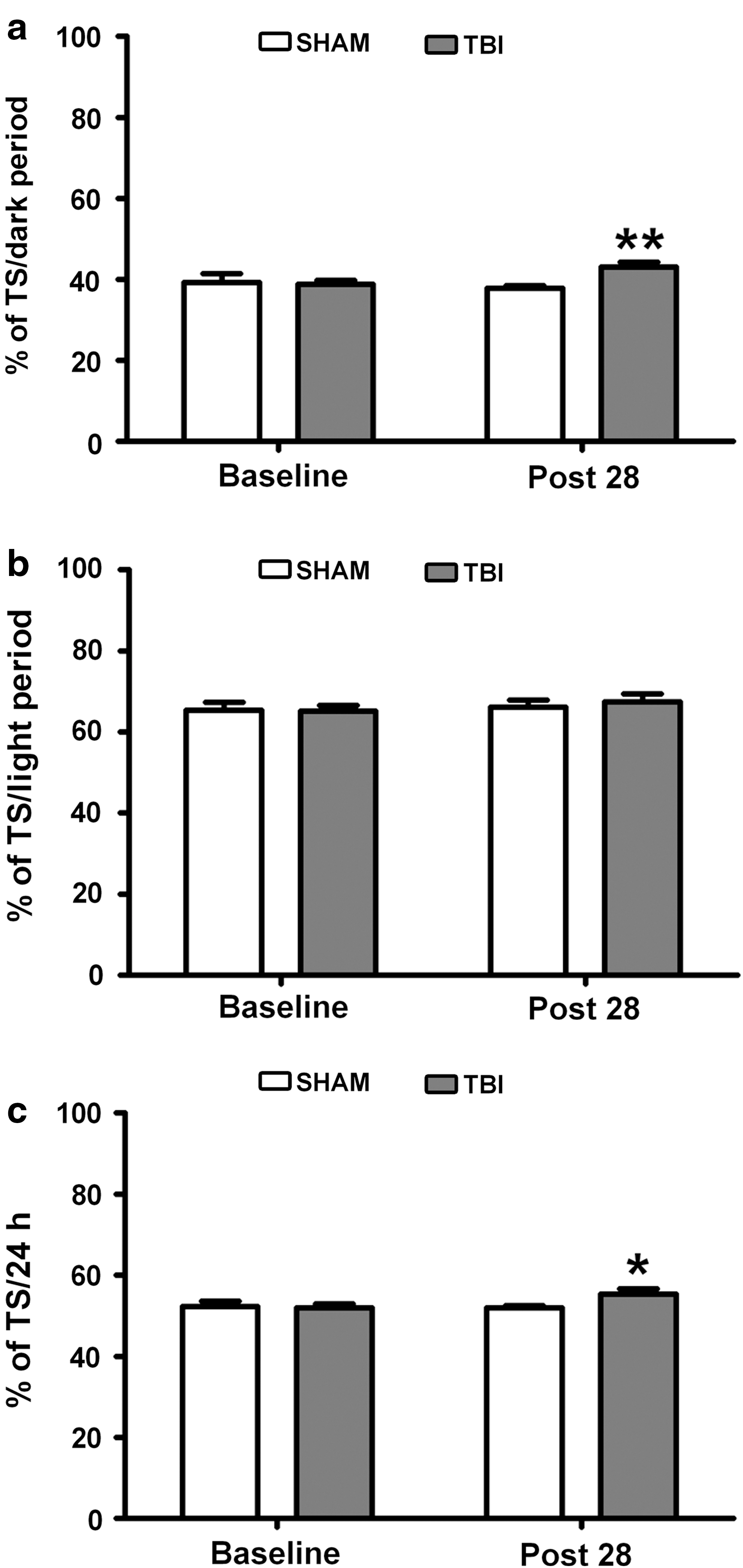
Sleep need 28 days after traumatic brain injury (TBI). Total sleep (TS, NREM+REM) need in the dark (
Sleep is more stable after TBI in rats
Sleep fragmentation index for NREM sleep 28 days after trauma in TBI animals was reduced when compared with the dark phase BL value (Fig. 4a; *p < 0.05, BL [SHAM n = 7, TBI n = 6] vs. P28 [SHAM n = 6, TBI n = 7], two-tailed paired t test), while we observed unchanged fragmentation indices for WAKE and REM and all states in SHAM animals. The number of WAKE, NREM sleep, and REM sleep bouts were invariable over time both in SHAM and TBI groups compared with the respective BLs (Fig. 4b). Finally, we evaluated delta EEG power, a measure of deep NREM sleep, per 24 h at all four time points in the TBI and SHAM groups. We observed a ∼25% transient increase of NREM relative delta power in the TBI group compared with SHAM controls at seven days after trauma, but no permanent long-term effect (Fig. 4c; group difference p = 0.008, linear mixed model ANCOVA, significant effect for P7 isolated *p < 0.05).
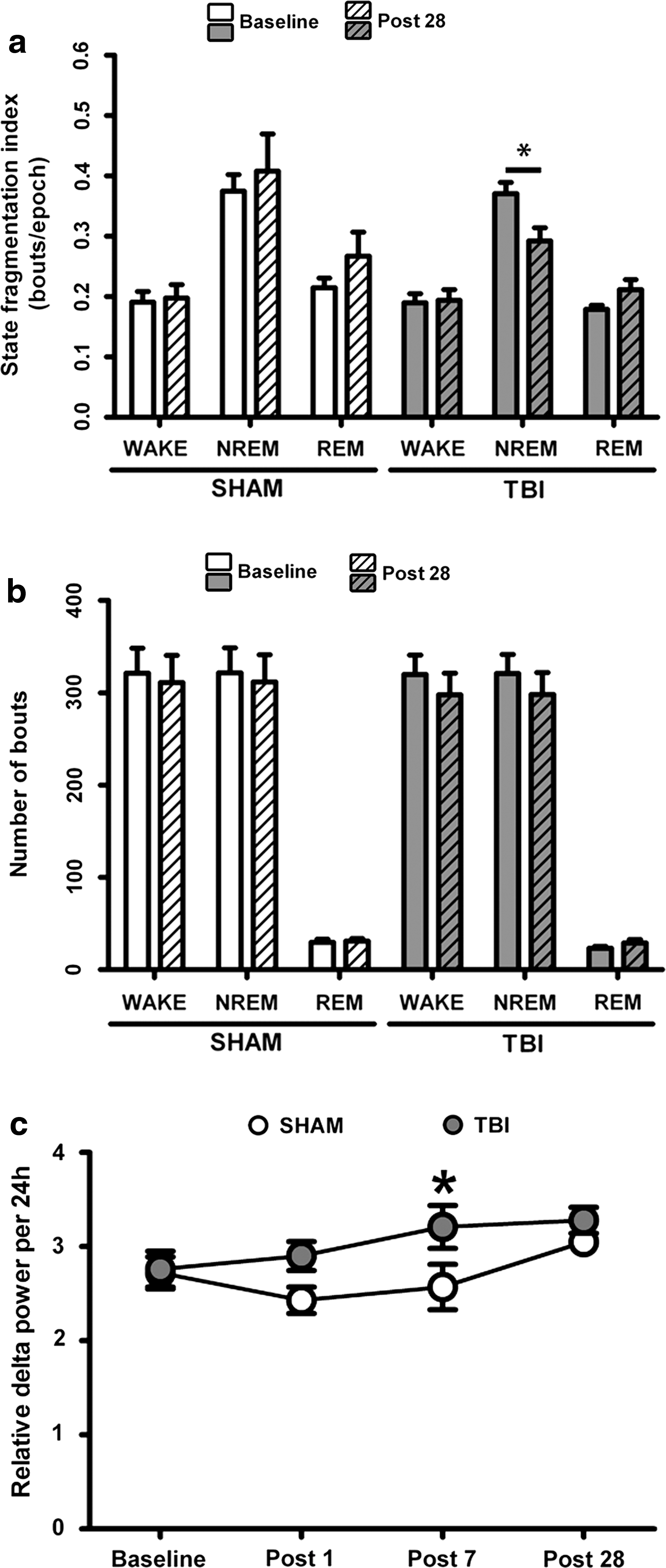
Behavioral state stability and relative delta power after traumatic brain injury (TBI). (
Loss of neuronal immunoreactivity in the histaminergic TMN after closed diffuse TBI
We performed immunohistochemistry studies in SHAM and TBI brains (Fig. 5a). We found that the number of MCH, Hcrt-1, and noradrenergic TH neurons was unchanged after TBI. We observed, however, a significant ∼36% decrease in the number of HA immunoreactive neurons in TBI brains (Fig. 5a; SHAM [n = 6] vs. TBI [n = 7], ***p < 0.001, two-tailed unpaired t test). Pearson correlation analyses including all SHAM and TBI animals (n = 13) evidenced both positive and negative associations between the number of immunopositive HA neurons in the TMN and the amounts of WAKE and NREM sleep, respectively, in the dark period 28 days after TBI (Fig. 5b, correlation HA cell number and WAKE time in the dark period: p = 0.008, Pearson r = 0.6931; correlation HA cell number and NREM time in the dark period: p = 0.01, Pearson r = −0.6384; Pearson correlation analysis). No significant correlations were found in SHAM (n = 6) and TBI (n = 7) groups individually (data not shown).
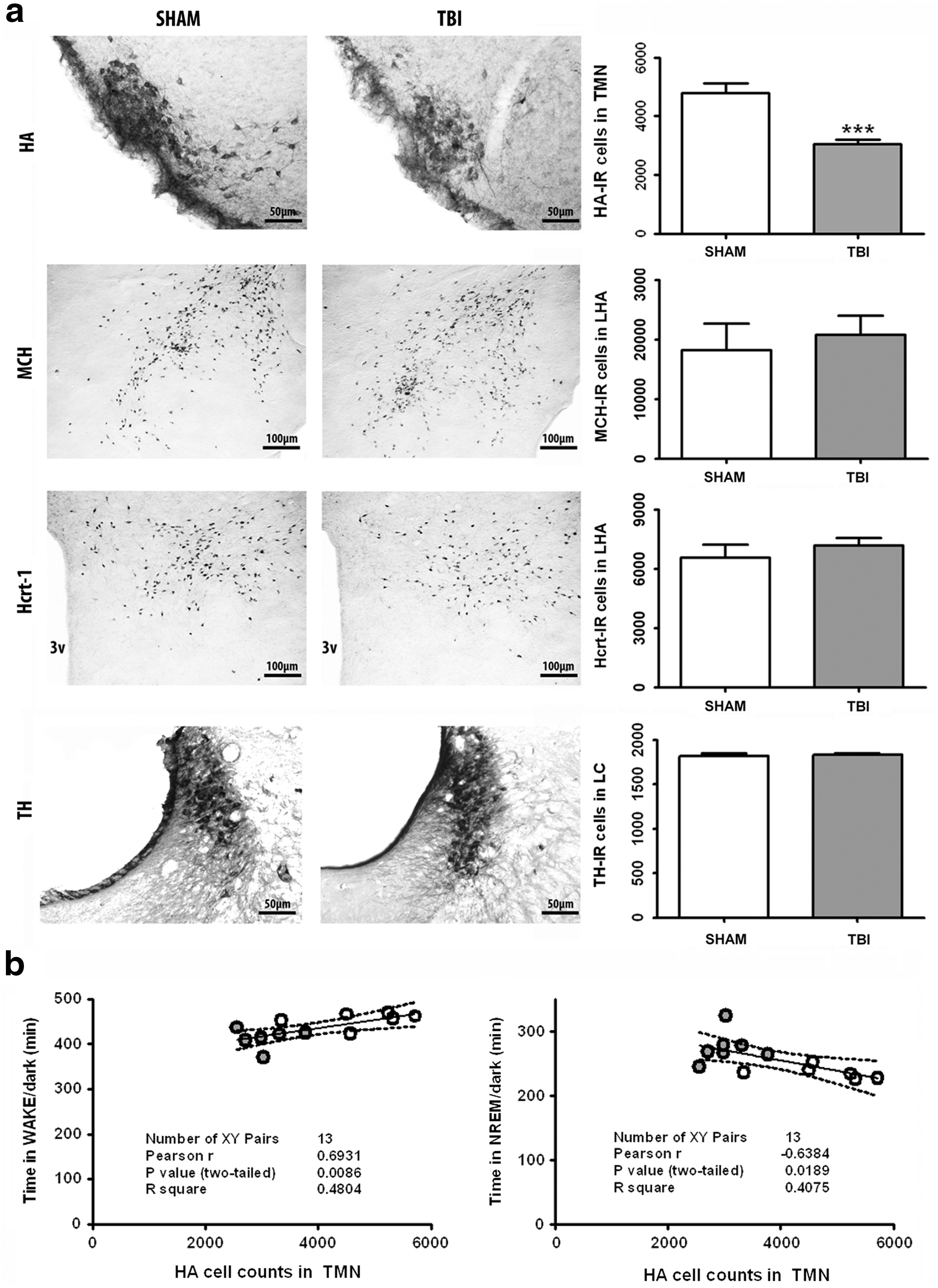
Immunohistochemical assessment of sleep-wake modulating neurotransmitter nuclei in hypothalamus and brainstem. (
To evaluate the potential mechanisms underlying decreased immunoreactivity of HA neurons, we performed colocalization studies of histidine decarboxylase (HDC) with markers of neuroinflammation (Ox42), astrogliosis (GFAP), and diffuse axonal injury (APP-1) in sections containing the TMN (Fig. 6a). There, we found increased neuroinflammation around the TMN after TBI compared with SHAM controls (Fig. 6b). Conversely, no salient differences were noted as far as astrocytic scarring and diffuse axonal injury are concerned (Fig. 6c,d). Regarding the latter, there was abundant somatic APP-1 immunoreactivity in colocalization with HDC, but only marginal colocalizing axonal staining, both in SHAM and TBI animals (Fig. 6d). This indicates that there is only little axonal accumulation of APP-1 in TMNs, irrespective of the presence of TBI.
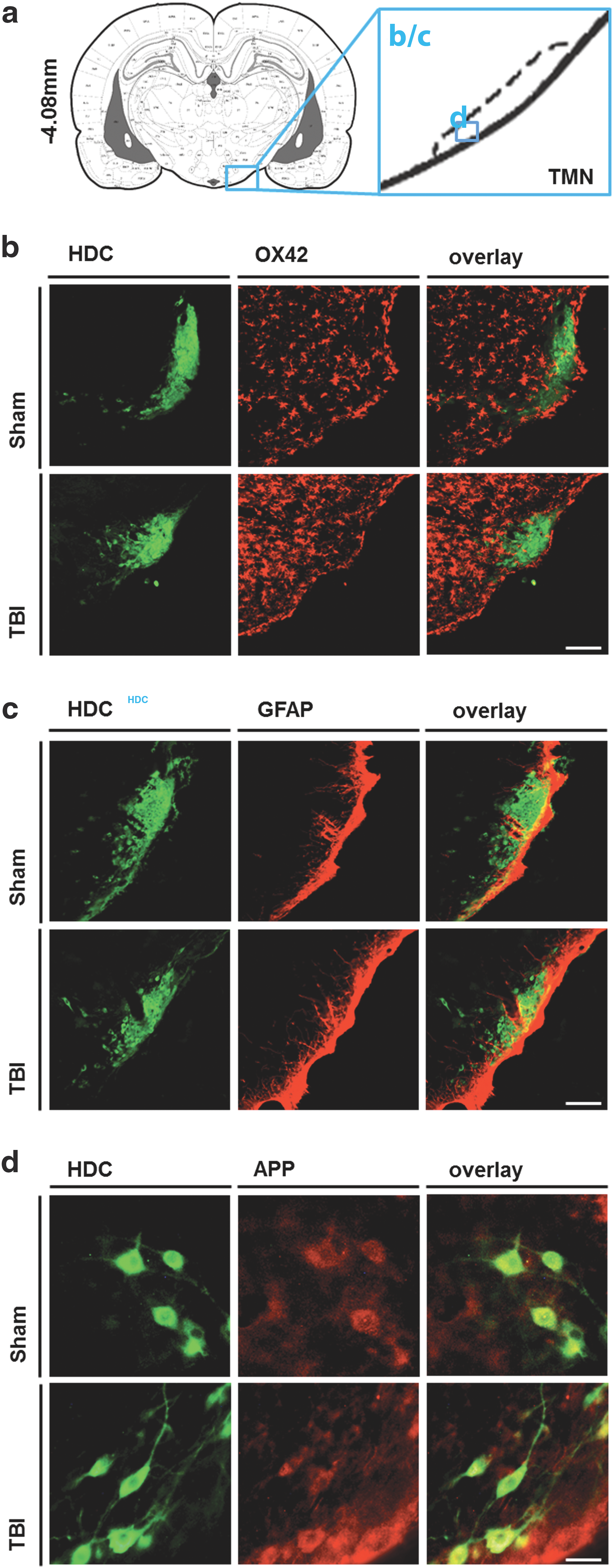
Immunohistochemical assessment of potential mechanisms underlying reduced histaminergic immunoreactivity. (
Discussion
In the present study, we provide evidence of increased sleep need one month after closed diffuse TBI in rats in the dark phase. These findings are strikingly similar to those observed in human patients with TBI, 4,5 but also to a previous rodent TBI study applying the lateral fluid percussion model. 22 In addition, we observed a ∼36% reduction of HA immunoreactive neurons in the TMN, and pooled data revealed an inverse correlation between the number of HA neurons and the amount of NREM sleep in the active dark period, suggesting that this loss might contribute to increased sleep need. Recent results in human autopsy material have suggested that TBI—which is very heterogeneous in human patients—causes a widespread loss of hypothalamic cells, particularly a >40% reduction of histaminergic neurons in the TMN of the posterior hypothalamus, and a less distinct damage to other hypothalamic and brainstem populations. 9,10
In an attempt to elucidate whether the observed increased NREM sleep reflects a higher consolidation of behavioral states, or whether it is associated with many short episodes of NREM sleep that would indicate acquired sleep instability, we determined the number of sleep bouts in the dark phase of TBI rats 28 days after trauma, and it was similar to the one assessed at BL. The average length of NREM bouts was increased, however, yielding a reduced fragmentation index. Thus, these results suggest that TBI induces a sleep-wake pattern of more consolidated sleep rather than a fragmented one, once more in agreement with our previous observations in patients 4 but in contrast with previous rodent reports. 22
The coincidence of present results with the human literature supports the notion that our TBI model reproduces human TBI and post-traumatic SWD in a very accurate manner. Interestingly, long sleep bouts are more likely to reflect excessive sleep need (i.e., pleiosomnia) than excessive daytime sleepiness (EDS), which is characterized by frequently intruding short sleep bouts, reflecting increased sleep pressure during wakefulness. 23,26 Again in agreement with human findings, 4 we only observed a rather mild and transient increase of delta power seven days after TBI, indicating that post-traumatic pleiosomnia is characterized by increased, consolidated sleep, but not by enhanced slow-wave activity, which would indicate chronic deeper sleep. The TMN neurons are proposed to have a wakefulness-maintenance function. 27 Thus, their loss or impairment could potentially cause or contribute to pleiosomnia, in line with the results of the present study.
We observed increased sleep need in TBI rats compared with SHAM animals only 28 days after trauma and not at earlier time points. This might be accounted for by two main reasons. First, unspecific stress reactions and higher anxiety levels could predominantly impact sleep-wake behavior within one and seven days after trauma. In this line, a marked and transient increase of NREM sleep with decreased WAKE time was observed in both SHAM and TBI animals one day after the surgical procedure. This might reflect an unspecific “surgery effect” related to surgical-anesthesia and/or post-surgical analgesia, as well as the general need of recovery acutely after the interventions. At seven days after operation, animals appear to have recovered normal sleep-wake behavior, as evidenced by vigilance time proportions similar to baseline levels, but anxiety levels might still be higher than normal. We have not performed corresponding anxiety measuring tests in our study, which must be noted as a limitation and should be performed in future approaches.
Second, the late occurrence of increase sleep need could be linked to a slow development of persistent and long-term trauma-induced damage to sleep-wake regulating neuronal populations. Long-term secondary injury after trauma has been studied in depth, and at present, post-traumatic injury is seen rather as a disease process than as a single event. 28 –30 For instance, it is permanent and caused by non-reversible pathological alterations, which fits with the current definition of the World Health Organization for chronic disease. 30
The distinct vulnerability of histaminergic neurons compared with other hypothalamic populations might be explained by their exposed anatomical position neighboring the base of the skull, both in humans and rodents. Thus, acceleration/deceleration mechanisms of the brain within the skull might cause direct superficial injuries in addition to tension and compression mechanisms in this area, ultimately leading to damage of the affected neurons. At the time point of study termination, we observed neither APP-1 accumulation in axons in the TMN nor enhanced gliosis around the TMN. This may indicate that the processes leading to diminished HA cell counts are others than APP-mediated degeneration or excessive scarring. On the other hand, we cannot exclude the possibility that the latter might play a role at earlier post-traumatic time points, and studies with longitudinal histological evaluations would be necessary for clarification.
The observation that sleep-wake disturbances only appear with significant delay after trauma, however, rather suggests more enduring mechanisms of action. As a possible explanation, we found enhanced neuroinflammation around the TMN after TBI. As the primary immune cells in the brain, microglia are known to release numerous factors, including pro-inflammatory and anti-inflammatory cytokines, chemokines, nitric oxides, prostaglandins, growth factors, and superoxide species, which could modulate secondary injury, also over a certain time span. 31 Yet, it remains unclear whether the findings of reduced histaminergic cell counts and increased neuroinflammation are only coincidental or also causally related. Thus, further studies including a more detailed characterization of the inflammatory response and anti-inflammatory treatment are warranted to answer this question.
Overall, consecutive follow-up studies are due to determine the exact nature and progression of the found histopathological changes after rat TBI and, moreover, determine their causal relationship with the observed altered sleep-wake phenotype after trauma. Further, the more restricted impairment observed in rodent tissue compared with human brains might well respond to a milder severity of trauma achieved in our animal model of closed TBI, which possibly does not reflect as high and uncontrolled trauma severity as observed in fatal human TBI cases. 9,10 For instance, our TBI model is a closed skull model with 100% survival rate and no long-term motor impairments, 25 which may well represent TBI in survivors with post-traumatic sleep-wake disturbances, but probably does not represent fatal human TBI cases. Further, our trauma induction allows a sensible amount of head movement in the vertical axis but, for the sake of reproducibility, there are almost no lateral movements during trauma induction because of a partial head fixation system, which may also account for a milder trauma outcome by reducing the number of angles in which the subjects' head can move toward and thus damage the brain.
Altogether, our study reports long-term post-traumatic sleep-wake disturbances with consolidated, excessive sleep need in association with reduced histaminergic immunoreactivity, possibly related to trauma-induced neuroinflammation. Because of its resemblance with the human condition, this rodent model could allow further studies and lead to novel and tailored therapies for post-traumatic pleiosomnia—a symptom that, so far, cannot be treated.
Footnotes
Acknowledgements
The authors would like to thank Tom Scammell for his constructive criticisms and Ertugrul Cam, Aleksandra Hodor, Amandine Valomon and Evangelia Symeonidou for their helpful assistance.
This project was supported by the Swiss National Science Foundation (SNSF, grant no. 125504) and by the Clinical Research Priority Program “Sleep and Health” of the University of Zurich.
Author Disclosure Statement
No competing financial interests exist.