
Abstract
Traumatic brain injury (TBI) induces a neuroinflammatory response resulting in astrocyte and microglia activation at the lesion site. This involves upregulation of neuroinflammatory genes, including chemokines and interleukins. However, so far, there is lack of knowledge on transcription factors (TFs) modulating this TBI-associated gene expression response. Herein, we analyzed activating transcription factor 3 (ATF3), a TF encoding a regeneration-associated gene (RAG) predominantly studied in peripheral nervous system (PNS) injury. ATF3 contributes to PNS axon regeneration and was shown before to regulate inflammatory processes in other injury models. In contrast to PNS injury, data on ATF3 in central nervous system (CNS) injury are sparse. We used Atf3 mouse mutants and a closed-head weight-drop–based TBI model in adult mice to target the rostrolateral cortex resulting in moderate injury severity. Post-TBI, ATF3 was upregulated already at early time points (i.e,. 1–4 h) post-injury in the brain. Mortality and weight loss upon TBI were slightly elevated in Atf3 mutants. ATF3 deficiency enhanced TBI-induced paresis and hematoma formation, suggesting that ATF3 limits these injury outcomes in wild-type mice. Next, we analyzed TBI-associated RAG and inflammatory gene expression in the cortical impact area. In contrast to the PNS, only some RAGs (Atf3, Timp1, and Sprr1a) were induced by TBI, and, surprisingly, some RAG encoding neuropeptides were downregulated. Notably, we identified ATF3 as TF-regulating proneuroinflammatory gene expression, including CCL and CXCL chemokines (Ccl2, Ccl3, Ccl4, and Cxcl1) and lipocalin. In Atf3 mutant mice, mRNA abundance was further enhanced upon TBI compared to wild-type mice, suggesting immune gene repression by wild-type ATF3. In accord, more immune cells were present in the lesion area of ATF3-deficient mice. Overall, we identified ATF3 as a new TF-mediating TBI-associated CNS inflammatory responses.
Introduction
T
Despite its significant socioeconomical impact, molecular and cellular mechanisms underlying TBI are less well understood compared to other central nervous system (CNS) injuries such as spinal cord lesion. Several TBI-inflicted processes observed in humans can be recapitulated in rodent TBI models. 8,9 A first injury result directly associated with the TBI is hematoma formation, frequently directly causing mortality. Further, cortical impact triggers mechanical shear forces, disrupting neuronal connections and eventually inducing neuron cell loss. 10,11 Injury induces a gene expression response in neuronal and non-neuronal cells. In neurons, mechanical injury induces upregulation of so-called regeneration-associated genes (RAGs). 12 –16 RAGs fall into genes encoding transcription factors (TFs) such as Atf3, Stat3, Sox11, and Smad1 or so-called effector RAGs such as neuropeptides (Galanin, Grp, Vip, Npy, and Ngf), Sprr1a, Gap43, or Timp1 that might directly modulate regeneration of injured axons. Of note, CNS and peripheral nervous system (PNS) neurons show differences in their potential of RAG propagation. In PNS injury, a typical RAG response encompasses induction of at least 200–500 genes and is suggested to partially account for the successful regeneration outcome after PNS in contrast to CNS injury. 13,16,17 In the CNS (e.g., in TBI or spinal cord injury), RAG induction has not been analyzed to the same extent. So far, induction of only a few RAG members such as Atf3, Sprr1a, and Timp1 has been reported on. However, induction levels of RAGs in the CNS are weaker and less persistent compared to PNS injury. 18 –21
Besides affecting neurons as a primary target, TBI induces a cascade of secondary injury responses. 8 For instance, TBI triggers a strong response of immune cells, resulting in neuroinflammation. 22,23 This includes infiltration of the lesion site by brain-resident (i.e., astrocytes, microglia) and peripheral immune cells, including macrophages. 22,23 From a molecular point of view, TBI-associated neuroinflammation includes transcriptional upregulation of several genes encoding for immune-modulatory proteins. The latter include genes encoding chemokines of the CCL and CXCL family (e.g., Ccl2, Ccl3, Ccl4, and Cxcl1), interleukins (Il6, Il1b, and Il33), TNF family members (Tnfa), lipocalin, and complement encoding factors (C3), which are upregulated by TBI, but also in other brain injury models. 24 –27 So far, detailed mechanistic insight of this TBI-associated transcriptional induction of immune genes and TFs involved is missing.
In this study we analyzed whether the TF, ATF3 (activating transcription factor 3), modulates transcriptional responses associated with TBI. As indicated above, ATF3 is a RAG-encoding TF strongly upregulated by PNS injury. 17,28 –30 In publications available so far, ATF3 induction was mainly described to occur in neurons. 31 –34 Nevertheless, other cell types, including immune cells, might also respond with ATF3 induction post-injury. 35 –37 Furthermore, astrocytes and microglia in the brain also upregulate ATF3 upon injury. 38,39 Both ATF3 loss- and gain-of-function studies in rodent models of PNS injury have shown proregenerative ATF3 functions. 31 –34,40 This might be accomplished through transcriptional activation of Sprr1a, neuropeptides such as Galanin, Vip, and Grp, or the heat shock protein, Hsp27, by ATF3. 32,34
In addition, ATF3 emerged as TF involved in regulation of inflammatory genes. 28 For instance, ATF3 acts as a transcriptional repressor of the CCL chemokine, Ccl2, in PNS, 32 but also in CNS injury. 41 Such negative regulation of immune genes by ATF3 also applies for other immune genes and was previously also reported outside the nervous system. 42 –44 In the CNS, increased inflammation and brain injury were observed after transient focal cerebral ischemia in Atf3 knockout mice. 41 Because TBI also results in strong activation of a neuroinflammatory response and ATF3 regulates expression of immune genes, we investigated in this study whether ATF3 might be involved in regulation of TBI-associated neuroinflammation.
ATF3 has not been studied to the same extent in CNS as in PNS injury. Previously ATF3 was shown to be induced by TBI, 19 but, so far, no function for ATF3 in TBI has been reported. However, evidence from other injury models (ischemia, amyotrophic lateral sclerosis, and epilepsy) suggests a neuroprotective function exerted by ATF3. 41,45,46
In this study, we used mice with a constitutive Atf3 deletion in all cells 47 in an established weight-drop–based TBI model. 48 Here, one cortical hemisphere is injured by a falling rod whereas the contralateral hemisphere is not directly injured. We show that ATF3-deficient mice have a slightly elevated mortality rate and weight loss. We identified ATF3 as a new TF contributing to the TBI-associated immune response. In ATF3-deficient mice, induction of several immune genes by TBI and abundance of immune cells was further augmented compared to the induction observed in wild-type (wt) mice.
Overall, we provide a first study of ATF3 in TBI and identified ATF3 as a novel transcriptional regulator affecting immune gene regulation upon CNS injury.
Methods
Atf3-deficient mice
Constitutive Atf3 mutant mice (Atf3–/– ) on a C57Bl/6 background were a kind gift of Dr. T. Hai (Ohio State University, Columbus, OH). The Atf3 mutant allele lacks exon B, which contains the AUG initiation codon and does not produce any ATF3 protein in the liver 47 or injured facial nucleus. 32 Genotyping followed a published protocol. 47 As control, we always used offspring harboring two wild-type (wt) Atf3 alleles (Atf3+/+ ). Wt and mutant animals were littermates derived from breeding two Atf3 heterozygous parents.
Traumatic brain injury model
We used a weight-drop–based TBI model. 48,49 For this, wt and mutant mice of both sexes were used at the age of 12–14 weeks after birth. The average weight of animals was: wt males (28.9 ± 3 g), Atf3 mutant males (28.3 ± 2 g), wt females (22.7 ± 1.5 g), and Atf3 mutant females (21.9 ± 1 g). Mice were anesthetized with sevoflurane inhalation, and the skin over the skull was shaved and cleaned with ethanol. Subsequently, mice were injected with Buprenorphin (Temgesic; 0.03 mg/kg), and a skin incision of the scalp was applied with a scalpel (approximately 1 cm in length) to expose the skull. The head of the animal was fixed manually on a modeling clay cushion to absorb some of the energy. The tip (3 mm in diameter) of a metal rod (weight, 333 g) was positioned over the left cortical hemisphere at a rostrolateral position (see previous works 48,49 ). The falling height of the rod was 2 cm for females and 2.3 cm for males. Immediately after hitting the closed skull, the rod was retracted to prevent a second hit on the cortex. Post-TBI, O2 was given for 1–2 min until regular breathing was restored. The scalp was sutured with three stitches. Buprenorphin was injected every 8 h for the first 24 h. All surgeries were performed on a heating pad pre-warmed to 37°C and lasted approximately 10 min for 1 mouse. For sham-treated animals, we performed exactly the same procedure (shaving, anesthesia, analgesia, skin incision, and skin suture), except for the actual TBI impact with the falling rod. Limb paresis was quantified by picking up mice by the tail and subsequently testing the potential of mice to grab a pole.
To estimate the size of the hematoma, a picture of the brain was taken directly after dissection using a Samsung NX1000 camera positioned vertical above the brain. For all mice with a hematoma, the size of the hematoma area in the ipsilateral cortex was circumscribed with the lasso tool in the graphics software
Mice had an apnea time for approximately 6–7 sec as described recently by us. 49 We scored TBI severity according to the neural severity score (NSS). 48,49 Mice had an NSS of approximately “5” at 2 h and “2” at 24 h post-TBI corresponding to a moderate TBI severity. 48,49
All experiments were in accord with regulations by the local veterinary authorities (Regierungspräsidium, Tübingen, Germany).
Quantitative real-time polymerase chain reaction
Cortical tissue (approximately 5–6 mm in diameter) of both hemispheres from sham- and TBI-treated animals was prepared from the rostrolateral cortex covering the impact site. In addition, both hippocampi from each animal were collected. Total RNA was isolated with the Isolate II RNA/DNA/Protein kit (Bioline, London, UK). For complementary DNA (cDNA) synthesis, reverse transcription was performed with approximately 0.75 μg of RNA using M-MLV reverse transcriptase (Promega, Madison, WI) and random hexamers. Quantitative real-time polymerase chain reaction (qPCR) was performed on a Roche LightCycler® 480 (Roche, Indianapolis, IN) with the SYBR Premix Ex Taq (Tli RNase H Plus) PCR Master Mix (TaKaRa, Tokyo, Japan). Expression was determined in relation to Gapdh RNA levels. Primer sequences are provided in the Supplementary Information (see online supplementary material at
Histology
ATF3 histology with rabbit anti-ATF3 antibody (1:500; Sigma-Aldrich, St. Louis, MO) was performed as previously described. 32 For all other immunostainings, we used the following protocol. Mice were anesthetized with ketamine/xylazine (100 and 16 mg/kg) and intracardially perfused with 50 mL of ice-cold phosphate-buffered saline (PBS) followed by 50 mL of 4% paraformaldehyde (PFA) in PBS. The brain was dissected and post-fixed in 4% PFA for 18 h at 4°C, washed in PBS, and cryoprotected in 30% sucrose in PBS. Brains were thereafter embedded in optimal cutting temperature on dry ice and sectioned at 40 μm in the coronal plane in a cryostat (Leica CM 1950; Leica Microsystems, Wetzlar, Germany). Floating sections were processed for immunostaining: Sections were blocked in 3% bovine serum albumin/3% donkey serum/0.3% Triton in PBS for 2 h at 24°C and then incubated with the appropriate primary antibody (mouse anti-GFAP [glial fibrillary acidic protein], 1:500, Millipore, Billerica, MA; mouse anti-CD11, 1:100, Millipore; mouse anti-CD45, 1:50, Millipore; mouse anti-Mac2 [galactose-specific lectin 3 {galectin-3}], 1:250, Millipore) for 48 h at 4°C; sections were thereafter washed 3 × 30 min in PBS and incubated in the appropriate secondary antibody mix (Donkey anti-mouse 568, Invitrogen, Carlsbad, CA; Donkey anti-rabbit 488, Invitrogen) for 2 h at 24°C, together with 4′,6-diamidino-2-phenylindole (DAPI; 1:1000) for nuclear counterstain, washed again (3 × 30 min in PBS), and finally mounted in Fluorogold-Prolong Antifade (Invitrogen).
Biochemistry
Protein lysates were prepared with the ISOLATE II RNA/DNA/protein kit (Bioline). 1 × PhosStop (Roche) was added to protein lysates. Samples were resolved on 8–10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, followed by transfer on polyvinylidene difluoride membranes (Amersham Biosciences, Foster City, CA). After 1 h of blocking, first antibodies were applied overnight at 4°C: rabbit anti-ATF3 (1:500, Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-GAPDH (glyceraldehyde 3-phosphate dehydrogenase;1:60,000; Acris, Herford, Germany) and goat anti-Lipocalin2 (1:100; R&D Systems, Minnesota, MN). Detection of first antibodies involved horseradish-peroxidase–conjugated secondary antibodies (1:2000; Santa Cruz) and the ECL Western Blotting Substrate (Pierce Biotechnology [Rockford, IL] or Millipore).
Microscopy and image analysis
Confocal images were acquired using an LSM-700 (Carl Zeiss AG, Jena, Germany) inverted microscope, fitted with a 20× oil objective. Low-magnification, wide-field images were acquired with a Keyence microscope fitted with a 10 × objective. All images were acquired in 12-bit format in tile-mode format; imaging parameters were set in order to obtain a fluorescence signal for the immunostained antigen >150 (according to the 12-bit intensity scale ranging from 0 to 4095) while avoiding saturation in high-intensity structures. In order to avoid fluorescence cross-bleed, all fluorescent channels were acquired independently. For each mouse, multiple brain sections (>3) were imaged. For quantification of cellular density, optical stacks composed of twenty 1-μm-thick optical sections were imported in ImageJ (NIH, Bethesda, MD) and collapsed separately in maximum-intensity-projection mode for each channel. A squared region of interest (ROI; 105 μm2 in size) was manually positioned in correspondence of layer II/III to have the vertical side in contact with the margin of the injury area and the horizontal axis parallel to the cortical contour. An intensity threshold was applied to bin the image into positive and negative cells; the value of the threshold was kept constant across images and replicated. The number of positive cells within the ROI was determined by visual inspection and manual logging; distinction of cells from staining artifacts was determined by morphology of cells and by the presence of a nucleus detectable in the DAPI channels. For assessment of astrogliosis, after thresholding, the total area occupied by GFAP-positive structures in the ROI was computed. In addition, the total area of astrogliosis was measured in 10 × wide-field images using the ImageJ (NIH) software suite. Low-magnification images encompassing the whole hemisphere subject to injury were imported in ImageJ (NIH). After thresholding, the contour of the area containing GFAP-positive astrocytes was manually traced and the surface computed.
Statistical analysis
Animal numbers, time points and distribution of sex are provided in Supporting Table 1 (see online supplementary material at
Results
Rapid but transient activating transcription factor 3 induction after traumatic brain injury
ATF3 was previously analyzed at selected time points post-TBI (e.g., an earlier work
19
). We studied ATF3 expression at several time points post-TBI. For this, adult wt mice (12–14 weeks) of both sexes were subjected to unilateral TBI targeting the cortex. Subsequently, we analyzed ATF3 expression on mRNA and protein level in the cortex (Fig. 1) and hippocampus (Supplementary Fig. 1) (see online supplementary material at
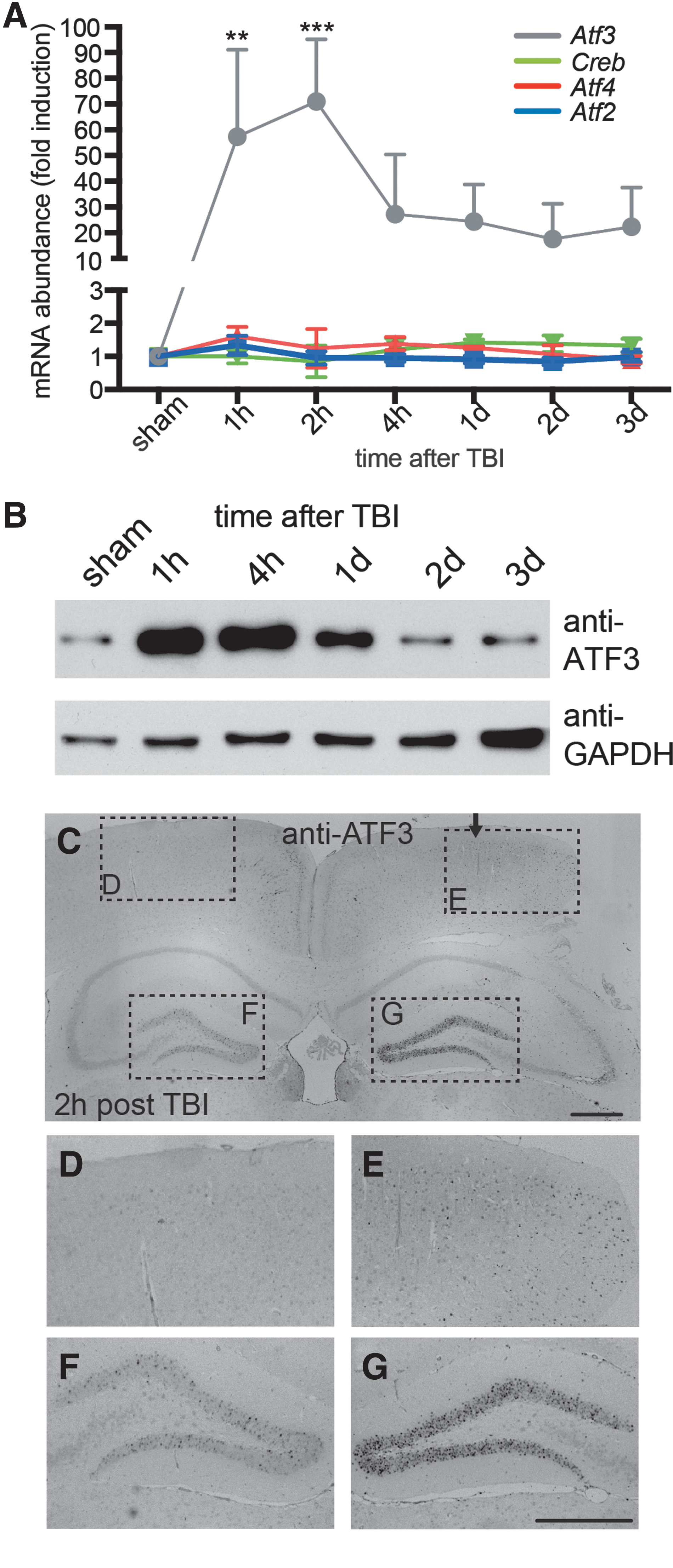
ATF3 is rapidly induced upon TBI. (
In all control brain areas, Atf3 was only weakly present. In contrast, Atf3 mRNA abundance was rapidly upregulated in the ipsilateral impact area post-TBI (Fig. 1A). Strongest increase in Atf3 mRNA levels was already observed 1–2 h post-TBI. At later time points, 2–3 days post-TBI, Atf3 levels were declining. Within the ATF/CREB family of TFs, TBI-induced upregulation appears restricted to Atf3 given that other members, including Atf2, Atf4, and Creb, were not modulated at the transcript level in (Fig. 1A), in line with a previous report.
32
Similar results to the cortex were also obtained for Atf3 in the hippocampus (Supplementary Fig. 1) (see online supplementary material at
Next, we analyzed ATF3 protein abundance (Fig. 1B–G). In western blotting experiments, ATF3 protein was upregulated at 1, 4, and 24 h post-TBI in the cortex and weak ATF3 induction was still present at 2 and 3 days post-TBI (Fig. 1B). Similar results were obtained in the hippocampus (data not shown).
Histological inspection revealed ATF3 protein abundance in neurons in the ipsilateral (Fig. 1C,E), but not in the contralateral (Fig. 1C,D) cortex, after 2 h in a TBI-treated animal. ATF3-positive cells also colocalized with the neuronal marker, neuronal nuclei (NeuN), in the ipsilateral cortex (Supplementary Fig. 2C,F) (see online supplementary material at
In summary, ATF3 is rapidly, but transiently, upregulated in the cortex and hippocampus post-TBI in neuronal cells, but also immune cells such as astrocytes.
Activating transcription factor 3 ablation modulates body weight, paresis incidence and hematoma formation after traumatic brain injury
Next, we analyzed constitutive Atf3 mouse mutants lacking ATF3 in all cells (previous work 47 ; Fig. 2). No Atf3 transcript or protein was observed in ATF3-deficient mice subjected to TBI (Fig. 2A,B). This is in line with other injury models analyzing ATF3-deficient mice. 32,41

Enhanced mortality, weight loss, paresis, and hematoma formation in Atf3 mutant mice post-TBI. (
The average mortality rate inflicted by TBI in wt mice was 9% (12% in males; 6% in females; N = 33). Mortality occurred in almost all cases within the first 5 min post-TBI. For ATF3-deficient mice, this mortality rate was approximately doubled. Now, 19% of mice (28% males; 11% females) died within the first 5 min after the TBI impact (Fig. 2C; N = 37).
We also measured TBI-associated weight loss (Fig. 2D; wt TBI, N = 18; knockout [ko] TBI, N = 18, wt sham, N = 5; and ko sham, N = 5). The average weight before TBI was indistinguishable between genotypes (see Methods). In wt mice, the maximum weight loss (approximately 9%) was observed at 1 day post-TBI (Fig. 2D). This was reduced to approximately 4% 3 days post-TBI in wt mice (Fig. 2D). In Atf3 mutant mice, we observed at all time points analyzed elevated weight loss post-TBI (Fig. 2D; N = 18 mice). For instance, 3 days post-TBI, mutant mice still showed a weight loss of almost 10%. This is 3 times as much as observed in wt mice at this time point (Fig. 2D). This finding is in accord with an enhanced weight loss of ATF3-deficient mice reported in an ischemia model. 41 Besides weight loss, we noted enhanced incidence of paresis in Atf3 mutant mice at 4 h post-TBI (Fig. 2E,F; wt, N = 23 mice; ko, N = 23 mice). For instance, hindlimb paresis was almost doubled in ATF3-deficient compared to wt mice (Fig. 2F).
In humans, hematoma formation is one the most frequent causes of TBI-inflicted mortality. In our rodent model, TBI induces hematoma in cortical tissue after impact. Dissecting brains of wt and ATF3-deficient mice at 3 days post-TBI, we noticed increased hematoma size on the cortical surface in Atf3 mutant mice (Fig. 2G–I). In wt mice, the hematoma area covered approximately 15% of the entire cortical surface (15.1 ± 11%; N = 17; Fig. 2G,I). This was significantly enhanced upon ATF3 deletion and now hematoma surface was almost doubled (26.3 ± 11.5%; N = 18; Fig. 2H,I). ATF3 was previously associated with bone formation.
50
Thus, differences in skull bone formation might account for enhanced hematoma formation upon ATF3 deficiency. However, the fracture prevalence was identical between genotypes (approximately 90%). Micro-computed tomography analysis revealed no significant differences in calvarial trabecular thickness and mineral density between wt and ATF3-deficient mice of both sexes (Supplementary Fig. 3) (see online supplementary material at
Overall, we observed slightly elevated mortality and weight loss in Atf3 mutant mice as well as larger hematoma formation.
Analysis of a traumatic brain injury–associated regeneration-associated gene response
RAG induction is a molecular hallmark of PNS injury.
16
In contrast, data available in CNS injury, such as spinal cord injury or a few reports on TBI, suggest generally a weaker RAG induction encompassing only a few RAG genes.
18
–21
In this study, we analyzed 15 prototypical RAG genes at different time points post-TBI in two brain areas, cortex (Fig. 3) and hippocampus (Supplementary Fig. 5) (see online supplementary material at
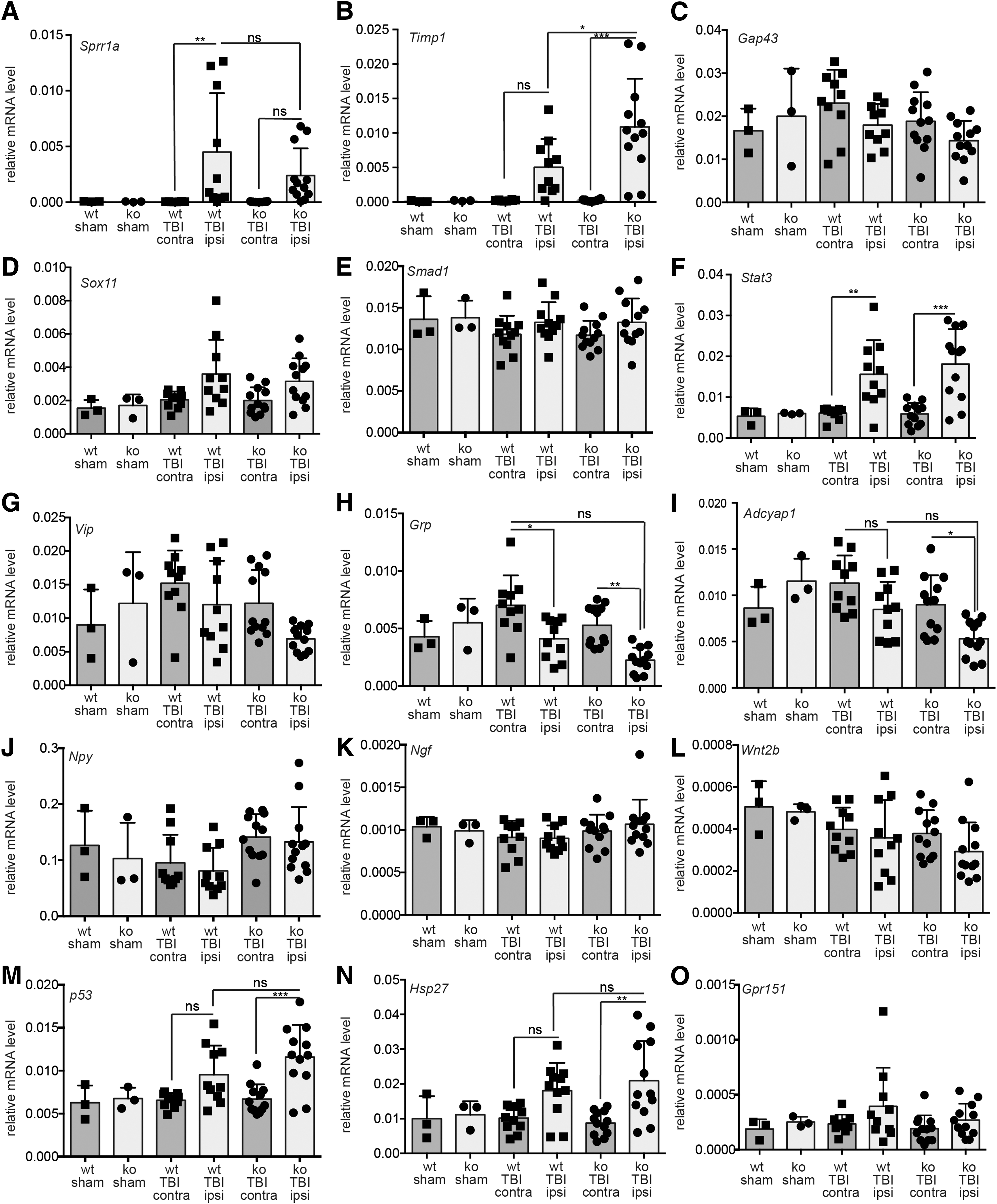
RAG gene induction in murine TBI. wt and Atf3 mutant (ko) animals were either sham operated or treated with TBI. In TBI animals, the ipsilateral cortex was targeted whereas the contralateral side serves as a further control. Three days post-TBI, cDNA derived from cortical tissue was subjected to qPCR analysis to quantify mRNA levels of genes indicated. Of the 15 RAGs tested, only Sprr1a (
Besides Atf3 (Fig. 1), we observed that only three (Sprr1a, Timp1, and Stat3) of the 15 RAGs analyzed were significantly induced in the cortex at 3 days post-TBI in wt mice (Fig. 3A,B,F). This is not attributed to this specific time point because RAG induction was also not observed at several earlier time points (Supplementary Fig. 6) (see online supplementary material at
qPCR results for selected RAGs obtained three days after CNS injury (i.e., cortical TBI, this study) and PNS injury (facial nerve injury 32 ) are summarized. Data are presented as fold induction in relation to uninjured tissue. Numbers below 1 indicate downregulation post-injury whereas numbers >1 indicate fold induction of the respective RAG. Galanin mRNA was not detectable in qPCR (“0”).
RAG, regeneration-associated gene; CNS, central nervous system; PNS, peripheral nervous system; TBI, traumatic brain injury; qPCR, quantitative polymerase chain reaction.
We also inspected the impact of ATF3 deficiency on this RAG response (Fig. 3). Previously, transcriptional induction of the aforementioned RAGs was demonstrated to partially require ATF3 in PNS nerve injury.
32
In line with these results, Sprr1a induction in the TBI-injured cortex also required the presence of ATF3 to some extent (Fig. 3A). In contrast, Timp1 mRNA induction was further elevated in the cortex of ATF3-deficient mice subjected to TBI (Fig. 3B), a finding in accord to results after PNS injury (Table 1).
32
Similar results for Sprr1a and Timp1 were obtained in the hippocampus 3 days post-TBI (Supplementary Fig. 5) (see online supplementary material at
In summary, we observed an overall weaker or even downmodulated RAG response in this CNS injury model compared to robust induction known after PNS injury.
Activating transcription factor 3 regulates induction of neuroinflammatory genes after traumatic brain injury
A widely recognized molecular hallmark observed in several rodent TBI models, but also other neurodegenerative diseases, is mRNA induction of neuroinflammatory genes.
18
–21
These include chemokines and several interleukins, among others.
53,54
ATF3 ablation was shown to regulate immune gene expression in several neuronal and non-neuronal injury models.
36,55
–57
Thus, we addressed whether ATF3 deficiency also impinges on the TBI-associated immune response. To accomplish this, we analyzed a potential upregulation of these immune genes at three days post TBI in the cortex (Fig. 4) or hippocampus (Supplementary Fig. 5) (see online supplementary material at
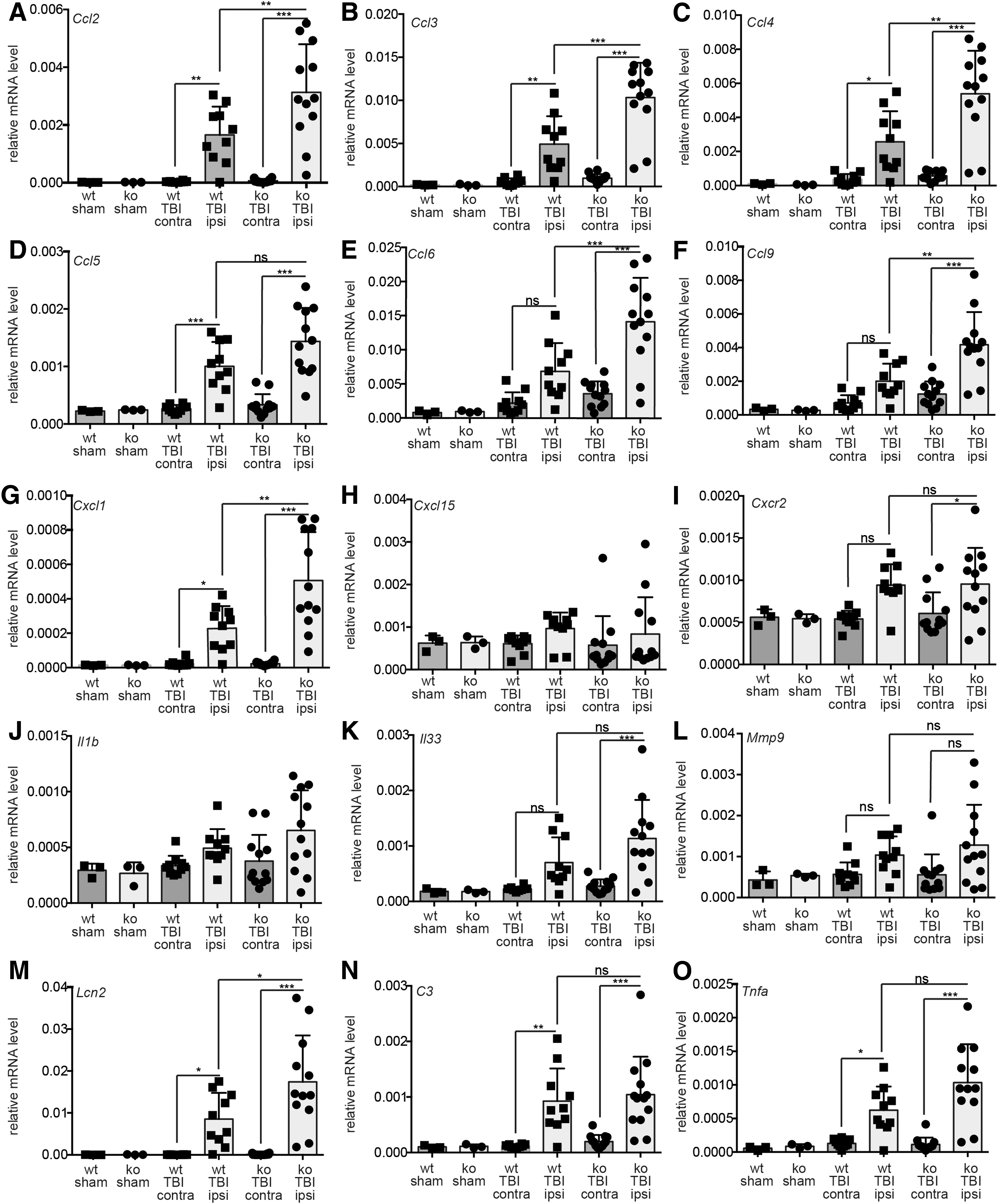
ATF3 ablation augments expression of inflammatory genes post-TBI. wt and Atf3 mutant (ko) animals were sham operated or treated with TBI. The ipsilateral cortex was targeted in TBI whereas the contralateral side serves as a further control. Three days post-TBI, cortical tissue was subjected to qPCR analysis for immune genes indicated. All six CCL family members, Ccl2 (
To start with, we tested several Ccl (Fig. 4A–F) and Cxcl (Fig. 4G–H) chemokine family members. In wt mice, Ccl2 (also named Mcp1; Fig. 4A), Ccl3 (Fig. 4B), Ccl4 (Fig. 4C), Ccl5 (Fig. 4D), Ccl6 (Fig. 4E), and Ccl9 (Fig. 4F), and Cxcl1 (Fig. 4G) were induced by TBI. Interestingly, this induction was, for almost all genes, significantly further augmented in ATF3-defcient mice (Fig. 4A–G). In fact, mRNA abundance of most of these chemokines was almost twice as high post-TBI as observed for wt littermates. Other chemokines, that is Cxcl15 (Fig. 4H), were not induced suggesting no general upregulation of all chemokines. Further, one receptor for these chemokines, Cxcr2, was upregulated by TBI, although with no difference between genotypes (Fig. 4I).
We also analyzed interleukin family members Il1b (Fig. 4J) and Il33 (Fig. 4K). Here, we noted a similar tendency as for chemokines. Interleukin mRNA levels were more strongly induced in Atf3 mutant compared to wt mice post-TBI, although not significant (Fig. 4J,K). Further, lipocalin-2 (Lcn2), a major proinflammatory factor,
58
was more strongly induced in ATF3-deficient mice than in wt mice (Fig. 4M). This result on mRNA abundance was also confirmed on the protein level (Supplementary Fig. 8) (see online supplementary material at
We noted enhanced hematoma formation upon ATF3 deletion at 3 days post-injury (Fig. 2). Hematoma size might be connected or be causally involved in enhancing immune gene induction in Atf3 mutant mice. To address this possibility, we correlated mRNA induction of several immune genes with hematoma size (Supplementary Fig. 9) (see online supplementary material at
Taken together, we observed enhanced expression of several immune-regulatory genes in ATF3-deficient mice post-TBI.
Elevated immune cell abundance in brain areas of traumatic brain injury–injured Atf3 mutant mice
Above, we observed enhanced transcript levels of several neuroinflammatory genes in Atf3 mutant brains (Fig. 4). Thus, as a consequence, a stronger recruitment of immune cells and a more pronounced glial reaction may take place in the injury site of ATF3-deficient compared to wt mice.
To address this experimentally, we histologically assessed the astroglial response and the immune cell infiltration in wt and Atf3 mutant mice at 3 days post-TBI (Fig. 5). Reactive astrocytes were identified by GFAP labeling, whereas inflammatory cells were identified by CD11b immunostaining (labeling both resting and activated microglia as well as leukocytes), CD45 (labeling activated microglia and other leukocytes), and Mac2-galectin3 (specific for macrophages; Fig. 5).
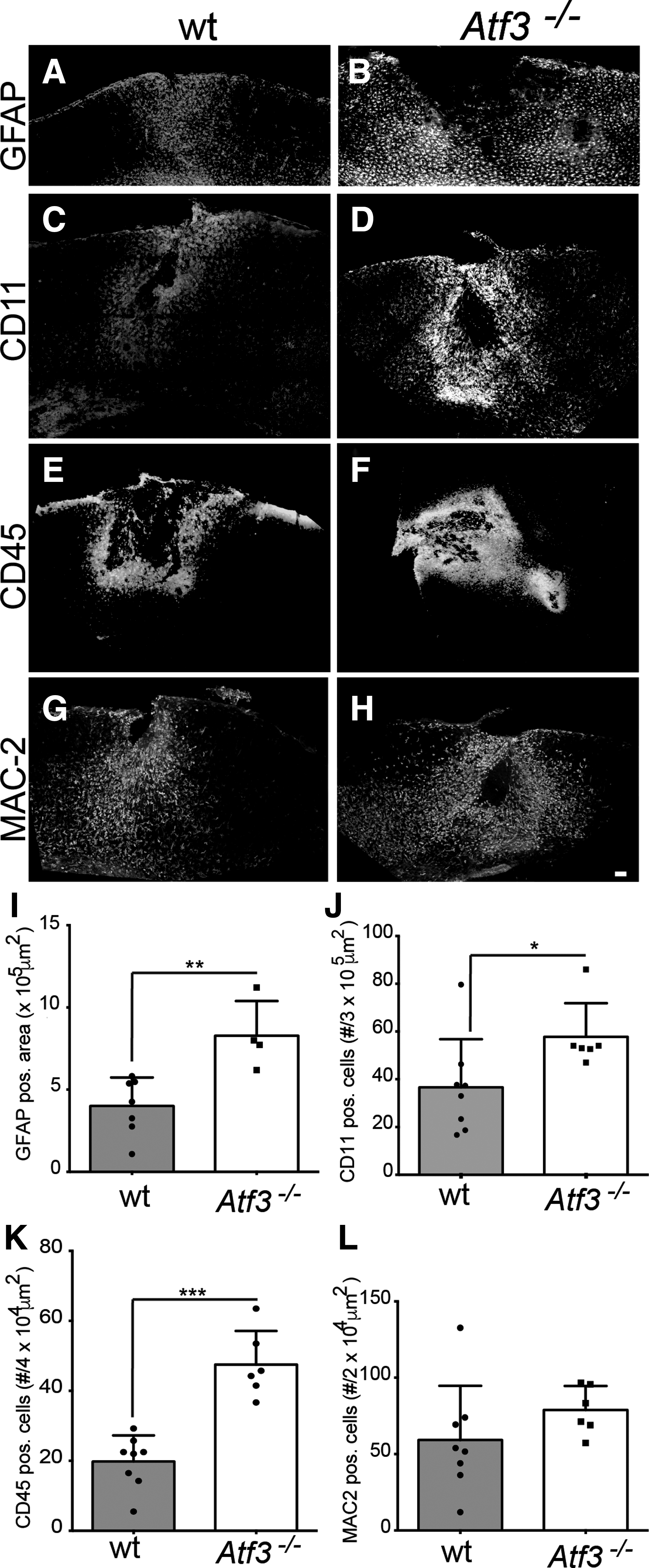
Enhanced abundance of immune cells post-TBI in ATF3-deficient mice. (
The area occupied by reactive astrocytes was significantly increased both in the cortex and hippocampus of the trauma ipsilateral side in wt animals at 3 days after TBI (Fig. 5A,I and data not shown) in comparison to the contralateral side. The area of reactive astrocytes was even further enhanced in Atf3 mutant brains; the overall area occupied by reactive astrocytes was significantly larger in the cortex of Atf3 mutant mice (Fig. 5B,I). We then analyzed the density of microglial cells and leukocytes. The density of CD11b+ cells in the cortical perilesional area was significantly larger in ATF3-deficient mice upon TBI (Fig. 5D,J) compared to wt mice (Fig. 5C,J). Likewise, recruitment of leukocytes and activated microglia, highlighted by the rich infiltrate of CD45+ cells (Fig. 5E,F,K) and macrophage-like cells (Fig. 5G,H,L) were more strongly present in the cortex of ATF3-deficient compared to wt mice. In comparison to the injured cortex, those peripheral immune cells were not present in the hippocampus that was largely spared by the TBI-caused lesion (data not shown).
We noted on brain sections that cortical lesion sites were generally larger in ATF3-deficient compared to wt mice (Supplementary Fig. 10) (see online supplementary material at
In summary, we observed pronounced presence of immune cells in brain areas of ATF3-deficient mice, in line with enhanced transcript levels of inflammation encoding genes in these mice.
Discussion
Comparison of the central nervous system versus peripheral nervous system injury regeneration-associated gene response and activating transcription factor 3's contribution
It was previously noted that RAG induction in CNS neurons is limited to individual RAGs and is generally weaker. This reduced potential of CNS neurons to propagate a RAG response may contribute to the limited regeneration potential of CNS in relation to PNS neurons. 13,17
In this study, we focused on 17 RAGs, including RAG encoding TFs and effector RAGs. Our results agree with the notion of a dampened RAG response in CNS compared to PNS neurons given that only four (Atf3, Sprr1a, Timp1, and Stat3) of the 17 RAG genes were robustly induced by more than a factor of 2 (Fig. 3; Table 1). Interestingly, for some effector RAGs encoding neuropeptides (Vip, Grp, and Adcyap1) and Gap43, we observed a downregulation post-TBI in wt mice (Fig. 3; Table 1). Here, it is worth mentioning that RAG levels are already low in the intact brain, making it difficult to observe further downregulation thereby impeding quantification on the protein level. Nevertheless, similar downregulations were shown for galanin 59 and Adcyap1. 60,61 Thus, in any case, in CNS injury there is a striking difference to PNS injury, which might, in part, be attributed to a loss of RAG-inducing neurons in the lesion site after TBI impact. This, for instance, is obvious when comparing RAG induction between cortical CNS neurons post-TBI and injured PNS neurons such as facial nerve motorneurons (Table 1). Facial nerve axotomy is—similar to sciatic nerve injury—a well-established model system of PNS injury resulting in a robust RAG response. 32,52 Thus, whereas RAGs in cortical TBI-injured neurons are induced 2-fold or less, many RAGs are induced 10-fold or more in de-afferented facial motor neurons (see Table 1).
Several of the RAG encoding TFs analyzed in this study are considered transcriptional hubs in a regeneration network, which individually or synergistically orchestrate the RAG response encompassing at least up to 500 genes in PNS injury. 14,16 However, during CNS injury, of the five RAGs encoding TFs (Atf3, p53, Stat3, Smad1, and Sox11), only Atf3 and, to a lower extent, Stat3 and p53 were induced by TBI in wt mice (Fig. 3; Table 1). This suggests no strong transcriptional activity of these hub TFs upon TBI with the exception of ATF3, whose activity is largely regulated by expression level. However, it should be kept in mind that some TFs, such as STAT3, are regulated by both transcriptional and post-translational mechanisms. In any case, the ATF3 abundance, alone or together with some increased STAT3 levels, was apparently not sufficient to induce the majority of RAGs tested in this study in a TBI model. Thus, insufficient abundance and/or activity of RAG encoding TFs might be one reason for an overall weaker RAG response in the CNS.
Activating transcription factor 3 as transcriptional regulator of neuroinflammation in central nervous system injury
Herein, we show, for the first time, a role of ATF3 in TBI-associated hematoma formation, immune gene regulation, and cellular neuroinflammatory response (Figs. 2, 4, and 5). We also analyzed male and female wt and Atf3 mutant animals separately (Supplementary Fig. 11) (see online supplementary material at
What might be the functional implication for limiting an injury-associated immune response? Currently, beneficial and detrimental roles of neuroinflammation are reported in several CNS injury models. Excess neuroinflammation might prevent neuronal regeneration and result in tissue fibrosis at the lesion site, whereas immune responses could be beneficial by removing cell debris at the injury site. From a cellular point of view, this might involve immune cells (e.g., microglia) switching between a proinflammatory M1 to an anti-inflammatory M2 phenotype,
71,72
although such strict phenotypic categorization is under dispute.
25,73
In such a scenario, a balanced neuroinflammatory response with initial activation and subsequent repression might support neuronal repair. Our data suggest that ATF3 upregulation in wt mice might restrict the proinflammatory immune response by repressing immune genes, thereby allowing for TBI-associated repair processes. In keeping with such a scenario is our finding that most genes upregulated in ATF3-deficient animals (Fig. 4) fall into the M1 proinflammatory group of genes (Tnfa, Il1b, Cxcl1, and Ccl2) and other CCL family members.
71,72
Similarly, lipocalin-2, also found to be upregulated upon ATF3 deficiency (Fig. 4, Supplementary Figs. 5, 7, and 8) (see online supplementary material at
Thus, our first analysis of ATF3 in a rodent TBI model shows a novel function of this TF in restricting the TBI-associated immune response and supports a general important function of ATF3 in immune responses in neuronal and non-neuronal injury models.
Footnotes
Acknowledgments
The work by A.I., B.K., and F.R. has been supported by the Deutsche Forschungsgemeinschaft (DFG) as part of the Collaborative Research Center 1149 “Danger Response, Disturbance Factors and Regenerative Potential after Acute Trauma.” F.R. is also supported by the ERANET-NEURON initiative “External Insults to the Nervous System” as part of the MICRONET consortium and by the Baustein Program of Ulm University. B.K. is supported by an Ulm University-Bundeswehr Krankenhaus Ulm research initiative and Paul und Marlene Hepp-Stiftung.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.