
Abstract
Diagnosis of ongoing epileptogenesis and associated hyperexcitability after brain injury is a major challenge. Given that increased neuronal activity in the brain triggers a blood oxygenation level–dependent (BOLD) response in functional magnetic resonance imaging (fMRI), we hypothesized that fMRI could be used to identify the brain area(s) with hyperexcitability during post-injury epileptogenesis. We applied fMRI to detect onset and spread of BOLD activation after pentylenetetrazol (PTZ)-induced seizures (PTZ, 30 mg/kg, intraperitoneally) in 16 adult male rats at 2 months after lateral fluid percussion (FPI)-induced traumatic brain injury (TBI). In sham-operated controls, onset of the PTZ-induced BOLD response was bilateral and first appeared in the cortex. After TBI, 5 of 9 (56%) rats exhibited ipsilateral perilesional cortical BOLD activation, followed by activation of the contralateral cortex. In 4 of 9 (44%) rats, onset of BOLD response was bilateral. Interestingly, latency from the PTZ injection to onset of the BOLD response increased in the following order: sham-operated controls (ipsilateral 132 ± 57 sec, contralateral 132 ± 57 sec; p > 0.05) < TBI with bilateral BOLD onset (ipsilateral 176 ± 54 sec, contralateral 178 ± 52 sec; p > 0.05) < TBI with ipsilateral BOLD onset (ipsilateral 406 ± 178 sec, contralateral 509 ± 140 sec; p < 0.05). Cortical lesion area did not differ between rats with ipsilateral versus bilateral BOLD onset (p > 0.05). In the group of rats with ipsilateral onset of PTZ-induced BOLD activation, none of the rats showed a robust bilateral thalamic BOLD response, only 1 of 5 rats had robust ipsilateral thalamic calcifications, and 4 of 5 rats had perilesional astrocytosis. These findings suggest the evolution of the epileptogenic zone in the perilesional cortex after TBI, which is sensitive to PTZ-induced hyperexcitability. Further studies are warranted to explore the evolution of thalamo-cortical pathology as a driver of epileptogenesis after lateral FPI.
Introduction
A
To identify the epileptogenic molecular mechanisms leading to PTE, it is important to first determine the brain areas that become hyperexcitable after TBI and then the seizure-onset zone(s). Analyses of epileptogenic molecular mechanisms rely mainly on animal models, however, given that human brain tissue is not usually available. TBI induced by lateral fluid-percussion injury (FPI) in adult rats results in the development of epilepsy in up to 50% of animals over a period of 1 year, 9 –12 consistent with the rate of development in humans with TBI. 8 The FPI-induced TBI model provides a useful tool to assess the evolution of the epileptogenic tissue pathology in the injured brain. The site of seizure onset after lateral FPI has not been fully characterized.
Ex vivo electrophysiological studies using hippocampal slice preparations from rats with lateral FPI show increased hippocampal excitability within hours post-TBI (for review, see a previous work 13 ). More recent in vivo recordings revealed perilesional seizure onset with hippocampal involvement. 9,14,15 Although these studies indicate perilesional and hippocampal excitability, the location of the seizure onset has remained somewhat uncertain because of the limited number of electrodes in different brain areas used to assess seizure initiation.
Functional magnetic resonance imaging (fMRI) can detect blood oxygenation level–dependent (BOLD) changes in the seizure-onset zone in humans. 16 –18 Despite the methodological challenges related to fMRI studies in rodents, such as the need for sedation, small brain size, and magnetic susceptibility problems in a high magnetic field, BOLD activation in Wag/Rij rats with spontaneous absence seizures has been successfully detected with fMRI. 19 Previous studies also revealed the potential of fMRI for detection of network activation during and after chemically induced seizures in normal rats, such as those induced by pentylenetetrazol (PTZ), which triggers a bilateral cortical BOLD increase that matches the timing and evolution of electrophysiological epileptiform activity, expression of c-Fos, and distribution of metabolic activity. 20 –24 To date, fMRI has not been used to investigate the hyperexcitability developing after epileptogenic brain injury.
The present study evaluated our hypothesis that at 2 months after lateral FPI when a subpopulation of rats undergoes epileptogenesis, the onset and spread of PTZ-induced seizures is altered because of focal network modifications induced by TBI. In particular, we expected that PTZ-induced BOLD activation would be lateralized to the perilesional cortex rather than being symmetric like that observed in control animals.
Methods
Animals
Adult male Sprague–Dawley rats (n = 19; weight at the time of TBI, 362 ± 11 g) were used. Animals were housed in individual cages in a controlled environment (constant temperature 22 ± 1°C, humidity 50–60%, lights on 7:00
Rats were randomized into sham-operated experimental control (n = 8) and TBI (n = 11) groups. Acute mortality (<48 h) was 18% in the TBI group (2 of 11) and 13% in the sham-operated experimental controls (1 of 8). The surviving 16 rats underwent simultaneous local field potential (LFP)-fMRI measurement. In 2 sham-operated controls, the LFP recording failed because of technical difficulties.
Induction of lateral fluid-percussion brain injury
TBI was induced by lateral fluid-percussion injury (FPI), as described previously, 25,26 in 11 rats. Briefly, rats were anesthetized by intraperitoneal injection of a mixture of sodium pentobarbital (58 mg/kg), chloral hydrate (60 mg/kg), magnesium sulfate (127.2 mg/kg), propylene glycol (42.8%), and absolute ethanol (11.6%; 6 mL/kg). Craniectomy (ø 5 mm) was performed with a trephine between bregma and lambda on the left convexity (craniotomy center: 4.5 mm posterior to the bregma, mediolateral 2.5 mm from the midline), leaving the dura intact. The injury was induced with a brief (21- to 23-ms) pressure fluid pulse impact against the exposed dura using an FPI device (AmScien Instruments, Richmond, VA). The pressure of the impact was 3.50 ± 0.04 atm, aimed at inducing a severe TBI. Sham-operated experimental controls underwent identical anesthetic and surgical operations, but were not exposed to lateral FPI.
Electrode implantation and monitoring of provoked seizures
At 2 months after TBI or sham operation, rats (n = 16; 439 ± 27 g at the time of fMRI) were anesthetized with isoflurane (induction 5% and 1.5–2.0% maintenance during surgery). To monitor blood gases and pH during the experiment, the femoral artery was cannulated using PE-10 tubing. An intravenous tube was inserted into the right femoral vein for administration of sedatives and muscle relaxants.
To record PTZ-induced epileptiform electrographic activity, a tungsten wire electrode (50-μm diameter; California Fine Wire, Grover Beach, CA) was inserted into the right (contralateral) frontal cortex (3.2 ± 0.7 mm anterior from bregma, 2.1 ± 0.2 mm lateral from the midline, 0.9 ± 0.4 mm ventral from the pial surface 27 ). Chloridized silver wire reference and ground electrodes were placed subcutaneously in the neck.
After electrode implantation, rats were tracheotomized (with a 13G tube) and transferred to a non-magnetic stereotaxic frame. Rats were secured in the animal holder with earplugs and a bite bar. The tracheal tube was connected to a mechanical ventilator (Harvard Apparatus, Holliston, MA), and animals were ventilated with a mixture of 70% N2 and 30% O2. Breathing rate was set between 60 and 65 breaths/min, and breathing volume was approximately 2.7–3.1 mL per breath depending on the weight of the rat. Arterial blood samples for blood gas analysis were obtained before (baseline) and at the end of the fMRI measurements.
To prevent motion artifacts during fMRI, rats were sedated with a bolus of medetomidine (0.05 mg/kg, intravenously [i.v.]; Orion Pharma, Espoo, Finland) and paralyzed with pancuronium bromide (1 mg/kg, i.v.; Organon, Oss, The Netherlands). Ten minutes after bolus injection, sedation and paralysis were maintained with a continuous i.v. infusion of a mixture of medetomidine (0.1 mg/kg/h) and pancuronium bromide (2 mg/kg/h). Five minutes later, the isoflurane was discontinued.
Simultaneous recording of field potentials and blood oxygen level–dependent functional magnetic resonance imaging
Simultaneous LFP and fMRI measurements were started approximately 45 min after switching the anesthesia from isoflurane to medetomidine sedation. During this period, the medetomidine sedation reached a steady state, and the effects of isoflurane on brain activity became insignificant.
MRI measurements were performed with a 9.4-Tesla horizontal scanner interfaced using a Varian DirectDrive™ console (Varian Inc., Palo Alto, CA). For signal transmission and reception in fMRI experiments, an actively decoupled volume radiofrequency coil and four-channel array receiving coil were used (RAPID Biomedical GmbH, Rimpar, Germany). A fast-spin echo multi-slice sequence was used to collect anatomic images (repetition time [TR] 5.4 sec, effective echo time [TE] 48 ms, echo spacing 16 ms, eight echoes/excitation, field of view [FOV] of 5.0 × 5.0 cm2, image matrix of 512 × 512, resolution 98 × 98 μm2, slice thickness 0.75 mm, 40 slices).
fMRI data were collected using a single-shot spin-echo echo-planar-imaging sequence (TR 4 sec, TE 40 ms, FOV of 2.5 × 2.5 cm2, image matrix 64 × 64, resolution 391 × 391 μm2, slice thickness 1.5 mm, number of slices 15). fMRI data from 15 slices were collected during the first second of the TR, leaving a 3-sec uncontaminated LFP electrophysiological signal between the MRI artifacts. LFPs were monitored from the contralateral cortical electrode during the entire fMRI measurement using a BrainAmp MR plus magnet compatible system (Brain Products GmbH, Munich, Germany). The electrographical signal was low-pass filtered at 1000 Hz (sampling rate, 5000 Hz).
Image acquisition started with 1000 images captured as a baseline. Seizures were then induced by intraperitoneal injection of PTZ (30 mg/kg; 1,5-pentamethylenetetrazol, 98%; Sigma-Aldrich, YA-Kemia Oy, Finland) dissolved in sterile 0.9% saline (4 mg/mL solution). Each rat received a single injection of PTZ. 28 Image acquisition was continued for 1000 images, resulting in a total of 2000 images per rat.
Histology
After completing the electrophysiological and fMRI recordings, rats were deeply anesthetized with isoflurane and decapitated. Brains were removed from the skull, immersion-fixed in 4% buffered paraformaldehyde for 4 h, and cryoprotected in a solution containing 20% glycerol in 0.02 M of potassium phosphate-buffered saline (pH 7.4) for 24 h. Brains were blocked, frozen in dry ice, and stored at 70°C until cut. Brains were sectioned in the coronal plane (30 μm, 1-in-5 series) using a sliding microtome. Sections were stored in a cryoprotectant tissue-collecting solution (30% ethylene glycol, 25% glycerol in 0.05 M of sodium phosphate buffer) at 20°C until processed.
Adjacent series of 1-in-5 sections were stained for thionin to assess lesion location and extent, myelin (0.2% gold chloride method 29 ), astrogliosis (glial fibrillary acid protein [GFAP] immunohistochemistry 30 ), microgliosis (OX-42 immunohistochemistry 30 ), iron deposits (Prussian blue 31 ), and calcifications (Alizarin red 32 ).
The extent of cortical lesion and the location of BOLD onset were reconstructed from coronal MRI slices as previously described.
33
The extent of perilesional astrocytosis was quantified by placing a counting grid on top of the section under a light microscope, and by counting the number of 100 × 100 μm squares with immunostained activated astrocytes along the edges of the lesion cavity throughout its rostrocaudal extent. Density of myelinated fibers in the S1 cortex was quantified ipsi- and contralaterally using ImageJ software (version 1.41o;
Analysis of functional magnetic resonance imaging data to determine the onset region of the provoked seizures
fMRI data were converted from native imaging software formats to NifTI using Aedes (
To assess the location of the provoked seizure onset, a short boxcar block analysis was performed. Because of the long baseline period, 150 baseline images captured before PTZ administration were used for the analysis. A sliding window of three images was used for the activation period. Timing was based on visual inspection of the BOLD time series. Statistical analysis of those 153 images was performed using the general linear model on a voxel-by-voxel basis. 37 Activated brain areas were assessed using a one-sample t-test thresholded at p < 0.05 (false discovery rate [FDR]-corrected). To detect the center of the activated region, a weighted average was calculated from the thresholded T-map for all activated regions with a region size >4 pixels in plane. The location(s) of the BOLD response(s) evoked by the PTZ-induced seizure was then normalized to the anatomical images of the reference brain.
Voxel-vise onset times were visualized by shifting a three-volume-long positive boxcar block over a time series of 400 sec after the onset of PTZ activation with 20-sec interval (Fig. 3). Every block was tested separately from the others. In order to correct for multiple comparisons, we performed FDR correction for all p values from all the tests and all the animals.
Statistical analysis
Statistical analysis was performed using SPSS for Windows (v. 23; SPSS, Inc, Chicago, IL) and Excel. Comparisons between animal groups were made with the non-parametric Kruskal–Wallis test followed by a post-hoc analysis with the Mann–Whitney U test. Interhemispheric differences were analyzed using Wilcoxon's test. Difference in astrogliosis along the rostrocaudal extent of lesion between the animals with ipsi- and bilateral BOLD onset was analyzed using repeated-measures analysis of variance, followed by post-hoc analysis with Dunnett's test. Correlations were calculated using Spearman's rho test. A p value <0.05 was considered significant. All data are presented with mean ± standard deviation or standard error of the mean as indicated in the text.
Results
Acute mortality, apnea time, and occurrence of post-impact seizures
Rats with TBI were divided into two subgroups based on whether the onset of the PTZ-induced BOLD response was bilateral or ipsilateral (Figs. 1 –3). In the whole group, mean duration of post-impact apnea was 31 ± 10 sec. Time in apnea did not differ between rats with bilateral (29 ± 3 sec; n = 4) and ipsilateral (33 ± 13 sec; n = 5) onset of PTZ-induced BOLD activation. Of 9 rats included in the final analysis, 5 had acute post-impact seizures lasting an average of 36 ± 11 sec. Occurrence or duration of post-impact seizures did not differ between subgroups.
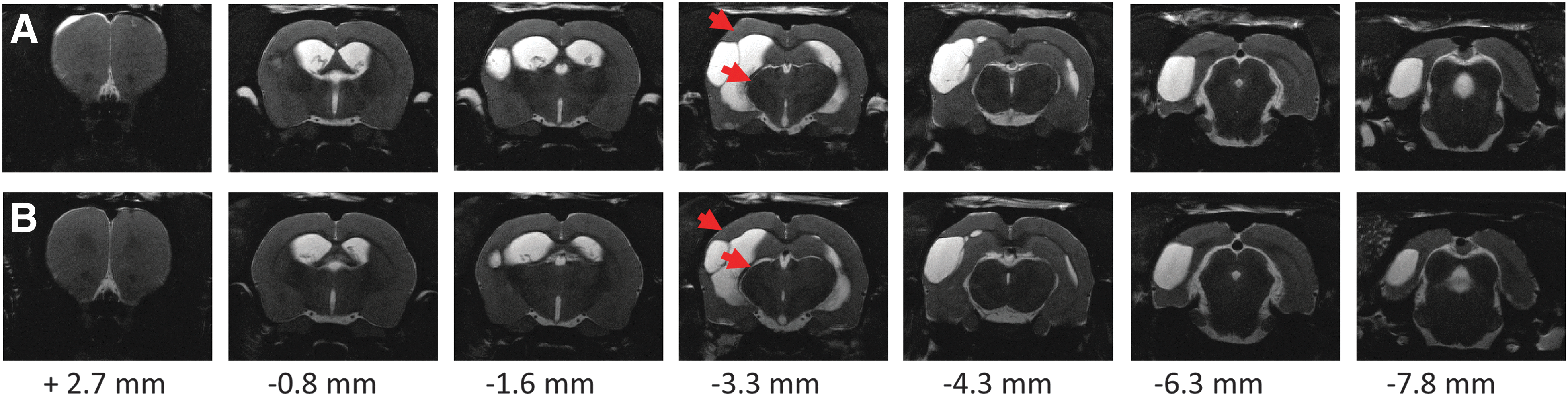
Anatomical T2-weighted images of two representative cases demonstrating variability in extent of brain damage at 2 months after lateral fluid-percussion–induced traumatic brain injury. Note the atrophy in the ipsilateral cortex and thalamus (red arrows). Anteroposterior level is shown relative to bregma. (

Location of onset of pentylenetetrazol (PTZ) activation overlaid on the anatomical reference brain. The center of the seizure-onset volume was calculated as a weighted average from the thresholded T-map for first activated regions. Each animal is shown with a colored symbol where the coloring represents onset time in seconds (please see the color bar) when the location was activated relative to PTZ administration. (
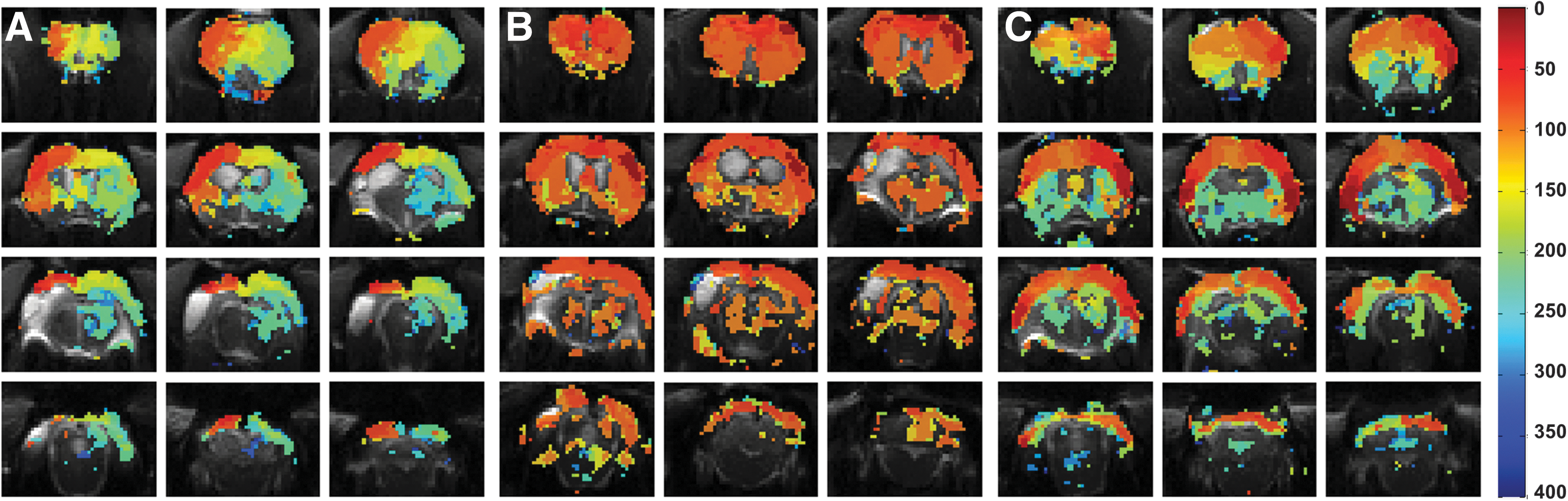
Map of blood oxygenation level–dependent (BOLD) onset times induced by pentylenetetrazol (PTZ) at voxel level. PTZ first activated the cortical areas, and later, the activation extended to the subcortical areas (e.g., striatum and thalamus). (
Physiological parameters during functional magnetic resonance imaging monitoring
Physiological data are summarized in Table 1. All physiological parameters were within the normal range in all animals during the experiment. In the TBI group, pCO2 and pH were higher and O2 saturation lower at the end of the fMRI compared with that at baseline (p < 0.05). Also, pH at the end of the fMRI session was lower in the TBI group than in sham-operated rats (p < 0.05). Physiological parameters also did not differ between rats with an ipsi- or bilateral onset of PTZ-induced BOLD response (data not shown).
Values are shown as mean ± standard deviation. Statistical significances: * p < 0.05 (sham vs. TBI group, Mann–Whitney U); †p < 0.05 (baseline vs. fMRI sample in the same rat, Wilcoxon).
TBI, traumatic brain injury; fMRI, functional magnetic resonance imaging.
Characteristics of the blood oxygen level–dependent response after pentylenetetrazol administration in functional magnetic resonance imaging
Onset of pentylenetetrazol-induced blood oxygen level–dependent response
In sham-operated experimental controls, onset of the PTZ-induced BOLD response was bilateral cortical. In 1 control rat, onset of bilateral cortical BOLD response coincided with thalamic activation ipsilateral to the craniotomy (Fig. 2A).
In TBI animals with bilateral onset of BOLD response, BOLD onset occurred bilaterally in the somatosensory cortex and extended throughout multiple functional slices. In 3 of 4 bilaterally responding rats, also the thalamus became activated bilaterally. In TBI rats with ipsilateral onset of the BOLD response, BOLD onset was located in the perilesional cortex, typically rostral to the lesion core. Thalamic involvement was delayed or not present (Fig. 3).
Delay to the onset of the blood oxygen level–dependent response
In sham-operated experimental controls, time delay between PTZ administration and onset of cortical BOLD response was comparable between hemispheres (left 132 ±57 sec vs. right 132 ± 57 sec; n = 6). Also, in TBI rats with bilateral onset of activation, time from PTZ injection to onset of cortical BOLD activation was comparable ipsilaterally (176 ± 54 sec) and contralaterally (178 ± 52 sec; n = 4). In rats with ipsilateral onset of cortical activation, time to onset of cortical BOLD activation was faster in the perilesional cortex than in the contralateral cortex (406 ± 178 sec vs. 508 ±140 sec; p < 0.05). Interestingly, delay from PTZ injection to onset of BOLD activation was longer in TBI rats with an ipsilateral onset (406 ± 178 sec than in TBI rats with a bilateral onset (176 ± 54 sec), or in sham-operated experimental controls (132 ± 58 sec; all p < 0.05).
Duration of the cortical blood oxygen level–dependent response
Duration of ipsilateral (relative to craniotomy) cortical BOLD response did not differ between sham-operated controls (614 ± 196 sec), TBI rats with a bilateral cortical onset (559 ± 132 sec), or TBI rats with an ipsilateral cortical onset (497 ± 133 sec). Duration of the contralateral cortical BOLD response also did not differ between groups (data not shown).
Pentylenetetrazol-induced thalamic blood oxygen level–dependent response
Delay to activation of the ipsilateral (left) thalamus was 268 ± 93 sec in sham-operated controls (6 of 6 rats had ipsilateral thalamic activation), 237 ± 71 sec in rats with a bilateral BOLD onset (4 of 4 rats), and 516 ± 136 sec in rats with an ipsilateral bold onset (2 of 5 rats).
Correlation of local field potential activation and the blood oxygen level–dependent response
Latency to seizure onset in the contralateral cortical LFP electrode strongly correlated with latency to the contralateral cortical BOLD onset (n = 14; r = 0.960; p < 0.001).
Post–traumatic brain injury brain pathology
Photomicrographs of representative cases summarizing the cortical and thalamic pathologies are shown in Figure 4 and Supplementary Figure 1. (see online supplementary material at

Photomicrographs of histological sections from a rat with traumatic brain injury (TBI) and ipsilateral onset of pentylenetetrazol (PTZ)-induced blood oxygenation level–dependent (BOLD) activation. (
Analysis of glial fibrillary acidic protein (GFAP)-stained preparations revealed that 4 of the 5 rats with an ipsilateral onset of the BOLD response showed widespread perilesional inflammation, particularly rostrally, at 2 months post-TBI (Fig. 4G), whereas only 1 of the 4 animals with a bilateral onset had comparable GFAP staining. When quantifying the perilesional astrocytosis along the rostrocaudal edge of the lesion core, we detected a trend toward more-intense GFAP immunopositivity in rats with an ipsilateral rather than a bilateral onset of the BOLD response, particularly rostrally (glial score 217 ± 69 vs. 176 ± 32, respectively; p = 0.33; Fig. 4G).
All injured animals showed calcifications in the ipsilateral thalamus (Fig. 4D and Supplementary Fig. 2). (see online supplementary material at
Density of myelin fibers in the ipsilateral S1 cortex (layers V–VI) relative to contralateral side did not differ between rats with an ipsilateral (79% of that contralaterally) or bilateral (67%) onset of the BOLD response (p > 0.05).
Discussion
The present study assessed the onset zone of PTZ-provoked fMRI BOLD activation at 2 months after lateral FPI-induced TBI to define the hyperexcitable area during the time period when some of the animals were expected to be undergoing epileptogenesis. Our data indicated that the perilesional cortex was hyperexcitable in a subpopulation of rats at 2 months post-TBI.
Pentylenetetrazol test for the diagnosis of epileptogenesis
Epileptogenesis refers to the development and extension of tissue capable of generating spontaneous seizures, resulting in the development of an epileptic condition and/or progression of established epilepsy. 38 Thus, an increase in seizure susceptibility precedes the appearance of spontaneous seizures, and should be detectable by methodologies used, for example, to determine the localization of the seizure onset in patients evaluated for epilepsy surgery.
Chemoconvulsant PTZ (Metrazol®) in combination with single-photon emission computed tomography (SPECT) is used to image hyperperfusion areas concordant with the cortical seizure onset zone in drug-refractory patients evaluated for epilepsy surgery.
39
–41
In Supplementary Material, (see online supplementary material at
Functional magnetic resonance imaging revealed the perilesional cortex as a hyperexcitable area in a subpopulation of rats with traumatic brain injury
PTZ is commonly used to test for seizure susceptibility in genetically modified mice, inbred mice and rats with spontaneous seizures, and animals exposed to various epileptogenic brain injuries, including TBI. 28,43 –46 The combination of PTZ administration with fMRI is used to image spatiotemporal evolution of seizure-induced BOLD responses and their suppression by an antiepileptic drug, ethosuximide, in brains of normal animals. 47 The present study is the first to apply PTZ-fMRI to localize the BOLD onset zone in animals undergoing epileptogenesis attributed to brain injury.
As the previous and present data show, lateral FPI induces cortical damage with an epicenter in the polymodal auditory cortex, extending laterally down to the perirhinal cortex and caudally to the postrhinal cortex. 33 Available video electroencepalography (EEG) monitoring data suggest that approximately 15% of animals exhibit spontaneous seizures at 2 months, 25–30% at 6 months, and 50% at 12 months post-TBI, and the evolution of pathology and epileptogenesis is largely variable between animals. 9 –12,48 Thus, we expected that some, but not all, animals in the cohort investigated at 2 months post-TBI would show cortical BOLD activation when exposed to systemic PTZ.
In approximately 50% of our rats with TBI, PTZ-evoked seizures induced a BOLD response in the perilesional cortex rostral and medial/lateral to the lesion cavity, suggesting ongoing excitability and development of a seizure focus adjacent to the cortical lesion core. In all TBI cases, the BOLD onset was in the rostral aspect of the lesion, independent of the exact lesion location on the cortical mantle. Our data using noninvasive fMRI corresponds with recent electrophysiology studies using chronic intracortical recordings, showing a perilesional cortical onset of spontaneous seizures after lateral FPI-induced TBI. 11,15 Importantly, Bragin and coworkers also demonstrated that rats destined to develop PTE exhibited pathological high-frequency oscillations as well as repetitive pathological high-frequency oscillations and EEG spikes in the perilesional cortex even before the appearance of spontaneous seizures (i.e., during epileptogenesis). 15 In 50% of our TBI rats and in sham-operated experimental controls, onset the BOLD response appeared bilaterally in the cortex, corresponding to the pattern of PTZ-induced BOLD activation in normal rats. 22,24,49 These observations suggest that the evolution of epileptogenesis within the study cohort was variable, as expected based on previous studies. 9 –12,28
In addition to the pattern of activation, latency from PTZ injection to onset of BOLD response varied between the animal groups. In particular, latency was longer in TBI rats with a unilateral onset of the BOLD response compared to TBI rats with a bilateral onset. Interestingly, there was no correlation between lesion severity and hyperexcitablity assessed by correlating the cortical lesion area with latency to PTZ-induced BOLD activation or its duration. Pharmacokinetic studies indicate that lipophilic PTZ rapidly penetrates the blood–brain barrier after systemic administration, suggesting rapid bilateral engagement of gamma-aminobutyric acid receptors. 50,51 Therefore, we consider it unlikely that the delayed onset of the unilateral BOLD response in a subgroup of TBI animals could be explained by a reduced ipsilateral cortical vascular density or blood–brain barrier penetration. Given that the BOLD response in the seemingly normal contralateral cortex was also delayed, we do not consider that a misplaced PTZ injection into the intraperitoneal cavity could explain the group differences either, because 4 of the 5 animals with an ipsilateral BOLD response had a delayed response onset compared with other animals. A slower spread of seizure activity in the thalamo-cortical pathways and corpus callosum attributed to white matter damage could slow down the spread of PTZ-induced BOLD activation in the injured brain.
Therefore, we next assessed the pathological substrate for unilateral onset of BOLD activation. We did not observe any apparent differences in the lesion area or in the location or number of perilesional iron deposits between TBI rats with an ipsi- or bilateral onset of the BOLD response (data not shown). Severity of myelin damage in layers V–VI in the perilesional cortex also did not differ between groups, which could have affected the thalamo-cortical spread of the seizure activity and consequent BOLD activation. 34 In contrast to our expectations, animals with an ipsilateral onset of the PTZ-induced BOLD response had almost no calcium deposits in the ipsilateral thalamus. Instead, rats with a bilateral BOLD onset had robust “mature” stone-like thalamic calcifications ipsilaterally, suggesting that thalamic calcifications were not responsible for the asymmetry in the cortical BOLD responses in this subgroup of animals. Assessment of GFAP-positive astrocytosis indicated that 4 of the 5 animals with an ipsilateral BOLD response had perilesional astrocytosis, whereas only 1 of the 4 rats with a bilateral BOLD response had a similar degree of perilesional GFAP immunopositivity. Moreover, the width of astrogliotic rim appeared wider, particularly rostrally, in rats with ipsilateral BOLD onset as compared to that in rats with bilateral BOLD onset. Even though further studies with larger animal cohorts are needed, our findings suggest that the prolonged presence of an inflammatory milieu could enhance the evolution of perilesional hyperexcitability, resulting in focal activation of the BOLD response in the perilesional cortex when rats were exposed to systemic PTZ.
Taken together, TBI animals with a faster bilateral onset of BOLD activation had large cortical lesions, white matter damage, and their areas of thalamic calcification were even larger than that in rats with an ipsilateral onset of the BOLD response. Therefore, further studies are required to elucidate the mechanisms of delayed asymmetric perilesional BOLD activation in a subpopulation of rats with TBI. Also, the inter-relationship between focal hyperexcitability and -connectivity, which was recently described by Harris and colleagues after unilateral controlled cortical impact in rats, 52 warrants future studies. Interestingly, a delayed hippocampal BOLD response was observed in 1 rat only, opposing the idea of a primary epileptogenic focus in the hippocampus.
Methodological issues
fMRI with submillimeter spatial resolution and full brain coverage provides a unique approach to localizing the hyperexcitable zone during epileptogenesis in rats. The temporal resolution of 4 sec, which is approximately twice larger than often used in fMRI, was used to guarantee a long enough artefact-free period of LFP recordings between volumes, and to improve the stability of data in long scans. This temporal resolution does not allow for detailed tracking of seizure spread, but it was sufficient to localize the onset zone in the perilesional cortex and differentiate subgroups of animals with either bi- or unilateral onset. Probably the most significant experimental compromise in animal fMRI experiments is the need for anesthesia to immobilize the animals and alleviate stress. In the present work, we sedated rats by medetomidine infusion. Although any anesthesia or sedation is likely to have some modulatory effect on brain activity and the BOLD fMRI response, we recently demonstrated that the fMRI response to chemically induced seizures under medetomidine sedation is similar to that in an awake state. 53
Previous fluorodeoxyglucose and c-Fos activation studies in normal uninjured rats 23,54 –56 showed that systemic administration of PTZ triggered bilateral activation of the pathway from the hypothalamic mammillary nucleus to the anterior thalamic nucleus that spread to the somatosensory cortex. We were unable to detect a mamillo-thalamic spread of the BOLD response in sham-operated or injured animals, which may be attributed to the 4-sec temporal resolution of our fMRI signal.
We have previously shown that cerebral blood flow, as measured by arterial spin labeling, is consistently reduced in the chronic phase 8–9 months after TBI in the perilesional cortex, 57 which may potentially influence the amplitude of BOLD response. However, we have previously shown that neurovascular coupling is preserved in the ipsilateral cortex outside the primary lesion, 34 so we are confident that fMRI is able to detect robust activation caused by PTZ.
Conclusions
Our findings demonstrated that the PTZ-induced BOLD response begins in the perilesional cortex in a subpopulation of rats at 2 months post-TBI, suggesting the ongoing evolution of an epileptogenic cortical focus. Similarly, PTZ-SPECT in a patient with PTE indicated perilesional hyperexcitability. Further analyses using PTZ-fMRI or PTZ-SPECT to reveal the evolution and localization of the epileptogenic zone after experimental TBI are warranted. Moreover, the contribution of chronic perilesional cortical inflammation and evolution of thalamic pathology to post-traumatic epileptogenesis requires further exploration.
Footnotes
Acknowledgments
We thank Merja Lukkari, Maarit Pulkkinen, and Jarmo Hartikainen for their excellent technical help and Raimo Salo for statistical advice. This study was supported by the Academy of Finland (A.P., O.G., X.E.N.E., A.S.L.) and the FP7-HEALTH project 602102 (EPITARGET; A.P., O.G.).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.