
Abstract
Excitotoxicity contributes to epileptogenesis after severe traumatic brain injury (sTBI). Demographic and clinical risk factors for post-traumatic seizures (PTS) have been identified, but genetic risk remains largely unknown. Thus, we investigated whether genetic variation in astroglial glutamate transporter genes is associated with accelerated epileptogenesis and PTS risk after sTBI. Adults (n = 267) 18–75 years old were assessed over a three-year period post-TBI. Single nucleotide polymorphisms (SNPs) throughout the SLC1A2 and SLC1A3 genes were assayed. Kaplan-Meier estimates and log-rank statistics were used to compare seizure frequencies by genotype. Multivariate Cox proportional hazards regression was used to estimate hazard ratios (HRs) for genotypes significant in Kaplan-Meier analyses. Thirty-nine tagging SNPs were examined (SLC1A2: n = 21, SLC1A3: n = 18). PTS developed in 57 (21.4%) individuals. Of those with PTS, n = 20 (35.7%) had an immediate/early seizure within the first seven days, and n = 36 (64.3%) had a late seizure occurring between eight days and three years post-TBI. When adjusting for multiple comparisons, rs4869682 genotypes (SLC1A3, GG vs. T-carriers) were associated with time to first seizure (p = 0.003). Median time until first seizure was 20.4 days for individuals with a GG genotype and 44.8 days for T-carriers. After adjusting for covariates, rs4869682 GG-homozygotes had a 2.05 times increased PTS risk versus T-carriers (aHR = 2.08, 95% confidence interval: 1.20, 3.62, p = 0.009). Variation within SLC1A3 is associated with accelerated epileptogenesis and clinical PTS development after sTBI. Future studies should validate these findings and examine how genetic variation at rs4869682 may be a target for PTS prevention and treatment.
Introduction
Traumatic brain injury (TBI) is a significant public health problem that decreases life expectancy by an average of nine years. 1 More than 2.5 million TBIs occur annually in the United States, and an estimated 3.2 million–5.3 million Americans live with a disability as a result of TBI. As such, TBI is beginning to be recognized as a complex disease process with a wide array of resulting chronic health conditions, including secondary neurological disorders such as post-traumatic seizures (PTS). 2 Individuals with TBI have a 50-times increased risk for dying from a seizure compared with healthy age-, sex-, and race-matched controls. 1 There is also evidence that seizures are associated with other commonly occurring secondary TBI complications, such as depression and anxiety. 3
The PTS can be classified by temporal onset: immediate seizures (<24 h), early seizures (1–7 days), and late seizures (>7 days post-injury). 4 These temporal distinctions are thought to represent differences in pathophysiology and seizure reoccurrence risk. 5 Demographic and clinical factors, such as subdural hematoma (SDH) and craniectomy, can increase risk for incident PTS; however, there remains ∼25% of unexplained variance in contemporary prognostic models. 6 Among a cohort of individuals with severe TBI (sTBI), we have identified biological and genetic factors that contribute to increased PTS risk, including variation in adenosine regulatory genes, 7 interleukin (IL)-1b genes, 8 gamma-aminobutyric acid genes, 9 and neuronal glutamate transport genes. 10
Glutamate is the main excitatory neurotransmitter in the mammalian brain. Glutamate is released into extracellular spaces immediately after a TBI, and these levels can remain elevated for at least one week post-injury. 11 This excitotoxic exposure leads to increased ion channel activation and calcium influx, resulting in cellular injury or death. 5 We have shown that the controlled cortical impact (CCI) model of experimental TBI results in regional reductions in glutamate excitatory amino acid transporter (EAAT) expression that may contribute to this pathology. 12 Together, this work suggests that excitotoxicity, and associated reduced glutamate transporter expression, could influence PTS risk.
There are five glutamate transporters in the human central nervous system (CNS), including two astroglial and two neuronal transporters. Previously, we investigated genetic variations in neuronal glutamate transporters: EAAT 3 and 4, encoded by the genes SLC1A1 and SLC1A6, respectively. We identified two single nucleotide polymorphisms (SNPs) in gene SLC1A1 associated with increased PTS risk. 10 Similarly, astroglial glutamate transporters EAAT1 (GLAST), and EAAT2 (GLT-1) are encoded by genes SLC1A3 and SLC1A2, and both are found throughout the brain. EAAT1 is expressed in the cerebellum, hippocampus, and cortex in higher mammalian species; EAAT2 is expressed predominantly in the hippocampus and motor cortex. 13
EAAT1 and EAAT2 are responsible primarily for extracellular glutamate uptake in most brain regions. Previous studies involving GLT-1 (EAAT2) knockout mice show increased susceptibility to cerebral edema after cold-induced injury and increased lethal seizure activity. 14 Only astroglial GLT-1 (EAAT2), and not neuronal GLT-1 (EAAT2), loss was associated with increased death, weight gain, and seizure activity. 15 Interestingly, identified human mutations in the EAAT1 gene (SLC1A3) can result in reduced glutamate uptake and contribute to both ataxia and seizures in clinical populations. 16
Because astroglial glutamate transporters EAAT 1 and 2 are the major CNS proteins by which to regulate extracellular glutamate levels and excitatory neurotransmission, and gene deletion and mutation can be associated variably with seizure activity, we investigated specific genetic variations and PTS development. For this study, we identified genetic markers of epileptogenesis within the astroglial glutamate management pathway that may have significant prognostic and precision care value in preventing PTS throughout a patient's rehabilitation and recovery.
Methods
Study design and population
Participants were recruited as a part of a larger study examining genetic associations with TBI outcomes, some of whom have been included in previous candidate gene associated studies evaluating PTE. 7,8,10 As presented in Figure 1, patients aged 18–75 from consecutive admissions to a level 1 trauma center with sTBI (Glasgow Coma Scale [GCS] score ≤8), positive head computed tomography findings, and requiring extraventricular drainage catheter placement for intracranial pressure management were screened for participation. Exclusion criteria included penetrating head injury, prolonged cardiac or respiratory arrest before admission, or inability to obtain legal proxy consent.
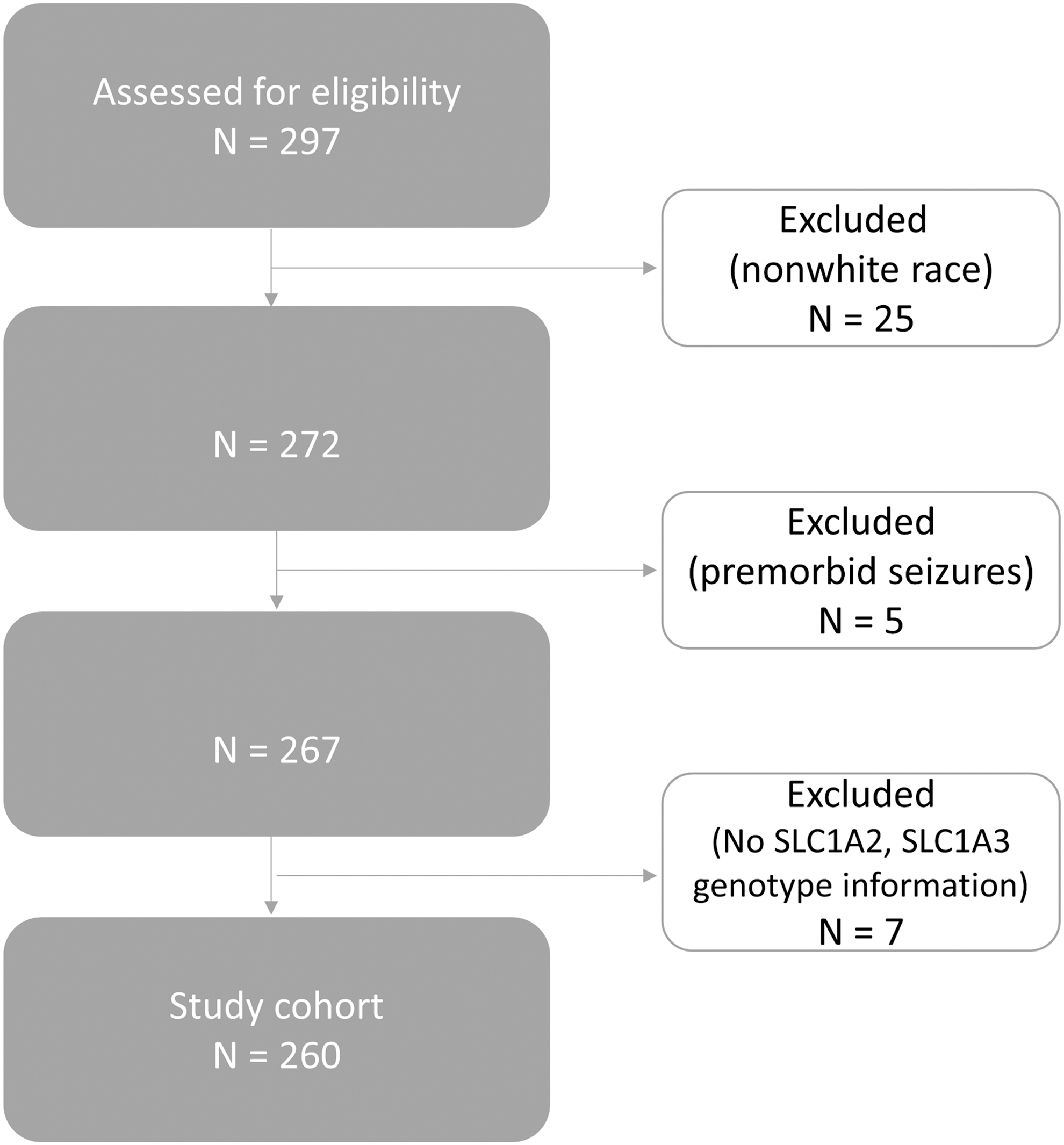
Consort diagram representing the cohort used for analysis.
Analyses were limited to individuals categorized as White by self/proxy-report (n = 25 participants excluded) because of racial differences in allelic frequency obtained from the database of Single Nucleotide Polymorphisms (dbSNP:
Critical care management
Participants were admitted to the neurotrauma intensive care unit at our level 1 trauma center and received treatment consistent with the Guidelines for the Management of Severe Head Injury. 17 Standard care consisted of early extraventricular drain (EVD) placement and central venous and arterial catheters. Further surgical intervention was pursued when clinically indicated. Intermittent electroencephalograms were obtained by treating physicians as part of standard-of-care when there was clinical concern for non-convulsive seizures. Most participants received anti-epileptic drugs (AEDs) for the first week post-injury.
Demographic and injury related data
Demographic and injury data were documented at enrollment. Intracranial pathology type was assigned one of seven distinct categories based on radiologic findings using the International Classification of Diseases, Ninth Edition. These categories were divided by injury type (present or absent) and were not mutually exclusive. Admission GCS score was used to establish study eligibility; however, the best GCS score during the first 24 h after admission was used as a covariate in analyses. Injury Severity score (without injury to head and neck) was obtained from medical records to determine overall body injury measure of extracerebral trauma, based on survivability of injuries within and across body regions. The electronic medical records (EMR) were reviewed to identify AED use for early seizure prophylaxis during acute care.
Genetics protocol
Deoxyribonucleic acid (DNA) was extracted either from cerebrospinal fluid (CSF), collected via passive drainage, using a QIAamp DNA extraction protocol (Qiagen) or from whole blood using a salting out procedure. DNA samples were genotyped using iPLEX Gold SNP Assay (Sequenom). Tagging single nucleotide polymorphisms (SNPs) for SLC1A2 (n = 21) and SLC1A3 (n = 18) were evaluated based on data available from the National Center for Biotechnology Information, HapMap Build 36. The major and minor allele designations, as well as the minor allele frequency, are presented in Supplementary Table 1 (see online supplementary material at

Haploview generated gene map displaying linkage disequilibrium (D′) for single nucleotide polymorphisms (SNPs) located on SLC1A2 (
Clinical and Demographic Characteristics by Seizure * Category
SE, standard error; GCS, Glasgow Coma Scale; IQR, interquartile range; CT, computed tomography; SDH, subdural hematoma; SAH, subarachnoid hemorrhage; DAI, diffuse axonal injury; EDH, epidural hematoma; IVH, intraventricular hemorrhage; ICH, intracerebral hemorrhage; MVA, motor vehicle accident.
Row totals are reported for categorical variables.
Seizure categories are for time until first seizure; groups are mutually exclusive.
indicates statistical significance at p < 0.05.
Outcome measure: PTS
The primary outcome of interest was time to first seizure after TBI. We reviewed electronic inpatient and outpatient medical records available from our medical center to obtain PTS status. Date of first seizure was recorded based on ambulance and/or emergency department report, inpatient progress or nursing note, electroencephalography (EEG) report, patient history, or discharge and transfer summaries. Medical record notation referring to convulsions, seizures, status epilepticus, or seizure disorder was considered evidence of seizure occurrence. Notation of possible seizure activity that was either ambiguous or non-conclusive was categorized as no PTS. Date of death was obtained from medical records or from social security death data (
Post hoc analysis compared genetic associations with immediate/early PTS versus late PTS (i.e., post-traumatic epilepsy [PTE]). Late PTS was left censored at seven days post-injury to compare with immediate/early PTS. For this post hoc analysis, participants who had a seizure (n = 19) or died (n = 36) during the first week post-injury were also excluded, leaving N = 212 for analysis.
Statistical analysis
SAS-9.4 (Cary, NC) and R-3.0.2 were used to complete analyses. All genotyped participants who met eligibility criteria were included. Demographic and clinical characteristics were compared between individuals with and without PTS using chi-square and Kruskal-Wallis tests when appropriate. Descriptive statistics are presented in three groups: no seizure, immediate/early seizure (within the first seven days post-injury), and late seizure (beginning after seven days post-injury). Among individuals with PTS, chi-square analyses were conducted, using the Fisher exact test as appropriate, to determine whether genotype frequencies differed by time of first seizure (i.e., early and late).
To test the primary hypothesis regarding genetic variation and post-TBI epileptogenesis over a three-year period, time-to-event analyses were performed. Because of LD, or correlation among selected SNPs, the number of effective number of tests conducted was smaller than the number of SNPs screened. The minimum number of effective tests (Meff) was calculated using methods based on eigenvalues. 19 The Meff was calculated for each gene and generated a Meff of 9.9 for SLC1A2 and 6.7 for SLC1A3. A Bonferroni correction was then applied to the original α = 0.05 using the Meff as the number of independent tests for each gene for subsequent time-to-event analyses. This adjustment corresponded to a threshold for significance of α = 0.005 and 0.007 for SLC1A2 and SLC1A3, respectively.
On visual inspection of the data, there was a dominant pattern of seizure incidence for minor allele carriers; therefore, genotypes were grouped by minor allele carriers versus major allele homozygotes. All bivariate and multi-variate analyses are presented with significant SNPs classified by minor allele carrier status.
Seizure rates were estimated at three years post-injury, considering the full follow-up period (i.e., time of injury through three years post-TBI), for individual SNPs by minor-allele carrier status using Kaplan-Meier curves. Log-rank statistics were used to compare rates. Cox proportional hazards regression was used to estimate hazard ratios (HRs) for minor-allele grouped SNPs that had significant Kaplan-Meier estimated PTS rates based on Bonferroni corrected p values. Cox regression models were adjusted for demographic and injury characteristics that differed significantly by seizure status (no seizure, immediate/early, and late seizure) or that have been shown previously to impact PTS incidence. Proportionality over time assumptions were assessed for all variables by examining correlations of Schoenfeld residuals with time for all included independent variables in the Cox regression model. If a given covariate did not pass the proportionality assumption, it was stratified in the Cox regression model. Time-to-event analyses were repeated for post hoc analysis evaluating risk for PTE.
Results
Study population
There were N = 260 individuals who met all inclusion criteria and were genotyped for both SLC1A2 and SLC1A3. In Table 1, clinical and demographic characteristics of the cohort are presented and stratified by seizure status. Similar to other sTBI studies, the majority of our study population were men (80.2%), and the average age was 35.3 (standard error [SE] = 0.9) years old. The median best in 24 h GCS score for the total cohort was 6 (interquartile range [IQR] 4–7). The average hospital length of stay of the entire cohort was 23.6 days (SE = 0.8). Approximately 95.5% were receiving some form of acute seizure prophylaxis during their acute hospital stay, based on EMR review. Also, 37.6% of the cohort had EEG information recorded during their acute hospital stay in the EMR.
PTS developed in 56 (21.5%) individuals. Of those with a seizure, n = 20 (35.7%) had an immediate/early seizure within the first seven days, and n = 36 (64.3%) had a late seizure occurring between eight days and three years post-TBI. Depressed skull fracture and SDH occurred more frequently among individuals with PTS compared with those without PTS.
Evaluation of tagging SNPs and PTS risk
Tagging SNPs for SLC1A3 and SLC1A2, grouped by minor allele carrier status, were independently evaluated for associations with time to seizure using Kaplan Meier models (Table 2). The data indicate that multiple SNPs located within SLC1A2 and SCL1A3 were associated with time to PTS. On SLC1A2, rs1570226 (TT vs. G-carriers) was nominally (log-rank p = 0.021) associated with time to PTS when comparing Kaplan Meier Curves. On SLC1A3, rs1049522 (AA vs. C-carriers) was nominally associated with time to PTS (log-rank p = 0.027), while rs4869682 (SLC1A3, GG vs. T-carriers) met threshold criteria for statistical significance using the log-rank test and when adjusting for multiple comparisons (p = 0.003). PTS incidence for GG homozygotes was 29.7% versus 16.9% for T-carriers. Kaplan-Meier curve analysis shows that GG homozygotes had a median time to first seizure of 20.40 days (IQR 0.92–415.2 days), while T-carriers had a median time to first seizure of 44.8 days (IQR 1.74–274.98 days) (Fig. 3A).
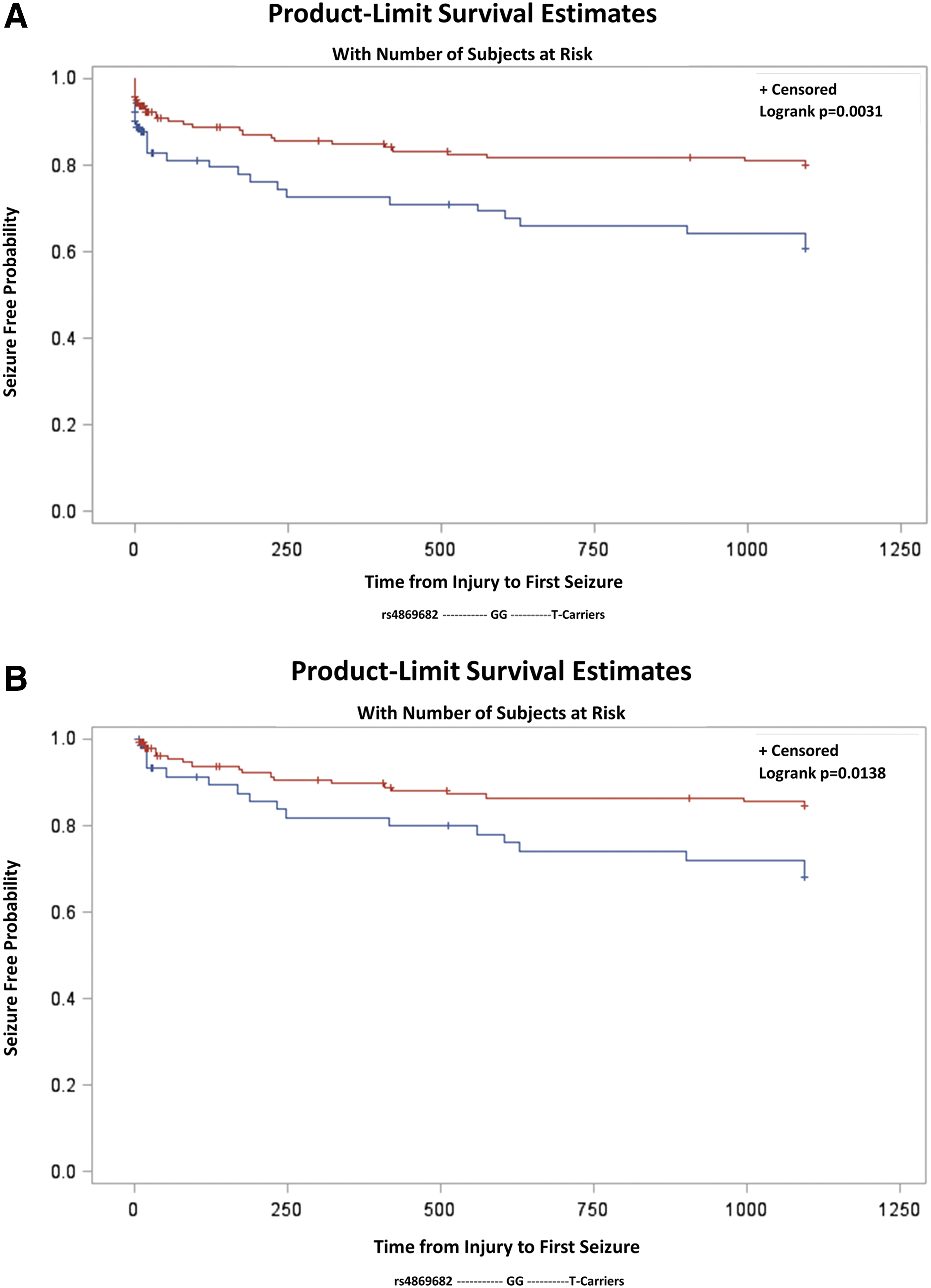
Kaplan-Meier curve estimates for time to first seizure by SLC1A3 single nucleotide polymorphism rs4869682 grouped genotype for follow-up beginning day 0 post-injury (
P-values for SLC 1 A 2 and SLC 1 A 3 Bivariate Associations with Post-Traumatic Seizure Grouped by Minor Allele Carrier Status
Bold represents statistical significance after multiple comparisons correction.
Italic represents nominal statistical significance.
SNP, single nucleotide polymorphisms; KM, Kaplan-Meier.
In Table 3A, the unadjusted and adjusted Cox regression models are presented for rs4869682 genotype, GG homozygotes versus T-carriers. Proportionality over time assumptions were assessed for all covariates, and it was determined that only SDH violated the assumption. Therefore, all adjusted Cox regression models were stratified by SDH. In an unadjusted model, GG homozygotes were at a 2.17 times increased risk for PTS compared with T-carriers (HR = 2.17, 95% confidence interval [Cl]: 1.28, 3.69, p = 0.004). After adjustment for age, best in 24 h GCS, and depressed skull fracture, and stratification by SDH, rs4869682 GG homozygotes had a 2.08 times increased PTS risk compared with T-carriers (aHR = 2.08, 95% Cl: 1.20, 3.62, p = 0.009). No other SNPs were significantly associated with PTS after multiple comparisons adjustment.
Results for Unadjusted and Adjusted Cox Proportional Hazards Regression Models for rs4869682 in SLC 1 A 3 for All Post-Traumatic Seizures
CI, confidence interval.
Indicates statistical significance at p < 0.05.
Adjusted for age, best in 24 h Glasgow Coma Scale, and depressed skull fracture; stratified by subdural hematoma.
PTE genotype associations: a post hoc analysis
Among individuals with PTS, there were no differences in rs4869682 genotype frequencies between individuals whose first seizure occurred within seven days post-injury versus those occurring day eight to three years post-injury. In the group with immediate/early seizures, genotype frequencies were 50%, 25%, and 25% for GG, GT, and TT, respectively. For the group with late seizures, the genotype frequencies were 48.6%, 40.0%, and 11.4%, respectively.
When excluding individuals who had a seizure or died during the first week after TBI, (N = 204) (Table 3B), PTE risk significantly differed between GG homozygotes and T-carriers (log-rank p = 0.014, data not shown). Kaplan-Meier curve analysis shows time to first seizure among GG homozygotes and T-carriers (Fig. 3B). Further, after adjustment for age, GCS, and depressed skull fracture, and stratification by SDH, GG homozygotes were at a 2.06 times increased risk for late PTS compared with T-carriers (aHR = 2.06, 95% Cl: 1.04, 4.08, p = 0.039).
Results for Unadjusted and Adjusted Cox Proportional Hazards Regression Models for rs4869682 in SLC 1 A 3 for Late Post-Traumatic Seizures *
CI, confidence interval.
Left censor individuals who died or seized in first seven days.
Indicates statistical significance at p < 0.05.
Adjusted for age, best in 24 h Glasgow Coma Scale, and depressed skull fracture; stratified by subdural hematoma.
Discussion
Glutamate is the main excitatory neurotransmitter in the mammalian brain, and its extracellular regulation is critical for maintaining homeostatic CNS function. The major glutamate clearance mechanism in the healthy brain is reuptake via astroglial glutamate transporters. 20 After a TBI, there is decreased astroglial glutamate transporter expression, possibly because of both the downregulation and degeneration of astroglial cells. 21 This injury response may cause the observed acute increase in extracellular glutamate, 11 leading to excitotoxicity and possibly facilitating epileptogenesis. Because variations in astroglial glutamate transporter genes could contribute to increased seizure risk after a TBI, we studied the time to first seizure among individuals after sTBI in relationship to SNPs within the SLC1A2 and SLC1A3 genes. We found that genetic variation in SLC1A3, and not SLC1A2, was significantly associated with time to first seizure. Homozygous (GG) individuals for the SLC1A3 SNP rs4869682 had a 2.08 increase risk of PTS versus T-carriers.
SLC1A2 and SLC1A3 encode excitatory amino acid transporters (EAAT) 2 and 1, respectively. EAAT1 is a major glutamate transporter found throughout the brain, with regional variability among species. Early studies involving rodent models show EAAT1 most heavily localized to the hindbrain and cerebellum. 22 In higher mammalian species, EAAT1 is concentrated in the cerebellum, hippocampus, and cortex. 13 This distribution correlates with our finding that SLC1A3 (which encodes EAAT1) is associated with PTS risk, because the pathogenesis of seizures is often localized to the cortex, including entorhinal cortex, and hippocampus. EAAT1 can have a larger distribution in a more varied cell type population in the human post-mortem brain. 23
EAAT1 is located primarily on astroglial cells, although it has also been described on neuronal processes. 23 It is the major mechanism for terminating excitatory transmission in the CNS 24 and is also involved in recycling of neuronal vesicular release via the glutamate-glutamine cycle. 22 In fact, Roberts and associates 23 describe neuronal EAAT1 protein expression, in the context of hypoxia, among those with severe TBI.
Our own experimental TBI work in the CCI injury model suggests both GLAST (human homologue EAAT1) and GLT-1 (human homologue EAAT2) expression in the frontal cortex are reduced up to 20 days post-injury. 12 There is a greater decrease in GLAST (human homologue EAAT1) compared with other glutamate transporters like GLT-1 (human homologue EAAT2) after cortical impact injury in the rat. GLAST significantly decreases throughout the brain within 15 min and up to two days after injury. 25 Also, there is conflicting research involving EAAT1 expression on microglia as a compensatory mechanism after injury or infection. Some rat 25 and human 26 studies have found that de novo EAAT1 expression in microglia reduces glutamate levels. In a TBI study involving human post-mortem brains, EAAT1 expression occurred in ramified microglia at earlier stages after TBI, and an increase in EAAT1 occurred among reactive astrocytes in later stages. 27 When taken with our clinical findings, these data suggest that genetic variation within the SLC1A3 temporally impacts cell-specific effects of EAAT1. Also, this body of work lends support for SLC1A3 as a candidate gene whose innate expression and function may be differentially impacted by TBI to independently influence PTS risk.
SLC1A3 encodes EAAT1, and is located on human chromosome 5p13, 28 and has a coding region 77,846 bp in length. rs4869682 is located in an intronic region of SLC1A3, and there are no known functional SNPs within the DNA block represented by this SNP. Because of low numbers of non-White individuals in our cohort, our analysis only includes self-reported White individuals. Allelic frequencies for this SNP are highly dependent on ancestry, with MAF reported to be Europeans: 46%; Asians: 26%; and Africans 72%, supporting the need for larger future studies involving a more diverse cohort and using a stratified analysis approach.
While no known specific functional SNPs are in significant LD with rs4869682, a multi-genetic model including the SNP rs4869682 has been used to predict seizure control and drug outcomes in studies focused on individuals with epilepsy in the general population. One study examined the hypothesis that a multi-SNP model would produce better classification of treatment outcomes than models using single SNPs. This study assessed 4041 SNPs in 115 individuals newly treated for epilepsy and prospectively followed them for one year to determine their outcome with respect to seizure control. A multi-SNP model including rs4869682 had a predictive accuracy of at least 80%, and a validation study using an independent cohort showed similar results. 29,30 These findings, along with our study results, support further work on if/how this SNP may be predictive of AED effectiveness after TBI for PTS prevention and effective PTS management.
Altered glutamate signaling and SLC1A3 genetic variations have been associated with other conditions, notably major depressive disorder. 31 The downregulation of SLC1A2 and SLC1A3 expression in the hippocampus, along with decreased expression of membrane glutamate transporters, may contribute to the development of major depressive disorder, 31 schizophrenia, 32 neurodegenerative diseases such as Alzheimer disease, 33 and possibly Tourette syndrome. 34 These shared biological relationships may contribute, in part, to known reciprocal relationships between epilepsy risk and mental health disorders. 3,6 When this literature is taken together with our findings, variation in EAAT1 may merit assessment of its association with mental health outcomes such as post-traumatic depression, particularly among those with PTE. 3
We also studied the time to first seizure in individuals after sTBI focusing on SNPs within the gene SLC1A2, which encodes EAAT2. EAAT2 is the main glutamate transporter of the forebrain 22 and is highly expressed in the motor cortex and hippocampus. 13 In rodent studies, EAAT2 was found to be the glutamate transporter most heavily expressed in the brain. 14,35 We did not find variability in the gene encoding EAAT2 (SLC1A2) to be associated with PTS despite its apparent robust expression, however. This negative finding could be because of the differences in the rodent and human brain, but could be an area of continued future investigation as new SNPs of interest on this gene are identified and reported in the literature.
Our findings provide clinical relevance and show potential translational value to understanding personal biology associated with PTS risk. The drug levetiracetam has been shown to improve cognitive outcomes in rodent TBI models, 36 while other experimental TBI studies have found this drug to decrease non-convulsive seizure frequency and duration, as well as delay time to first seizure. 37 We have shown that daily treatment, 12 but not abbreviated acute treatment, 38 with levetiracetam is effective in reducing inflammation and normalizing regional glutamate transporter expression in an experimental model of TBI. Levetiracetam acts in part by inhibiting synaptic vesicle protein 2A 39 and selectively inhibiting calcium channels, 40 as well as by upregulating astroglial glutamate transporter (EAAT1 and EAAT2) expression. 12
Levetiracetam is generally considered safe and well-tolerated in humans with TBI 41 and is one of many AEDs used in seizure prophylaxis and PTE treatment; similar data exist for craniotomy patients undergoing limited treatment after operation. 42 The recent Guidelines for Treatment of Severe TBI 17 however, suggest there is insufficient evidence to support a recommendation for or against the use of levetiracetam for early seizure prophylaxis among patients with TBI. Our findings of variable seizure risk among populations with genetic variations of SLC1A3 could, therefore, be relevant clinically when considering short/longer term levetiracetam treatment with TBI.
Some reports suggest that levetiracetam is well tolerated generally, yet adverse side effects, such as irritability and aggression, have been noted with ongoing levetiracetam therapy, particularly in the pediatric/young adult populations and those who may be neurogenetically vulnerable to these symptoms. 17,42 –47 Somnolence has been reported among older adults with ongoing levetiracetam use and concurrent dementia. 45 Thus, there is a need for well-conducted clinical trials assessing its efficacy, side effects, and safety with use beyond the typical prophylactic window in sTBI populations. Proactive treatment with levetiracetam in patients may pharmacologically modulate EAAT1 expression and treatment efficacy in these individuals, but may be associated with variable adverse event profiles after TBI. Other genetic variants associated with PTE 7 –10,48 and cognition/behavior after TBI 49 –52 may also warrant further study regarding efficacy and adverse event susceptibility.
Seizure status and time to first seizure may have been misclassified because of missing data involving healthcare provided outside our hospital system. Studies suggest that presentation to acute care is commonplace for both provoked and unprovoked seizures, 53,54 medical care that can then be captured through our EMR. Nonetheless, another limitation of this study is the passive nature of seizure follow-up from the EMR at our health system. Our health system is the predominant healthcare provider in the region, and our cumulative incidence does not differ much from the cumulative incident rate at five years post-injury in a recent multi-center prospective cohort of individuals with moderate to severe TBI. 55 It is possible, however, that PTS was underreported if a patient did not seek care for a seizure, or presented at a hospital outside of our health network.
Our results suggest that there is a SLC1A3 genetic contribution to PTS risk after TBI. Our results, however, included only individuals with sTBI, and our findings may not generalize to those with less severe injuries. To minimize racial and ancestral differences in allelic frequency, we limited our analyses to individuals who self-reported their race as White; therefore, other research must be performed to study allelic frequencies among other racial groups with TBI. Follow-up studies are also needed to replicate our findings. Continuing to increase our knowledge of genetic variants like SLC1A3 affecting PTS development may improve prognostic seizure models that currently contain only demographic and clinical risk factors. 6 To support and validate this endeavor, future work should also include the development of multi-SNP gene risk scores, to assess the cumulative impact of previous SNPs associated with PTE in existing and independent populations.
Footnotes
Acknowledgments
This work was supported by National Institutes of Health (NIH) R01 NR013342, DOD W81XWH-071-0701, NIH R01 HD048162, NIDILRR 90DP0041, and the CURE Foundation. Thanks to the subjects and their families for their generous participation. Thanks to the UPMC Trauma Registry for assisting with some elements of data collection.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.