
Abstract
Traumatic brain injury (TBI) and spinal cord injury (SCI) are critical medical conditions and a public health problem for which limited therapeutic options are available. The complement cascade is activated after TBI and SCI, and the resulting effects have been investigated in gene-knockout and pharmacological models. Multiple experimental studies support a net detrimental role of C3 and C5 activation in the early stages of TBI and SCI. Less firm experimental evidence suggests that, downstream of C3/C5, effector mechanisms, including the generation of membrane-activated complex and direct damage to membranes and neutrophils infiltration, may bring about the direct damage of central nervous system tissue and enhancement of neuroinflammation. The role of upstream classical, alternative, or extrinsic complement activation cascades remains unclear. Although several issues remain to be investigated, current evidence supports the investigation of a number of complement-targeting agents targeting C3 or C5, such as eculizumab, for repurposing in TBI and SCI treatment.
Introduction
T
Additionally, secondary brain damage post-TBI represents a multi-faceted danger that adversely affects the disease course, resulting in persistent neurological impairments. 8 –10 More precisely, neuroinflammation appears to be a major driver for the secondary pathology. Several studies provide evidence that complement activation in the central nervous system (CNS) is involved in the immune-mediated secondary neuropathology and may play a role in blood–brain barrier (BBB) dysfunction post-TBI. 11 –13 Therefore, specific inhibition of the highly interactive complement cascade seems to be a promising and clinically relevant therapy to prevent and/or minimize secondary damage in TBI patients. 14,15 However, given that the complement system plays an important role on the BBB immune defense, targeting the local activation site of the complement may be necessary to avoid a general systemic inhibition of complement and thus an overall infection risk. 16,17 The aim of this review is to highlight the current findings on complement activation in TBI and the potential of targeting the complement system as a future therapeutic option.
The Complement System
The complement system is part of the innate immune system and plays a central role in maintenance of tissue homeostasis and in the host's defense against pathogens. 18,19 It is composed of approximately 30 proteins, most of them synthesized in the liver, circulating as inactive precursors, so-called zymogens, or expressed—including their corresponding receptors—as membrane-bound proteins on the cell surface. 20,21 As active proteins, these components possess the ability to initiate, amplify, and regulate complement activation. 18,22
Generally, three canonical pathways for complement activation have been identified, known as classical, lectin, and alternative pathways, together with an extrinsic pathway that provides the cross-talk between coagulation and complement cascades (Fig. 1). Each pathway generates a C3 convertase, a serine protease able to cleave the central complement protein, C3. The cleavage products of C3 have a crucial impact in almost all the biological consequences of complement. 23
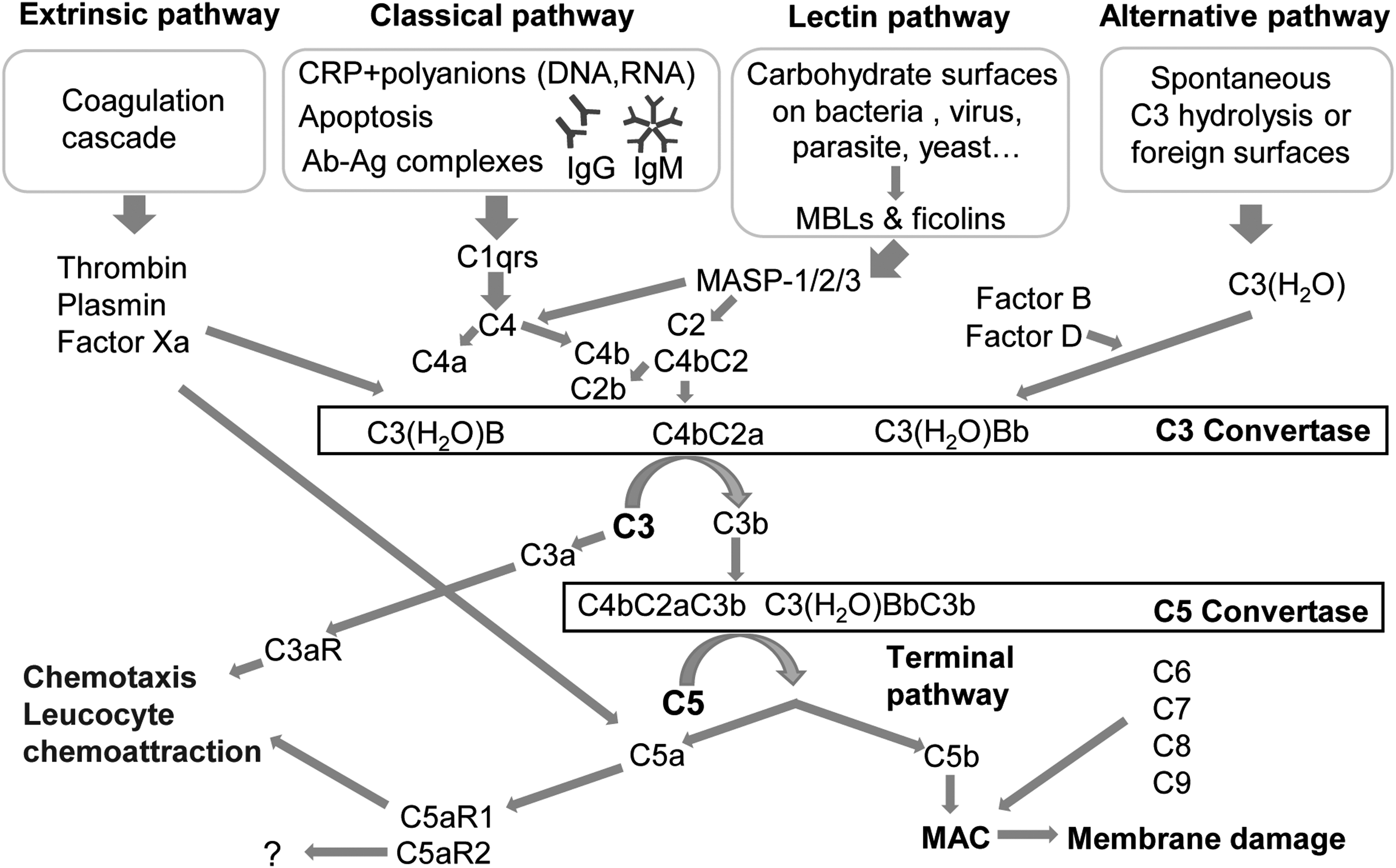
An overview of known pathways of complement activation. Ag-Ab complexes, antigen-antibody complexes; CRP, C-reactive protein; Ig, immunoglobulin; MAC, membrane attack complex; MASP, MBL-associated serine protease; MBL, mannose-binding lectins.
The classical pathway of complement activation involves the recognition molecule, C1q, which circulates in the serum. C1q can be activated after binding to pathogens, immunoglobulin (Ig) G and IgM antigen-antibody (Ag-Ab) complexes or C-reactive protein (CRP) bound to polyanions (bacterial polysaccharides, DNA, RNA). 24,25 Subsequently, C1q is able to activate C1r, which, in turn, cleaves C1s. C1s is then able to cleave C4 into C4a and C4b, which subsequently leads to the cleavage of C2 into C2a and C2b. C4b remains associated with C2a, forming the C3 convertase (C4bC2a). 26
The lectin pathway is based on protein-carbohydrate interactions by using mannose-binding lectins (MBLs) and ficolins to identify patterns of carbohydrate ligands that are found on the surface of a wide variety of microorganisms including bacteria, yeast, parasites, and viruses. 25,27 MBL structurally resembles C1q, and ficolins are homologous to MBL. After binding of MBLs or ficolins to carbohydrates on the pathogenic surface, MBL-associated serine proteases (MASP-1, MASP-2, and MASP-3) are activated, which, afterward, cleave C4 and C2 and generate C4bC2a in an analogous reaction compared to the classic pathway. 28 –30
The alternative pathway consists of the main components C3, factor B (fB), factor D, and properdin. This pathway is constitutively active at low levels in the normal host. 31 It is initiated by spontaneous hydrolysis of C3 to C3(H2O), which is able to bind with factor B. Association of factor B with C3(H2O) leads to conformational changes of factor B, which can then be cleaved by the serum protease factor D, generating Ba and Bb. Subsequently, the Bb fragment remains in a complex with C3(H2O), forming the C3 convertase, C3(H2O)Bb, of the alternative pathway. Properdin stabilizes C3(H2O)Bb by extending the half-life up to 10-fold, acting as a positive regulatory protein of complement activation. 32 –34
As already outlined, all three activation pathways generate a C3 convertase, which is able to split C3 into C3b and a smaller anaphylatoxin, C3a. C3b connects to the C3 convertase and forms the C5 convertase. Further, the C3b molecule is also able to act as an opsonin, which covalently binds to target surfaces. Thereby, C3b facilitates target recognition and their clearance through phagocytosis. 35 The C5 convertase initiates the last phase of the complement cascade, the formation of the lytic membrane attack complex (MAC). The C5 convertase splits the parental C5 molecule into C5b and the smaller anaphylatoxin, C5a. Association of C5b with C6, C7, and C8 leads to the subsequent binding of 10–15 C9 molecules, which combine to form, all together, the transmembrane pore, MAC. Consequently, the MAC leads to a disturbed ion flow and an imbalance of the concentration gradient, resulting in altered cellular signaling and eventually in lysis of the target or abnormal host cell. 36,37 The anaphylatoxins, C3a and C5a, enhance the inflammatory response of complement by interacting with their distinct receptors. 21 C3a interacts with the G-protein-coupled receptor, C3aR, and consequently is a mediator of proinflammatory events. For the anaphylatoxin, C5a, two receptors have been identified, C5aR1 and C5aR2. Whereas interaction with C5aR1 mediates proinflammatory effects, including the release of inflammatory cytokines, chemokines, or leukocyte activation, the role of C5aR2 still remains unclear. 38 –40 Besides these established pathways, other extrinsic pathways (Fig. 1) have been proposed, including active proteases of the coagulation system—among them, the serine protease, thrombin, which can directly cleave C3 and C5 molecules, independent of the C3 or C5 convertase. 41,42
Taken together, the complement cascade represents a complex and multi-level process, which is mediated through the coordinated action of diverse proteins (Fig. 1). Therefore, a tight regulation is indispensable for proper modulation of immune response intensity and adjustment of the biological effects, for example, by Complement receptor 1 (CD35), membrane cofactor protein (CD46), decay-accelerating factor (CD55), and the MAC-inhibitor, CD59. All of these biological effects lead to a restrained and sufficient inflammatory response, which is beneficial for the host but harmful for the invading pathogens. However, under certain circumstances, activation of complement is unfavorable and associated with harm to the host tissue, especially in situations where the host is already affected by a primary pathological event such as trauma. 43 The following sections on purpose omit the various roles of complement in neurological diseases or stroke, but rather focus on complement activation post-TBI.
Local complement activation after traumatic brain injury
Both human data and experimental investigations have established that the complement cascade is activated in cerebral parenchyma post-TBI (Fig. 2). In fact, in cerebrospinal fluid (CSF) samples obtained from TBI patients with different degree of severity and parenchyma damage, elevated levels of C3 and fB have been found, with large variability both in time and across patients; for a subset of patients, intrathecal synthesis of C3 and fB was detected. 44 C3-mRNA has been detected in the penumbra of cortical contusions in the human brain, confirming the instance that local synthesis of C3 may contribute to the blood-derived pool. Deposition of MBL has been detected in pathological brain samples obtained during surgical treatment of traumatic lesions. 45 Finally, CSF levels of the MAC were found to be elevated, with patients showing a variable time course. Interestingly, CSF MAC levels were significantly correlated with the extent of BBB disruption. 46 Together with the reproducible detection of complement factors and regulators in experimental models of TBI (see below for extensive references), these data establish the notion of complement activation in TBI. However, the role and the net effect (beneficial or detrimental) of each complement factor and regulatory protein are far less established.
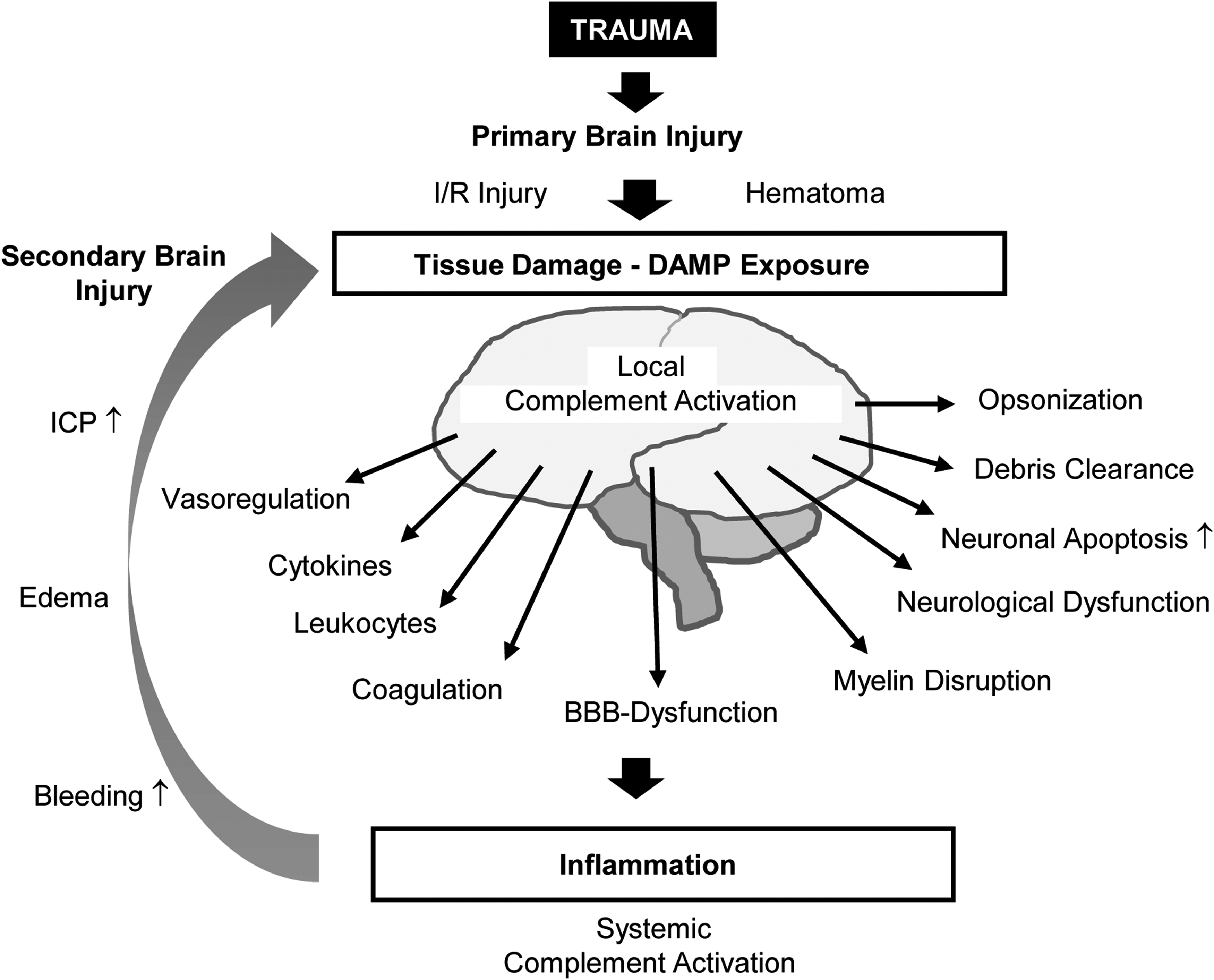
Involvement of complement in traumatic brain injury (TBI). Local complement activation post-TBI contributes to progression of damage to neurons and non-neuronal structures and, in combination with systemic complement activation, leads to secondary injury. Experimental evidences demonstrate how early local modulation of complement factors and regulators has to be considered as a viable therapeutic target for new therapeutic strategies. BBB, blood–brain barrier; DAMP, damage-associated molecular pattern; ICP, intracranial pressure; I/R, ischemia-reperfusion.
C3: A central hub of inflammation network after traumatic brain injury
Central to elucidating the overall role of complement in TBI, effort has been made to clarify whether the experimental manipulation of the terminal common pathway of complement affects behavioral and structural readouts of TBI (summarized in Table 1). Given that complement activation hinges upon the cleavage of C3 and C5, leading to the generation of several potent biologically active fragments and direct effectors, the effect genetic or pharmacological blockade of C3 and C5 have been investigated in brain and spinal cord injury (SCI). In a cryo-injury model of brain trauma, C3−/− mice displayed a 50% smaller lesion size at 4 and 7 days post-lesion, whereas the acute lesion size was comparable with wild-type (wt) animals. C3−/− mice showed a reduced inflammatory response, with 2-to- 4-fold decrease in the levels of chemokines (chemokine ligand [CCL] 2, CCL5, and CCL11), but unchanged expression of interleukin (IL)-6 and IL-12, and very limited (around 90% decrease) neutrophils extravasation. The effects of C3 deficiency are, at least in part, local given that local administration of C3 increased significantly (to 50% of control-lesioned mice) the neutrophil recruitment to the injury site. 47 Likewise, C3−/− mice have been reported to have a better neurological score already 2 days post-SCI (scoring 6 on the Basso, Beattie, and Bresnahan rating scale, whereas controls rated around 0.5 on average) and resulting in a much smaller residual consolidated neurological deficit (with a Basso, Beattie, and Bresnahan score of 19 of 21 in C3−/− mice at 21 days whereas control mice scored 11 on average) and display minor change in reduced gliosis (20% decrease at 1-h time point), but a 3 -to- 4-fold decrease in neutrophil infiltration, resulting in enhanced regeneration of axons. 48 These findings have been confirmed by another study using C3−/− mice to investigate recovery post-SCI, reporting improved acute (Basso, Beattie, and Bresnahan of 2 vs. 0 in wt at 3 days post-injury [dpi]) and long-term outcome (16 vs. 10 at 14 dpi). 49
BMS, Basso Motor Score; NSS, Neurological Severity Score; MWM, Morris Water Maze; BBB, Basso, Beattie, and Bresnahan motor score; wt, wild type; dpi, days post-injury TNF-α, tumor necrosis factor alpha; Bcl-2, B-cell lymphoma 2; NSE, neuron-specific enolase; SC, spinal cord; GAP-43, growth associated protein 43.
Nongenetic manipulation of C3 has largely confirmed the observations in the genetic models. Complement receptor type 1-related protein Y (Crry), an inhibitor of both the classical and alternative pathways of complement activation in mice (related to CD55 and CD46), has delivered beneficial effects both in brain and spinal cord traumatic injuries. In fact, transgenic mice with astrocyte-restricted overexpression of Crry displayed a less-pronounced overall neurological impairment after blunt TBI (between 1 and 1.5 points in the Neurological Severity Score [NSS] already at 1 h post-injury) and a reduced BBB disruption at the injury site (with Evans' blue extravasation reduced to half the amount of the wt controls). 50 Administration of Crry either as IgG-fused to Crry or as a fusion protein with Complement receptor 2 (CR2; [CR2-Crry]; in which the CR2 component targets the Crry to C3d-deposition sites) delivered beneficial effects in TBI: Mice administered with Ig-Crry displayed a significantly better motor performance after trauma (50% better performance at 4 h in the beam-walk test and normal performance at 24 h when control mice are still significantly impaired), although only a slight improvement in overall neurological score (at 24 h, 1-score difference between treated and control mice). In addition, histological analysis of hippocampal neurons revealed hints of preservation (no quantification is provided by the researchers) in Ig-Crry–treated mice compared to vehicle-treated ones. The impact of Ig-Crry administration on the expression of complement-related inflammation genes was limited (changes in C1q, CD55, and CD59 were significant, but with large deviations). 51 Similarly, wt mice intravenously injected with CR2-Crry 1 h post-SCI demonstrated improved functional (a Basso, Beattie, and Bresnahan score of 3 at 4 days, compared to 6 in C3−/− mice and 0.5 in control mice) and histological outcome (including a 3-fold decrease in neutrophils infiltration) compared to the control and similar to that obtained in C3−/− mice post-SCI. 48 A different strategy for C3 inhibition has been based on CR2-fH (mTT30), a chimeric CNS-restricted site-targeted pharmaceutical compound that link CR2 to complement inhibitor factor H in order to investigate alternative pathway inhibition by targeting C3 in the closed-head injury model. Results demonstrated attenuated microglial response, C3 deposition, and cellular death, suggestive of a targeted inhibition of the alternative pathway, resulting in neuroprotection (4-fold decrease in apoptotic neurons are reported). 52 In agreement with these data, sequestering C3 cleavage products with a soluble form of Complement receptor 1 (CR1) results in reduced neutrophils infiltration (measured as myeloperoxidase levels, around 40% decrease at 12 h in sCR1-treated mice) and in animproved outcome (Basso, Beattie, and Bresnahan score of 15 vs. 10 in control mice at 14 dpi) post-SCI. 53
Downstream of C3, further exploration of complement in TBI and SCI has focused on the role of C5 and its cleavage products. Deletion of C5 complement factor resulted in a cryo-injury-induced brain damage intermediate in size between C3−/− and wt (although the original publication does not provide precise percentages), implying that a large fraction, albeit not all, of C3 effects are mediated by the C5-dependent cascade; likewise, neutrophil recruitment was significantly reduced in C5−/− mice (60% decreased), but not to the extent observed in C3−/− mice (90% decreased). 47 In fact, C5a generation post-SCI has been found to be reduced, but not absent, in C3−/− mice subject to SCI (based on histochemical detection, the decrease appears to be around 50%), suggesting that C5a generation may be largely, but not only linked to the C3 step of the complement cascade. 54,55 The above-mentioned cross-talking between complement and coagulation cascade has been shown to generate C5a independently of C3 by various activated coagulation factors, such as thrombin, which certainly are present in both TBI and SCI. 42 Interestingly, nongenetic inhibition of C5 generation using the C5-binding protein, OmCI (which prevents both C5a and MAC generation), is sufficient to improve the trauma-associated weight loss if administered before or within 15 min after trauma (limiting weight loss to less than 10% vs. 20% in controls), whereas administration 30 min after trauma is completely ineffective (obtaining a weight loss in excess of 20%); the effect on neurological impairment was equal for 0, 15′, and 30′ OmCI-administered mice at 24 h, all groups showing a significantly lower NSS than in phosphate-buffered saline (PBS)-treated mice (between 2 and 3.5, with control mice at above 4), but at 72 h, 30′-delayed treated mice showed an NSS indistinguishable from PBS-treated control mice. 15 In agreement with these observations, systemic administration of C5a at the time of SCI significantly worsens the acute neurological deficit and delays recovery, with mice remaining at Basso, Beattie, and Bresnahan score 3 or lower until 28 dpi, when control mice have already recovered to a Basso, Beattie, and Bresnahan of 9. 55
Interestingly, delayed treatment with C5a (starting 24 h post-SCI) actually improves the recovery of neurological deficits, with delayed-treated mice reaching Basso, Beattie, and Bresnahan scores above 12. 55 Taken together, the data on C3 and C5 deletion or inhibition are consistent with a detrimental net effect of the activation of the complement cascade in the acute phase of TBI and SCI. The effectors of the C3/C5 stage of the complement cascade are either the factors C5b-9 for MAC formation or the release of the potent anaphylatoxins, C3a and C5a, which interact with a number of distinct receptors and modulate vascular tonus, leukocyte chemotaxis, glial activation, and cytokine release.
Important role of membrane attack complex in traumatic brain injury
There is evidence that direct MAC-mediated damage to neural structures contributes to the pathogenesis of TBI and SCI in the acute phase (summarized in Table 1). In closed blunt TBI, MAC has been immunolocalized in white and gray matter within 72 h post-injury, matching the time course of microglial activation. Systemic decrease in complement C6 protein resulted in the almost complete abrogation of MAC deposition after TBI and SCI, respectively. 15,56 Notably, the reduced accumulation of MAC was associated with a 3-fold decrease in microglial density and a lower number of apoptotic neurons and axonal damage. Recovery of neurological damage was significantly improved in mice with reduced systemic levels of C6: Their neurological severity score was lower than in control mice already soon after trauma (C6-depleted mice had an NSS score of around 1 already at 3 and 24 h, when control mice exhibited scores of 6 and 4, respectively), and the overall neurological recovery was more complete. 15 Together with the observed small temporal windows for effective pharmacological inhibition of C3, 55 the large effect is already detectable at 1 h after trauma, suggesting that MAC-mediated damage may take place in the first fraction of the hour after trauma and may contribute significantly to the acute neurological impairment after TBI.
Additional evidence of the involvement of direct effector mechanisms in complement-mediated damage has been acquired manipulating endogenous inhibitors of MAC. CD59a−/− mice displayed a significantly worse neurological recovery, which became evident only 7 days (with a persistent NSS score of 3.5, on average, against 1.5 in wt mice) after blunt TBI, whereas the earlier part of the neurological recovery was comparable to wt mice (at 24 h, 4 and 3.5 in CD59a−/− and wt mice, respectively). 57 Interestingly, CD59a−/− mice displayed a much larger elevation of neuronal damage serum biomarkers (serum neuron-specific enolase, a marker of neuronal damage, was 3 times higher in CD59a−/− mice than in wt ones) 57 and increased number of apoptotic neurons at the injury site. 57 Likewise, in comparison to wt mice, CD59a−/− mice exhibited aggravated neurological damage and poor recovery after SCI (at 7 days after SCI, CD59a−/− score 2 in the Basso Mouse Scale [BMS], whereas wt mice score 4 and sham mice 9); in these mice, modest additional loss of myelin has been observed after SCI (when qualitatively quantified) associated with larger and more persistent inflammatory infiltrates (histological inflammation score of 7 vs. 5 in wt mice at 72 h post-SCI). 58 On the other hand, an interesting strategy to enhance CD59-dependent inhibition of MAC has been developed using a chimeric protein based on the Complement receptor of the immunoglobulin (Ig) superfamily (CRIg) as a homing domain to deliver the CD59a domain (linked to the CRIg by an IgG hinge domain). 17 After closed TBI, the CRIg domain was effectively recruited into C3b-positive areas within the injury site; whereas in control animals C3b-positive sites (opsonized surfaces) corresponded to the localization of MAC deposition, delivery of CD59a by the CRIg successfully prevented local MAC formation. 17 This resulted in a significant decrease in acute neurological damage after TBI (at 3 h post-injury, NSS score was reported to be, on average, 3.75 in CRIg-treated mice and 7.5 in control mice) and improved overall recovery (body weight loss was above 10% for control mice, but around 5% for CRIg-treated mice), with a decreased neuroinflammatory response (microglial activation was reduced to one third of vehicle-treated mice in cortex and hippocampus) and reduced amyloid precursor protein levels in neurons. 17 The marked effect on microglial activation suggests that terminal complement cascade components are critical mediators in triggering and amplifying neuronal damage after TBI. Nevertheless, it has been proposed that the activation of the terminal complement cascade, leading to MAC formation, may be restricted to TBI associated with a significant parenchymal damage such as neuronal and vascular lesions after penetrating TBI. 59 In contrast, in axonal-injury–restricted TBI such as the one obtained in rotational injury, MAC formation was not observed, despite comparable upregulation of C3 in both penetrating and nonpenetrating TBI. 59
Taken together, these data suggest that a significant part of the detrimental effects of complement activation in TBI or SCI is mediated by a direct attack to CNS structures by MAC, although a significant heterogeneity may exist in terms of the relative contribution of different complement effector pathways in TBI.
Role of complement receptors in traumatic brain injury
Complement receptors provide the interface through which the activation of the complement cascade, a cornerstone of innate immunity, amplifies and modulates cellular responses. In fact, binding of complement fragments to CR1/2 activates neutrophil migration, increases T-cell proliferation and survival, and the complement receptors are also involved in microglia activation. 60 –62 Therefore, complement receptors have been investigated for their role in TBI and SCI using genetic and pharmacological approaches (summarized in Table 1).
The role of complement C3 receptors in TBI has been investigated in CR2−/− mice. Because in mice (but not in rats or humans) the same gene gives rise to CR1 and CR2 by alternative splicing, these mice reflect a pan-complement-receptor-knockout model. CR2−/− mice subject to closed weight-drop TBI displayed a decrease in acute mortality down to 10% from 18% in wt littermates and a significantly better neurological score already 1 h after injury (on average, 3.75 in CR2−/− compared to 6 in wt littermates), which resulted in a faster overall recovery. 63 These findings were matched by the strong decrease in neuronal loss (a 6-fold decrease in terminal deoxynucleotidyl transferase dUTP nick end labeling–positive cells in cortex) and the overall decrease in C3 deposition in cerebral parenchyma in CR2−/− mice after trauma. Activation of microglia and astrocytes (3 and 2 times lower in CR2−/− mice, respectively), as well as the appearance of peripheral inflammatory markers such as IL-6, was significantly suppressed in CR2−/− mice. 63 Thus, CR2−/− data show that C3-caused detrimental effects after TBI are also mediated by receptor-dependent activation of cellular responses.
The roles and net effect of anaphylatoxin C5a-C5aR1 receptor interactions after TBI is less established. C5aR1 was found upregulated in neuronal and non-neuronal cells after TBI. 64 In fact, administration of a C5a antagonist small peptide sufficiently reduced neutrophil infiltration (in cortical cryo-injury) to levels observed in C5−/− mice and close to sham animals. 47 Interestingly, in SCI models, a biphasic effect of C5aR has been reported, although the absolute magnitude of C5aR contributions appears comparatively small. In fact, C5aR1−/− mice displayed a faster initial recovery after injury (median BMS score at 7 days after trauma is around 3 in wt animals and around 3.75 in C5aR1−/− mice, in a scale ranging from 0 to 9), followed by a lack of continuing progress, which resulted in a worse long-term performance (median BMS score at 35 days after injury was 4 in C5aR1−/− mice and 5 wt littermates). 65 The two phases could be dissociated using a pharmacological antagonist of the C5aR: The administration of the antagonist only in the first 7 days produced a sustained improvement in neurological recovery, without causing a delayed worsening (at 21 days after injury, mice treated for 7 days with the C5aR-antagonist had a median BMS score of approximately 5 as compared to 4 in vehicle-treated mice). The beneficial effect was associated with reduced inflammatory cytokine levels (although with variable effects, ranging from 80% decrease in IL-1β to 20% in IL-6 levels, with tumor necrosis factor alpha (TNF-α), monocyte chemoattractant protein-1 (MCP-1), and IL-10 barely or not affected) and a 50% decrease in macrophage recruitment. However, peripheral immune cells were not involved in the beneficial effects of the C5aR1 blockade, because chimeras with C5aR1−/− leukocytes were indistinguishable from nonchimeric animals. On this basis, it is conceivable that the effect of C5aR1 is attributed to the involvement of resident CNS cells and not attributed to circulating leukocytes. In fact, C5aR1 was shown to drive astrocyte (but not oligodendrocyte) proliferation through a pathway involving signal transducer and activator of transcription 3, with C5aR1−/− showing a 50% decrease in proliferating astrocytes. Thus, the net contribution of C5aR1−/− to SCI-related pathogenic cascades may be mainly linked to the regulation of gliosis, although the net effect on the motor performance may be comparatively small. 65
Likewise, the role of C5aR2 has been studied so far only in SCI models. Interestingly, C5aR2−/− mice displayed a worse recovery than wt upon SCI (BMS scores of 2.75 vs. 4 at 14 days and 3 vs. 5 at 35 days after injury in C5aR2−/− mice and wt littermates, respectively) and a significantly larger (around 75%) spinal lesion volume. The detrimental effect of the loss of C5aR2 was correlated with lower levels of TNF-α and IL-6 in the acute phase (reduced by 50%, whereas several other cytokines, including interferon-gamma and MCP-1 were unaffected). However, overall recruitment of inflammatory cells was unaffected by C5aR2 deletion, suggesting that the changes in the inflammatory response may be attributed to the impaired response of astrocytes or microglia. 66 Notably, the detrimental effect of C5aR2 loss could be compensated by the administration of a C5aR1 antagonist, suggesting that the balance between the two C5a receptors provides the set point regulating glial responses to trauma in the spinal cord. The existence of multiple roles, possibly temporally distinct, for C5a is further supported by the beneficial effects obtained with systemic administration of C5a starting 24 h after SCI, but detrimental effects observed for administration at the time of SCI. 55
Although the comparison of results from different reports may be fraught with biases, current literature seems to show a comparatively larger impact of C5aR2 than of C5aR1 on the overall neurological recovery after SCI.
Looking upstream after traumatic brain injury: Undefined role of the classical pathway
The observed massive activation of complement in TBI and SCI, and the overall detrimental effect attributed to the cascade downstream of C3 and C5, raises the question about the mechanisms leading to complement activation.
The role of classical pathway of complement activation in TBI was tested by assessing the effect of controlled cortical impact (CCI) in C4−/− and C1q−/− mice. Whereas C1q−/− mice were reported to be comparable to wt littermates in terms of neurological recovery and lesion volume, a statistically significant difference was reported in C4−/− mice. 67 Deposition of C4 in cortical microvessels was already detected 60 min post-impact. Interestingly, the deposition of C3b has been reported to be normal in this model, suggesting that C4 may be not critical for alternative C3 activation. Nevertheless, C4−/− mice performed significantly better in motor tests (wire-grip test score of approximately 0.75 in wt mice and 2.25 in C4−/− mice 1 day after trauma in a 0-to-5 scoring chart), but not in the spatial memory test (Morris Water Maze; MWM). Likewise, C4−/− mice showed a smaller overall lesion volume, although the absolute difference in volume was admittedly small (approximately 20%). 67 Although You and colleagues have reported no effect of C3−/− on the TBI outcome, suggesting therefore that the C4−/− effects are attributed to intrinsic biological activities of C4 fragments, other studies have suggested otherwise. 47 Thus, it is possible that the functional role of C4 may be partially bypassed by the nonclassical C3 activation, which would explain the small effect of C4−/− on behavioral and histological measures. It must be stressed that the redundancy of TBI models and readout measures makes the direct comparison of the studies complex and fraught with uncertainty.
In SCI, there is evidence for improved outcome in C1q−/− mice, associated with 40% reduced lesion volume and a 25% increase in macrophage infiltration at 3 dpi (but no change in acute neutrophils infiltration), although the absolute effect on the neurological recovery seems comparatively small and only appearing in early (at 2 days post-injury the BMS score was 3 in C1q−/− mice and 2 in wt littermates in a scale ranging 0–9), but not in late measurements of the motor performance (no difference in BMS scores from 7 days after injury onward). 68 Nevertheless, proregenerative roles of C1q in SCI have been reported, in particular with C1q interfering with myelin-associated inhibition of axonal growth. 69 Thus, the role of C1q in SCI may be multi-faceted and be more relevant for regeneration rather than for acute damage.
Taken together, these data seem to point toward a limited or controversial role of the classical pathway for complement activation in TBI and SCI.
Looking upstream after traumatic brain injury: Unsolved role of alternative pathway
The role of the alternative pathway in complement mediated TBI pathogenic cascades has been explored in factor-B knockout (fB−/−) animals. In fB−/− animals, peripheral generation of C5a post-TBI was completely blocked (a 5-fold decrease compared to wt animals), suggesting that the alternative pathway may be strongly recruited upon TBI. Interestingly, the number of apoptotic neurons was significantly reduced after blunt weight-drop TBI in fB−/− mice compared to wt littermates both 4 and 7 days post-injury (although no precise quantification is reported). This effect might easily be caused by the described upregulation (up to 4-fold compared to wt animals) of the prosurvival protein bcl2 and the downregulation of Fas apoptotic receptors in fB−/− mice. 70 The role of fB in TBI has been further explored using a blocking monoclonal antibody to sequester fB and thus to prevent the activation of the alternative pathway. However, the results were not conclusive: Despite the administration of 2 doses at 1 and 24 h, the neutralizing antibody reduced fB levels (and C5a generation) only up to 24 h post-injury. 71 Despite the transient decrease in fB levels, significant reductions in the number of apoptotic neurons and inflammatory cells in the brain parenchyma post-TBI were reported. However, no effect on the neurological severity score or other biometric measurements was observed, concluding that transient inhibition of the fB may be insufficient, transient, or take place before the administration of the blocking antibody to deliver clinically relevant beneficial effects. 71
Likewise, SCI recovery has been shown to be faster (BMS score at 7 days of 3 in fB−/− mice vs. 0.5 in wt littermates) and more complete in fB−/− mice (BMS score of 6.5 in fB−/− mice vs. 4.5 in wt littermates at 21 days post-injury, in a range between 0 and 9), together with an enhanced preservation of spinal cord myelin. 58 Notably, administration of anti-fB monoclonal antibody also resulted in improved motor performance only if administered in the 1- to 12-h window (with outcomes comparable to that observed in fB−/− mice), whereas late administration was completely ineffective. 58
Taken together, these data suggest that a significant part of complement activation in TBI and SCI may take place through the alternative, fB-dependent pathway. Further, these data suggest (together with the data on the C3 inhibition) that the temporal window for intervention on the complement pathway may be restricted to the first few hours. 15
Looking upstream after traumatic brain injury: Minor role of lectin pathway
Lectin-dependent activation of complement in TBI has been also investigated. Deposition of mannose-binding lectin (MBL) has been detected in human brain tissue obtained from contusive lesions. Deletion of MBL-C resulted in a minor improvement in neurological impairment after CCI (20% decrease in foot-fault rates in the beam walk, with large overlap between MBL−/− and wt) and only in a limited preservation of neuronal density in perilesional areas. 45 Notably, no difference in C3 activation was found in blood. 45 Likewise, the effect of CCI on MBL−/− was largely indistinguishable to that on wt littermates, both in terms of behavioral performance and histology (no change in neutrophils infiltration, 10% decrease in injury volume), with some limited protective roles reported only in the mildest CCI protocol (detectable in a subset of MWM tests, but not in vestibulomotor tests such as the wire-grip test). 72 Thus, the evidence available, albeit limited, seems to point to a minor role of lectin-dependent complement activation in TBI.
Complement in traumatic brain injury: Beyond inflammation
The evidences available to date point toward an overall negative effect of complement cascade after TBI and SCI (summarized in Table 1). The detrimental effects seem to hinge over the triggering of C3/C5 terminal cascade with damage to brain and spinal cord structures and may be attributed to at least three effectors: opsonization and subsequent clearance of neuronal cells by C3 cleavage product deposition; direct membrane disruption brought about by MAC; and enhancement of the neuroinflammatory response triggered by C3a/C5a and their cognate receptors. Intriguingly, most of the nongenetic interventions on complement factors seem to show a striking time dependency, with efficacy shown only for early interventions; although further data would be needed to settle this point, these observations suggest that the complement-mediated damaging cascades may be set in motion quickly after the damage and rapidly become difficult to control. The precise targets of complement-caused damages remain largely speculative. Both TBI- and SCI-induced loss of myelin seem to be sensitive to complement manipulation and direct MAC-induced damage, as shown in other conditions (such as autoimmune myelin disorders), may be the principal culprit. 73 Given that demyelination may produce acute conduction deficits, blockade of this mechanism may be responsible for a significant part of the beneficial effects of complement inhibition. However, recently multiple lines of evidence suggest that complement may act at synaptic level and a C3-dependent pathway can induce synaptic loss with the contribution of microglial cells. 74,75 Notably, in these and other instances, C1q is considered a key player, and, in particular, it has been implicated in synaptic loss triggered by spinal cord compression. 76,77
The extent of complement-mediated synaptic loss in TBI and how much this may contribute to the onset of acute deficits remain to be further investigated.
Likewise, the contribution of Complement receptors to direct neuronal damage or modulation is unclear. No studies with subpopulation-restricted deletion of Complement receptors are available, and therefore the effects on neurons and glial cells versus inflammatory cells remain to be ascertained. Expression of complement receptors on neurons and direct modulatory effects on neurogenesis and development have been reported. 64,78 –80 To date, the role of complement receptors on neurons in acute settings are unclear, and very little is known of the C5aR2 in this context. However, the expression of C5aR1 on pre-synaptic terminals allows the speculation that acute effects on the synaptic network may be involved in C5a effects post-TBI. 81
Thus, it is possible that noninflammatory mechanisms, such as anaphylatoxin-induced alterations in cellular electrophysiological features, may contribute to the onset of complement-dependent dysfunction in brain (or spinal cord) soon after acute trauma. Given that loss of neuronal activity has been identified as an unexpected consequence of acute trauma to brain and spinal cord, disturbances at synaptic levels may be a new aspect of complement involvement in TBI and SCI. 82
Clinical implications of complement interventions after traumatic brain injury
Although protective effects and therapeutic potential of specific complement inhibition has been postulated in the clinical setting of trauma a present screening of registered clinical studies revealed no current randomized, controlled trial using complement inhibitory strategies in the case of TBI (complement targeting approaches and molecules currently approved, in clinical trials or under study, are summarized in Fig. 3). 83 There is one trial investigating the effects of multiple concussions in retired National Hockey League players, which addresses cognition as a primary readout and as a second readout—among multiple others—C3b in blood, urine, and saliva (NCT02746523) just as a descriptive marker of general inflammation. More than a decade ago, general inhibition of complement activation by heparin-coated extracorporal circuits in cardiac-bypass surgery (indicated by decreased C5b-9 plasma concentrations) resulted in reduced brain cell injury as assessed by enhanced S100B levels. Of note, complement blockade resulted also in mid-term preservation of neuropsychological function, such as attention, flexibility, or executive performances. 84 However, in the context of major heart surgery or in the setting of traumatic-hemorrhagic shock with occult or evident hemodynamic depression, S100B serum concentrations may not reliably reflect neurological damage. 85 Based on the above-mentioned extensive cross-talking between the complement and coagulation/kinin system, it is likely that post-TBI in humans any early activation and disturbance of the coagulation cascade by direct tissue injury or by hemorrhagic shock may result in a local and systemic complement activation with a subsequent enhanced inflammatory response contributing to second brain damage. 42,86 Therefore, it remains of most importance to control any extracerebral organ and system dysfunction to avoid any additional systemic complement activation and inflammation.
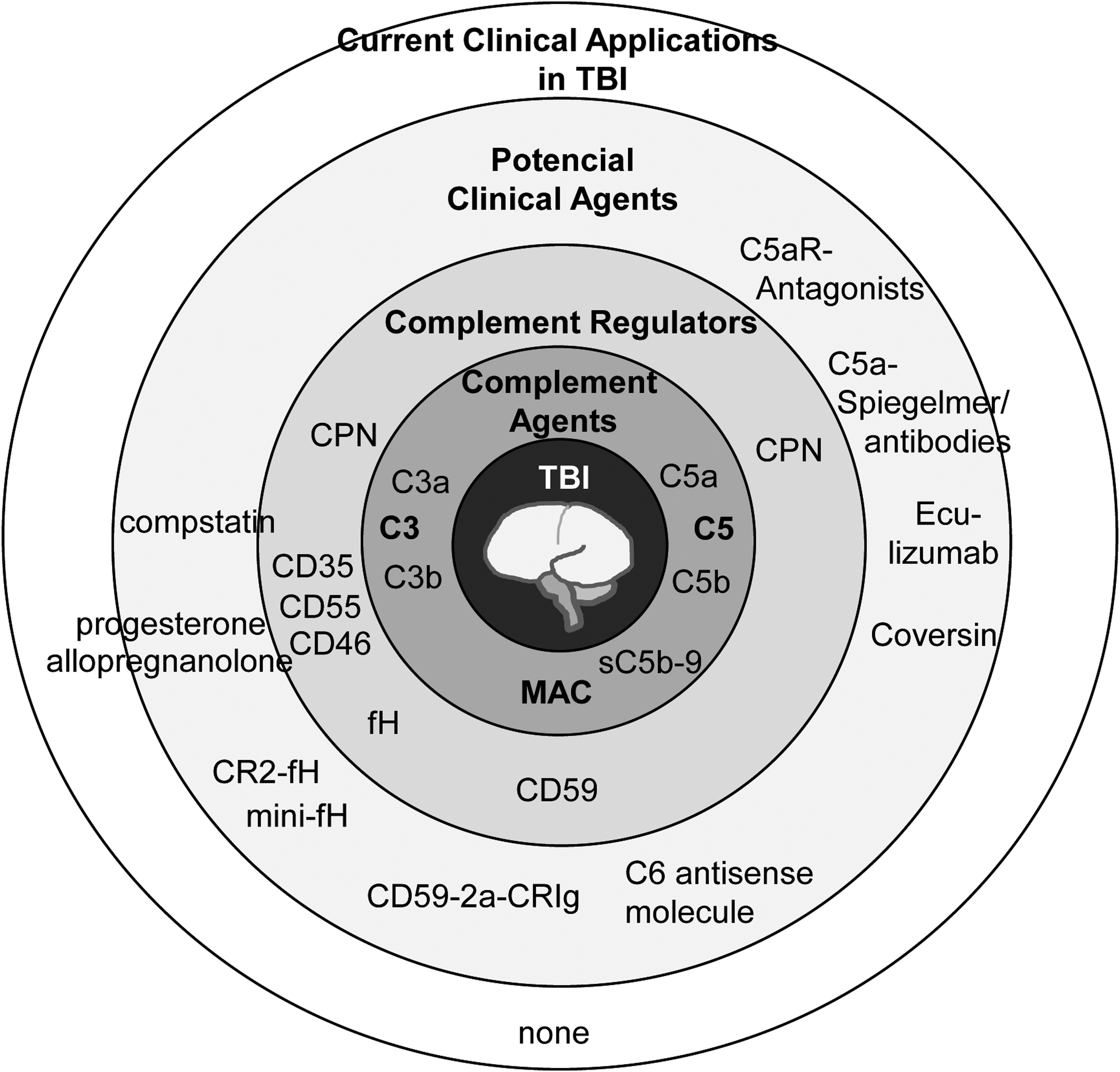
Therapeutic strategies to targeting complement in traumatic brain injury. Although no therapeutic agent aimed at the complement cascade is approved for CNS trauma treamtment, a number targets have been investigated in proof-of-concept in transgenic or knockout models. A number of small molecules, antibodies, chimeric proteins and anti-sense oligonucleotide all interfering with the complement system have demonstrated promising effects in preclinical models. CNS, central nervous system; CPN, carboxypeptidase N; MAC, membrane attack complex; TBI, traumatic brain injury.
In this context, some therapeutic approaches may also indirectly modulate the complement response, as shown for the neurosteroid allopregnanolone, which, in a rodent TBI, was able to enhance the production of CD55 and thereby inhibit central complement components. 87 Clinical studies focusing on neurosteroid interventions after acute or chronic TBI are limited. As reviewed by Marx and colleagues, two phase III trials on progesterone administration after acute, moderate, and/or severe TBI recently reported negative results. Currently, a phase II clinical trial (NCT01673828) utilizing intravenous allopregnanolone for acute TBI is in progress.
However, based on the lack of complement intervention studies in humans post-TBI, we currently completely rely on experimental TBI data and can only speculate on promising future clinical candidates for reducing the inflammatory consequences of TBI. Based on the experimental considerations, targeting C3 (e.g., by compstatin) or C5 (e.g., by eculizumab) and, more specifically, the anaphylatoxin C5a (e.g., by C5a Spiegelmer; or small peptide C5aR antagonist) may be (as alternative approaches) rational for testing in a clinical study. 65,88,89
To date, the peptide, compstatin, and its analogs are the only inhibitors that act on native C3; by preventing activation of C3 by convertases, compstatin also blocks C3b opsonization and generation of effector molecules. Despite specificity for primate as well as for human C3, compstatin analogs have been tested in models ranging from sepsis to hemodialysis-induced thromboinflammation. Cp40 is one of the new derivatives of compstatin and is characterized by improved inhibitory potency and pharmacokinetic profiles. Studies have revealed that it is able to prevent hemodialysis-induced complement activation in cynomolgus monkeys. 88 –91 However, as a central component within the complement cascade, targeting C3 remains questionable. A major concern represents that complete shutdown of C3 activity will generally increase susceptibility to infections as the result of a loss of opsonic capacity of C3, which has been observed in C3-deficient patients. 92
Given that experimental data indicate a crucial impact of MAC formation for the cell-damaging effects in the CNS, targeting of the terminal pathway may represent a promising concept for further clinical studies on TBI. In consideration to target C5, eculizumab is a first-generation C5 inhibitor and currently is approved for paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. This humanized antibody binds to a site on C5 that prevents its activation by C5 convertases, which, in turn, impairs the release of C5a and formation of the MAC while leaving opsonic pathways intact. 93,94
Further, eculizumab was tested in a clinical study (NCT00904826) with patients suffering from neuromyelitis optica. This disease is characterized by simultaneous inflammation and demyelination of the optic nerve and the spinal cord, and complement activation after binding of an IgG autoantibody to aquaporin 4 is thought to be a major determinant of CNS inflammation and astrocytic injury in neuromyelitis optica. Treatment with eculizumab resulted in significantly decreased complement activity in serum. This finding encourages further investigation of eculizumab as a potential therapeutical agent for TBI or SCI. 95
However, it has to be taken into consideration that therapeutic efficacy of eculizimab is prevented in patients with polymorphisms of the C5 molecule. Coversin represents a second-generation recombinant protein inhibitor, and its mode of action on C5 is similar to that of eculizumab. It seems to be a promising alternative therapeutic approach for patients resistant to eculizumab. A phase I clinical trial in human subjects has just been successfully completed and demonstrated a nearly total blockade of complement C5 activity, followed by remaining activity below 50% for 48 h after subcutaneous injection of coversin. 96
Targeting the potent proinflammatory anaphylatoxin, C5a, may represent another important concept in order to reduce neurouinflammation. In this context, C5a mirror-image aptamers (Spiegelmer) seem to be promising drug candidates for clinical studies. Spiegelmers are mirror-image structured oligonucleotides (l-oligonucleotides) that bind and antagonize their target in a manner conceptually comparable to monoclonal antibodies. A big advantage of Spiegelmers is their non-natural chirality, which makes them resistant to nucleases. 97 One promising drug candidate may represent NOX-D20, which binds to mouse and also human C5a. In vitro experiments with both a CD88-expressing cell line and primary human polymorphonuclear cells revealed that NOX-D20 competed with C5a receptor binding and blocked C5a-induced cellular activities. Further, in in a rodent model of sepsis, it was able to reduce multi-organ failure and improve survival. 98
Given that C5a receptor is expressed on astrocytes and seems to play an important role in controlling the inflammation in the brain and may be a central component of complement-mediated brain injury, targeting C5aR represents an important therapeutic concept. 99 Therefore, another important compound for future clinical trial in TBI may be CCX168, an orally administered small molecule inhibitor of C5aR, which showed ability to block C5a-mediated chemotaxis, neutrophil margination, and CD11b upregulation in a variety of in vitro and pre-clinical models. 100 Currently, it will be evaluated in a phase III clinical trial (NCT02994927) for patients with a type of rare autoimmune inflammation caused by autoantibodies, referred as antineutrophil cytoplasmic antibody–associated vasculitis. In consideration to target complement-mediated neuroinflammtion, these studies may represent important concepts for future clinical trials focusing on outcomes post-TBI.
A very promising future approach are damaged tissue targeted complement inhibitors. For example, the described CD59-2a-CRIg dimer assembles at the site of increased iC3b/C3b deposition after cellular injury and subsequently blocks complement attack. 17 Further, in another study with a mouse model of TBI, treatment with a C6 antisense oligonucleotide reduced C6 synthesis and serum levels and inhibited MAC deposition in the injured brain. 15 Another targeted therapy is represented by CR2-fH (mTT30), which targets as a chimeric construct damage-specific C3 and has revealed protective effects in TBI. 52
An overview about the compounds and the respective studies are summarized in Figure 3 as well as in Table 2. Experimental evidence suggests that targeting C3 and C5 activation may be worth investigating as a treatment option in the acute phase (possibly in the first few hours) of treatment of TBI and SCI patients. Thus, complement modulation may add to the efforts to improve the still poor outcome after CNS trauma.
The compounds are listed with their respective study.
TBI, traumatic brain injury; SCI, spinal cord injury; MAC, membrane attack complex; CRIg, Complement receptor of the immunoglobulin superfamily; CLP, cecal ligation and puncture; IL-10, interleukin-10; CSF, cerebrospinal fluid; Ref, reference.
Footnotes
Acknowledgments
F.R., E.K., and M.H. are supported by DFG as part of the Collaborative Research Center 1149 “Danger Response, Disturbance Factors and Regenerative Potential after Acute Trauma” at Ulm University (Ulm, Germany). F.R. is also supported by the ERANET-NEURON “European Research Projects on External Insults to the Nervous System” initiative as part of the MICRONET consortium and by the Baustein Program of Ulm University.
The authors thank Akila Chandrasekar and Rida Rehman for the critical review of the manuscript.
Author Disclosure Statement
No competing financial interests exist.