
Abstract
The antifibrinolytic agent, tranexamic acid (TXA), an inhibitor of plasmin formation, currently is evaluated to reduce bleeding in various conditions, including traumatic brain injury (TBI). Because plasmin is implicated in inflammation and immunity, we investigated the effects of plasmin inhibition on the immune response after TBI in the presence or absence of induced pneumonia. Wild-type mice treated with vehicle or TXA or mice deficient in plasminogen (plg-/-) underwent TBI using the controlled cortical impact model. Mice were then subjected to Staphylococcus aureus induced pneumonia and the degree of immune competence determined. Significant baseline changes in the innate immune cell profile were seen in plg-/- mice with increases in spleen weight and white blood cell counts, and elevation in plasma interleukin-6 levels. The plg-/- mice subjected to TBI displayed no additional changes in these parameters at the 72 h or one week time point post-TBI. The plg-/- mice subjected to TBI did not exhibit any further increase in susceptibility to endogenous infection. Pneumonia was induced by intratracheal instillation of S. aureus. The TBI did not worsen pneumonia symptoms or delay recovery in plg-/- mice. Similarly, in wild type mice, treatment with TXA did not impact on the ability of mice to counteract pneumonia after TBI. Administration of TXA after TBI and subsequent pneumonia, however, altered the number and surface marker expression of several myeloid and lymphoid cell populations, consistent with enhanced immune activation at the 72 h time point. This investigation confirms the immune-modulatory properties of TXA, thereby highlighting its effects unrelated to inhibition of fibrinolysis.
Introduction
Traumatic brain injury (TBI) is the main cause of death in adults younger than 45 years. 1 Even after surviving the first impact, patients are at risk of life-threatening complications such as coagulopathy, leading to enhanced or prolonged bleeding 2 and immunosuppression, resulting in an increased risk of infection. 3 The antifibrinolytic agent tranexamic acid (TXA) recently has been demonstrated to improve survival in bleeding trauma patients if administered within 3 h after injury in the Clinical Randomisation of an Antifibrinolytic in Significant Haemorrhage (CRASH) 2 trial. 4,5 A nested trial within CRASH-2 on intracranial hemorrhage, however, was not conclusive, 6 and large randomized controlled trials are currently assessing TXA treatment in this indication. 7,8 As a lysine analogue, TXA inhibits plasmin formation and subsequent clot dissolution, thereby reducing blood loss. 9
In addition to degrading fibrin, however, plasmin has also been well characterized as a potent modulator of inflammation and immunity. 10,11 Moreover, various pathogens harness the plasminogen activation system to enhance their virulence. 12 Previously we have reported an immune-dampening effect of plasmin via binding to dendritic cells (DC) and reducing their ability to mount an allogeneic immune response. 13
Hence, the observed benefit of TXA treatment in trauma might not only be a result of reduced blood loss, but also an improved immune response against invading pathogens. This might be of particular relevance for brain injury patients who are not only at increased risk of infections associated with ventilators, lines, and other medical devices, but also susceptible to development of a central nervous system (CNS) injury-induced immune deficiency syndrome (CIDS) because of activation of the hypothalamus-pituitary-adrenal axis and hyperactivation of the sympathetic nervous system resulting in release of immunosuppressive glucocorticoids and catecholamines. 14 In addition, a translocation of bacteria from the intestinal tract into the lungs has been described in other models of CNS injury. 15
We hypothesized that plasmin contributes to CIDS in the setting of experimental TBI, and, hence, plasminogen deficiency or TXA treatment would improve immune function and promote bacterial clearance in a model of post-TBI pneumonia.
Methods
Animals
Male C57BL/6 wild type mice at 8 weeks of age and 20–24 g weight were obtained from the Animal Research Council. Sex- and age-matched plasminogen-wild type (plg+/+) and plasminogen-deficient (plg-/-) mice were obtained from a heterozygote breeding colony maintained at AMREP Animal Service and the Monash Animal Research Platform. All experiments were approved by the AMREP animal ethics committee, and mice were kept under pathogen-free conditions with uninterrupted access to food and water.
Controlled cortical impact (CCI) model of TBI
The CCI model induced TBI. This procedure is well established in our group and induces a reproducible brain trauma with low mortality. 16,17
After anesthesia with avertin, 0.5 g/kg, injected intraperitoneally (IP), mice were placed in a stereotaxic frame. An incision was made in the midline of the head to expose the skull, followed by a 5 mm diameter craniotomy over the left parietal cortex. A single blunt force trauma was inflicted to the exposed brain area with an impact depth of 2 mm, 5 m/sec velocity, and a dwell time of 400 msec inducing a moderate to severe brain trauma. The exposed brain was then sealed using bone wax, and the skin incision was sutured and treated with a local anesthetic and an antiseptic.
For the sham procedure, only anesthesia and the scalp incision, without craniotomy and TBI, was performed. A sham group including craniotomy was used only for the experiments on endogenous infection (see below) to rule out contamination during the TBI surgery as a contributor to detected bacterial growth.
Drug administration
The TXA (Cyklokapron, Pfizer, New York, NY) (100 mg/kg) or vehicle (0.9% sodium chloride) in an equivalent volume was administered to mice intravenously (IV) 20 min after induction of TBI or sham procedure. After 6 h, mice were injected IP with TXA or vehicle. On days 2 and 3, mice were again injected with TXA IP twice daily (8 h apart).
The 100 mg/kg concentration of TXA selected has been used in cardiac surgery, although the dosage is sometimes reduced to 50 mg/kg to reduce seizure rates. 18 Also, TXA administration 20 min after the injury reflects usual clinical practice and is an achievable practical scenario. The TXA is also being used currently in the pre-hospital setting in recent clinical trials. 19
Assessment of endogenous infection
To assess the rate and extent of endogenous infection in our mouse model, we plated the lysate from the right lung, liver, and mesenteric lymph node of each mouse (all 100 mg/mL, ground through a cell strainer in sterile phosphate buffered saline (PBS), pH 7.4) on Heart Infusion (HI) agar plates (3.7% HI broth, 0.5% yeast extract (both Oxoid), 1.5% granulated agar (Difco), 0.1% L-cysteine, 0.375% glucose) to evaluate bacterial growth, which was measured as counts of colony forming units per gram of tissue (CFU/g).
The TBI mice were compared with both a sham group where only the skin incision was performed and another sham group, in which mice were craniotomized, to account for any false positives from the potential exposure to bacterial contamination during the invasive TBI surgery. To assess the presence of both aerobic and anaerobic bacterial species, each sample was plated in duplicate onto HIS agar and incubated at 37°C for 48 h either aerobically or anaerobically (10% H2, 10% CO2 and 80% N2) in an anaerobic chamber (Coy Laboratory Products, Inc., Grass Lake, MI).
Preparation of Staphylococcus aureus for in vivo infections
The S. aureus strain ATCC25923 (American Type Culture Collection [ATCC]) was cultured in HI broth or on HI agar (3.7% HI broth [both Oxoid, Lenexa, KS] and 1.5% granulated agar [Difco]). Bacterial cultures for infections were grown in 20 mL HI broths at 37°C overnight. To obtain ∼1 × 108 CFU/mL for the infections, the overnight cultures were then used to inoculate fresh broths at a starting OD600nm of 0.2. The cultures were then grown to an OD600nm of 0.8 and 1 mL samples centrifuged at 13,000 × g for 3 min to pellet the cells. The pellets were then washed twice in PBS (137mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) and each pellet resuspended in 1 mL of PBS. Viable counts were determined by plating dilutions of the cultures onto HI agar, incubating plates at 37°C overnight, and counting colonies.
Post-TBI pneumonia model
Pneumonia is a common infection after TBI, 3,14 and hence it is a suitable infection model to assess relevant functional changes to the immune response. We selected S. aureus for this challenge because this species is among the most frequent pathogens associated with ventilator and line-related infections in trauma patients. 3 The C57BL/6 wild type mice were infected intratracheally with 1 × 107 CFU/mL of S. aureus (ATCC25923). The dose of 107 CFU was chosen because it allowed an appropriate clinical manifestation of illness in the mice, whereas 106 CFU did not induce clinically significant sepsis and 108 induced a too severe model of infection (see results).
Infections were performed 48 h, 72 h, or six days after induction of the sham or TBI procedure, and animals were euthanized 24 h thereafter. The right lung from each animal was ground through a 70 μm cell strainer (BD Biosciences, Franklin Lakes, NJ) in sterile PBS (100 mg/mL), serially diluted, plated on HI agar, and incubated overnight at 37°C. The following day, bacterial numbers were counted and are presented as CFU/g of lung tissue.
Scoring of disease severity
After infection, mice were monitored for disease severity, using the mouse clinical assessment score for sepsis (M-CASS) as described previously.
20
Using eight clinical parameters, this score allows the classification of mice into four groups, whereby “1” represents a healthy mouse, and “4” the most severe form of disease (Supplementary Table 1; see online supplementary material at
Euthanasia and harvesting of organs
Mice were euthanized with an IP injection of urethane (3.3. g/kg dissolved in distilled H2O), and whole blood was drawn from the inferior vena cava into a syringe containing sodium citrate. Differential white blood cell count was assessed by analyzing citrated blood samples with the Hemavet 950FS analyser (Drew Scientific, Waterbury, CT). Lungs and spleens were harvested under aseptic conditions and processed for assessment of bacterial load and flow cytometry, as described above, and below.
Assessment of lung edema
We assessed the weight of the left lung of infected mice or control animals 24 h post-infection immediately after extraction and after incubation at 60°C for 24 h to then calculate the wet-dry ratio and thereby the fluid content as a proportion of the overall tissue mass. This reflected a readout for lung inflammation and fluid extravasation. 21
Plasma cytokines
Plasma was isolated from citrated whole blood samples. A cytokine antibody array kit, Mouse ProcartaPlex™ Panel (eBioscience, San Diego, CA), detecting eight different analytes (interleukin [IL]-6, interferon [IFN]-γ, tumor necrosis factor [TNF]-α, IL-β, IL-13, monocyte chemoattractant protein [MCP]-1, IL-10 and transforming growth factor [TGF]-β) simultaneously on a Luminex platform was used, according to the manufacturer's instructions.
Briefly, antigens coated in beads were prepared for each analyte, a standard for each analyte was assessed, and the plates were mapped and marked for each analyte and the standards. The individual mouse plasma sample was thawed, and 25 μL of plasma was added to the designated wells, in duplicate. The plate was then incubated at 4°C overnight, then washed to remove any unbound antibody, and 50 μL of detection antibody was added, followed by streptavidin-PE. The reading buffer was added and the plate was read to acquire data on a Luminex platform MAGPIX.
The median fluorescence intensity (MFI) was calculated for each analyte including the standard, and data were analyzed using Procartal Plex Analyst software. The concentration of each analyte was performed by comparing MFI of the known concentration of the standards. This assay was performed by Crux Biolab (Melbourne, Australia).
Plasmin-antiplasmin (PAP) complex enzyme-linked immunosorbent assay (ELISA)
Citrated plasma samples were evaluated for levels of the PAP complex. Briefly, 96-well flat-bottom plates (Nunc MaxiSorp) were coated with 100 μL polyclonal rabbit antibodies against mouse α2-antiplasmin (4 μg/mL) in PBS at 4°C overnight. The following day, wells were blocked with 200 μL per well 1% bovine serum albumin (BSA)-PBS for 2 h at room temperature.
After four washing steps with PBS-0.004% Tween standards and samples diluted in PBS/Tween +1% BSA and 0.5 M ethylenediaminetetraacetic acid (EDTA) were added and incubated for 2 h at room temperature. After four further washing steps, 100 μL of biotinylated polyclonal rabbit antimurine plasminogen antibody (12.5 mg/mL) were added and incubated for 2 h at room temperature. Wells were again washed four times and then incubated with 100 μL Streptavidin-HRP at room temperature for 30 min. After additional four washing steps, 100 μL TMB per well were added, and the reaction was stopped with 50 μL of 2M H2SO4 after 5–10 min. The optical density (OD) of the reaction was assessed with the FLUOstar Optima microplate reader (BMG Labtech, Cary, NC) at 450 nm.
D-dimer ELISA
Citrated plasma samples were assessed for D-dimer levels with an ELISA according to the manufacturer's instructions (LifeSpan BioSciences, Seattle, WA) using 100 μL of sample (at a 1:100 dilution), standards, or blank. The OD of the reaction was assessed with the FLUOstar Optima microplate reader (BMG Labtech, Ortenberg, Germany) at 450 nm.
Flow cytometry
Flow cytometry of spleens was performed 72 h and one week after TBI or sham procedure aiming to characterize broadly the cellular innate immune response. It is now well accepted, however, that cells of the adaptive immune system, particularly Tregs, can regulate the innate immune response. 22 We were therefore interested in the identification and description of both myeloid and lymphoid cell populations, with a main focus on DC and T cell subsets. Further, neutrophils, macrophages, and monocytes as well as NK cells and B cells were assessed with our staining protocol.
After extraction of spleens, they were ground through a cell strainer to prepare a single cell suspension. Cells were centrifuged at 1500 g for 5 min and resuspended in PBS +2% foetal calf serum (FCS). Red blood cell lysis was then performed with a red blood cell lysis buffer (BioLegend, San Diego, CA) according to the manufacturer's instructions. Tissue samples were stained with three distinct cocktails containing fluorochrome-labeled antibodies.
One cocktail served the phenotyping of myeloid subsets and consisted of CD11c V450, MHC class II APCeFluor780, B220 AF700 (all BD Biosciences), CD11b PECy7, Gr1 FITC, CD86 PE (all eBiosciences, San Diego, CA), CD103 AF647, F4/80 PerCPCy5.5, CD8 BV650 and CD80 biotin (all BioLegend) + streptavidin PE-CF594 (BD Biosciences). A second fluorochrome-labeled antibody cocktail was used to identify and describe lymphoid subsets and consisted of CD3 APCeFluor780, CD69 V450, NK1.1 PE, CD19 FITC, CD44 PECy5 (all eBiosciences), CD8 BV650 (BioLegend), CD4 BV605, B220 AF700, and CD62L PE-CF594 (all BD Biosciences).
A third cocktail was used specifically for characterization of regulatory T cells (Treg) consisting of CD3 APCeFluor780, LAP PerCPeFluor710, (both eBiosciences), CD4 BV605, CD25 PECy7 (both BD Biosciences), CTLA4 BV421 (BioLegend), and the intracellular marker FoxP3 APC (stained after permeabilization) using a mouse regulatory T Cell Staining Kit (eBiosciences).
All samples were stained additionally for live/dead discrimination with the LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (Life Technologies, Carlsbad, CA) and were fixed in 1% paraformaldehyde (in PBS), except for permeabilized samples (stained with the Treg cocktail), which were resuspended in PBS only. The antibody panels are summarized in Supplementary Table 2 (see online supplementary material at
After the staining procedure, they were acquired on the LSR Fortessa flow cytometry analyzer (BD Biosciences) using the FACS DIVA software (BD Biosciences) at the AMREP Flow Cytometry Platform. Data analysis was performed with FlowJo software V10 (Tree Star Inc., Ashland, OR). A summary of identified cell populations and functional markers used is provided (Supplementary Table 3; see online supplementary material at
Statistics
Statistical analysis was performed using GraphPad Prism v 7.0 (Graphpad Software, Inc., La Jolla, CA). Data distribution was assessed with a D'Agostino & Pearson normality test. For normally distributed data, an unpaired two-tailed Student t test was used when two groups were compared. For comparison of more than two groups, one-way analysis of variance (ANOVA) with Dunnett correction was performed. For comparison of four groups differing in two factors, a two-way ANOVA with Sidak correction was used. Non-parametric data were analysed using a Mann-Whitney U test for comparison of two groups, and more than two groups were compared with a Kruskal-Wallis test and Dunn correction test for multiple comparisons. In all instances, p < 0.05 was considered significant.
Results
Development of a model to evaluate the effect of TBI on infection rates
To study the impact of the plasminogen activation system and antifibrinolytic therapy on CIDS after TBI, we first investigated whether the CCI model of TBI resulted in endogenous infection from the intestinal tract, as described previously in a mouse model of stroke. 15 Aerobic and anaerobic bacterial growth could not be detected in the majority of sham and TBI mice in the right lung, liver, and the mesenteric lymph node at the 24 h, 72 h, and one week time point post-TBI (data not shown). Hence, isolated TBI using the CCI model does not promote endogenous infection.
Therefore, we proceeded to introduce a bacterial challenge by intratracheal inoculation of a sublethal dose of S. aureus. We evaluated three bacterial doses for intratracheal inoculation in naïve mice (Fig. 1A). While delivery of 1 × 106 CFU did not induce sickness as assessed by the M-CASS score, and resulted in complete clearance of bacteria within 24 h, a dose of 1 × 107 or 1 × 108 CFU induced profound clinical signs of sickness within 6 h after infection (M-CASS of 2 or 3). With a dose of 1 × 107 CFU, marginal recovery was observed at 20 h post-infection, whereas the majority of mice infected with 1 × 108 CFU did not recover after 20 h.
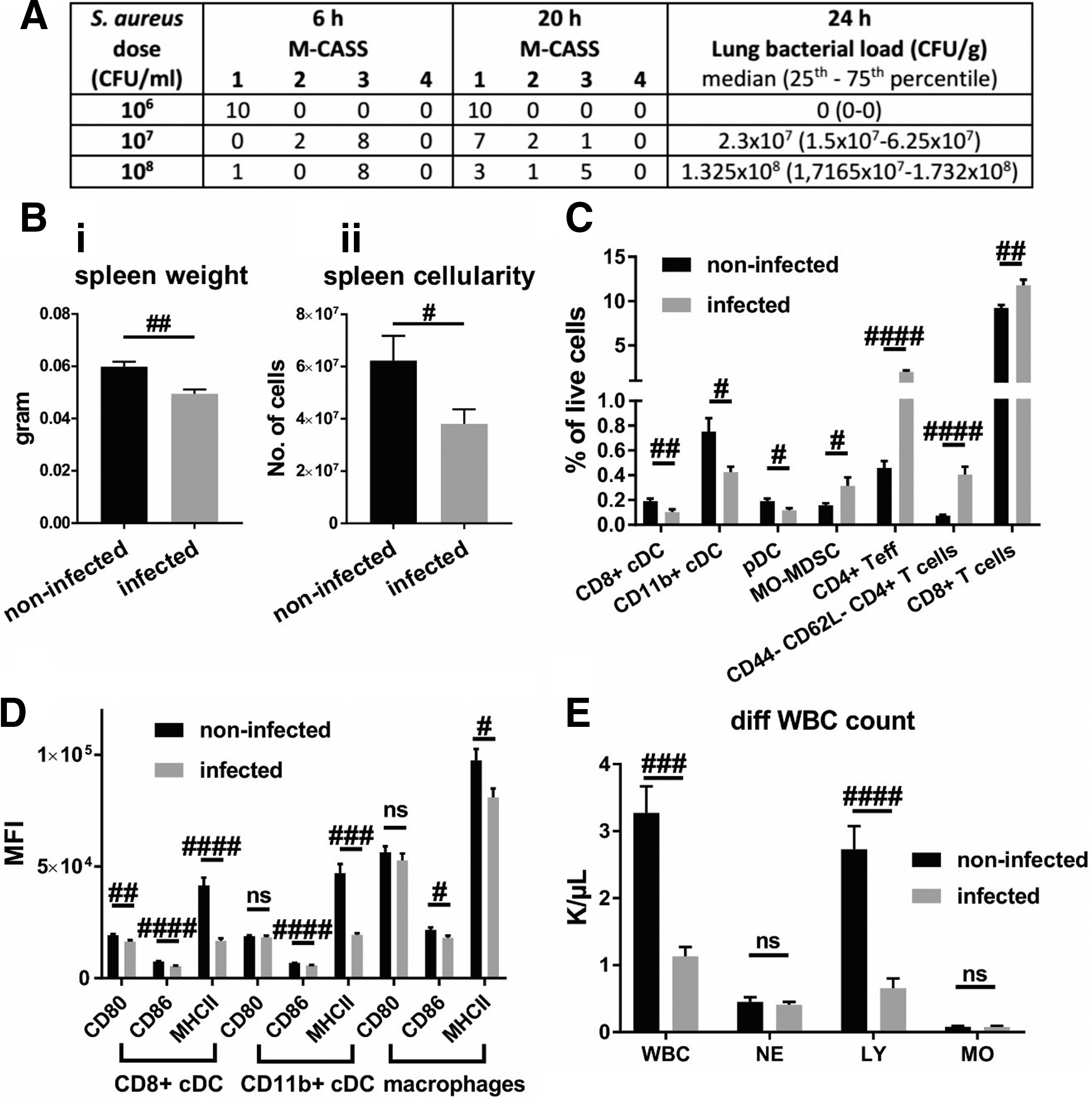
Effect of pneumonia: Staphylococcus aureus infection induces profound immunological changes within 24 h. (
The S. aureus colonies could be detected in the homogenized right lungs of all mice for both of the higher doses, confirming the onset of pneumonia (Fig. 1A). All splenic suspensions were devoid of bacteria (data not shown), indicating a lack of dissemination of bacteria from the lung. Mice inoculated with 1 × 108 CFU were overtly sick and unable to clear bacterial infection within the desired time frame. Therefore, the dose of 1 × 107 CFU was selected for further experiments because it seemed amenable to experimental therapeutic intervention, given the rapid onset of sickness followed by a strong recovery over a 24 h period, likely because of clearance of the instilled bacterial load. Lung injury in infected mice was, moreover, confirmed by the significant increase in liquid content of the left lung, indicating inflammation and fluid extravasation (Suppl. Fig. 1).
S. aureus pneumonia induces a profound systemic immune response
Instillation of 107 CFU of S. aureus into the lungs of naïve mice resulted in a significant reduction of spleen weight (Fig. 1Bi) and cellularity (Fig. 1Bii). Reduced splenic cell counts in infected animals was underscored by the observed reduction in proportions of CD8+ cDC, CD11b+ cDC, and pDC, yet a relative increase in monocytic myeloid-derived suppressor cells (Mo-MDSC), CD4+ T cells (particularly CD44- CD62L- CD4+ T cells) as well as CD8+ T cells (Fig. 1C). Expression of the activation marker CD80 on CD8+ cDC was reduced significantly by infection. Expression of CD86 and MHCII on CD8+ and CD11b+ cDC as well as macrophages was significantly lowered (Fig. 1D). Infection reduced overall white blood cell counts, specifically by reducing lymphocytes, but not neutrophils or monocytes (Fig. 1E). Other parameters assessed by flow cytometry or counts of other blood cells were not altered significantly by the infection (not shown).
Plasminogen deficient mice do not show differences in lung bacterial clearance
Having established a model to evaluate bacterial clearance, we assessed the role of plasmin(ogen) on bacterial clearance itself, independent of TBI. The plg-/- mice displayed remarkable and significant baseline differences in various immune parameters including a 1.5-fold increase in spleen weight, a threefold increase in plasma IL-6 levels, and up to twofold increases in white blood cells overall, neutrophils, lymphocytes, and monocytes, as well as a reduction in frequencies of T cells overall and a further reduction of CD8+ cells within the T cell population (Fig. 2A–C). Nevertheless, there were no changes in the bacterial load of lungs 24 h post-infection (Fig. 3A) or M-CASS scores 6 h and 20 h post-infection (Fig. 3B).
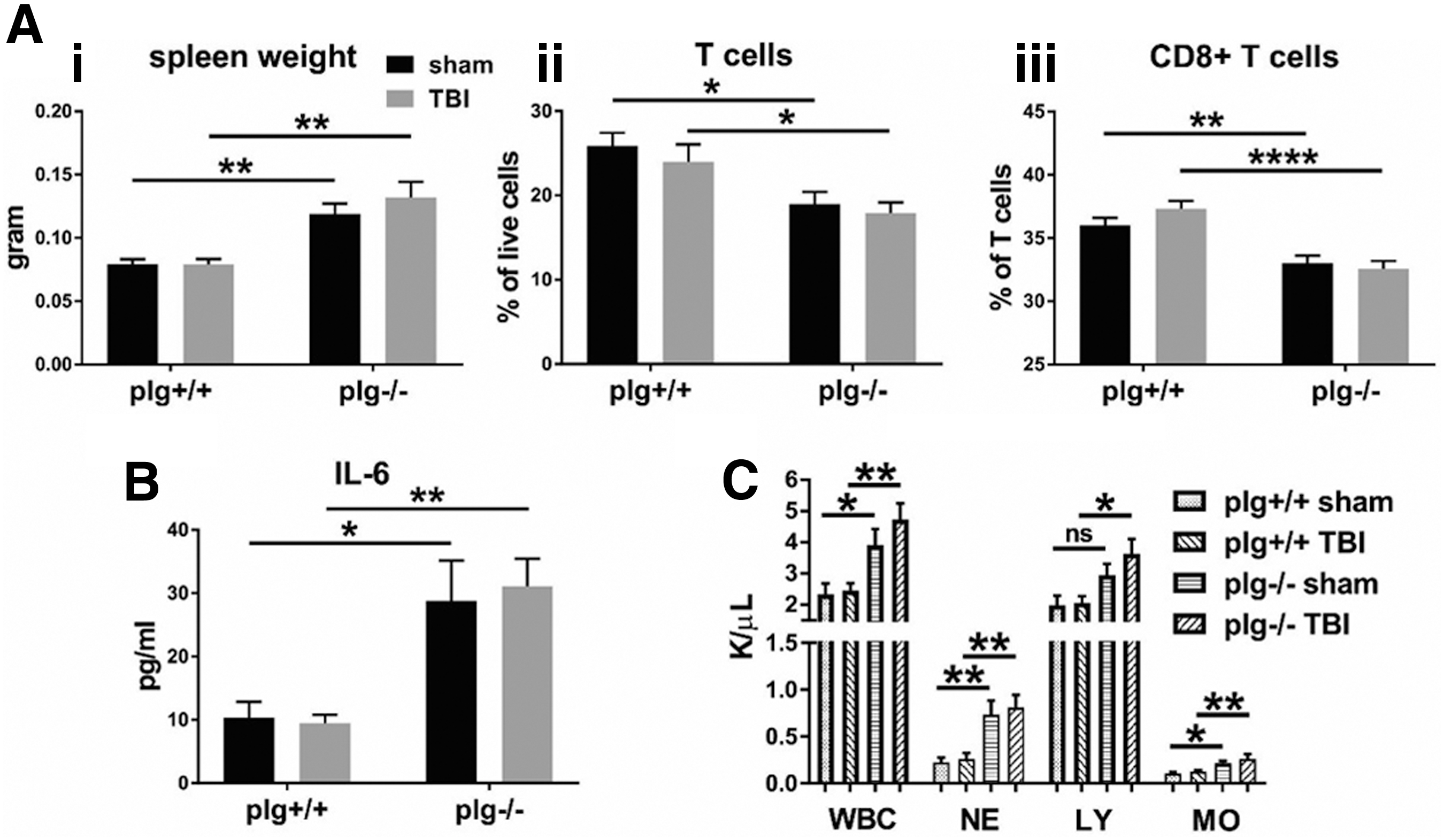
Immune response after traumatic brain injury (TBI) in plg-/- mice: plg-/- mice display significant differences in baseline immune parameters in spleen and blood compared with their plg+/+ littermates, which are unaffected by TBI. (
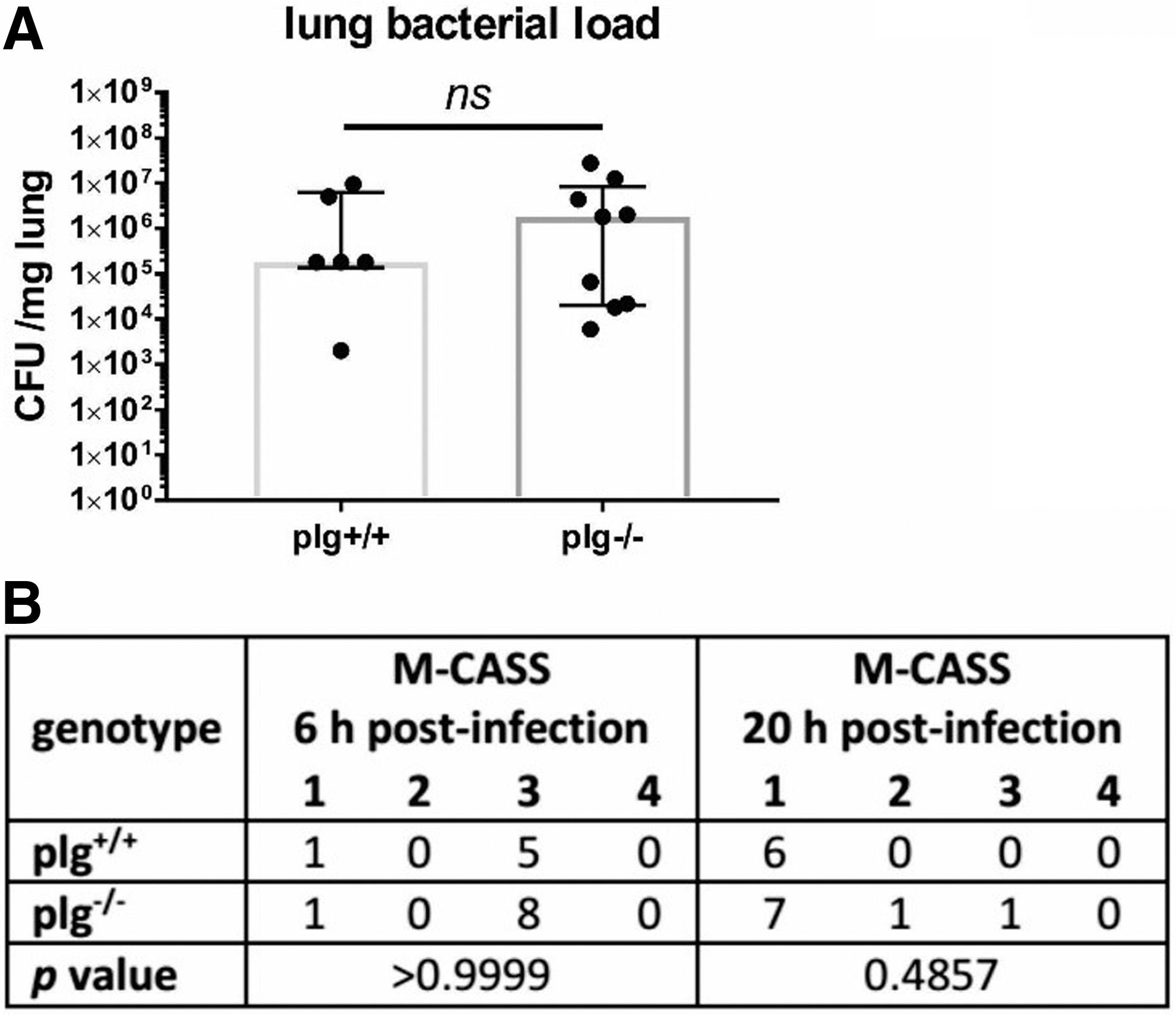
Effect of plasminogen deficiency on infection alone: plasminogen deficiency does not alter the ability of mice to clear bacteria from the lung 24 h post-infection (n = 6–9).
Other immunological parameters assessed with flow cytometry or cytokine ELISA than those shown in Fig. 2 were not significantly altered in plg-/- mice. Also, TBI in these strains did not alter any of the parameters being studied, relative to sham mice at the 72 h time point (Fig. 2) or one week time point (data not shown). In summary, plasminogen deficiency alone does not alter susceptibility to pneumonia.
TBI does not affect bacterial clearance and recovery from infection
The TBI did not influence the recovery from pneumonia at the 72 h (Fig. 4A), 96 h (Fig. 4B), or one week time point (Fig. 4C). Infection in sham operated or injured mice resulted in a comparable disease burden as represented by an increased M-CASS score at 6 h post-infection in both cohorts and at all time points with a recovery to some extent (Fig. 4D). Similar to the infection alone group, in the TBI + pneumonia group, bacteria did not disseminate because spleens were devoid of bacteria at all tested time points (data not shown). Lung edema assessed by wet/dry ratio of the left lung was also unchanged in these cohorts (data not shown). Therefore, it is unlikely that post-TBI immunosuppression occurs after a focal injury in this CCI model of TBI.
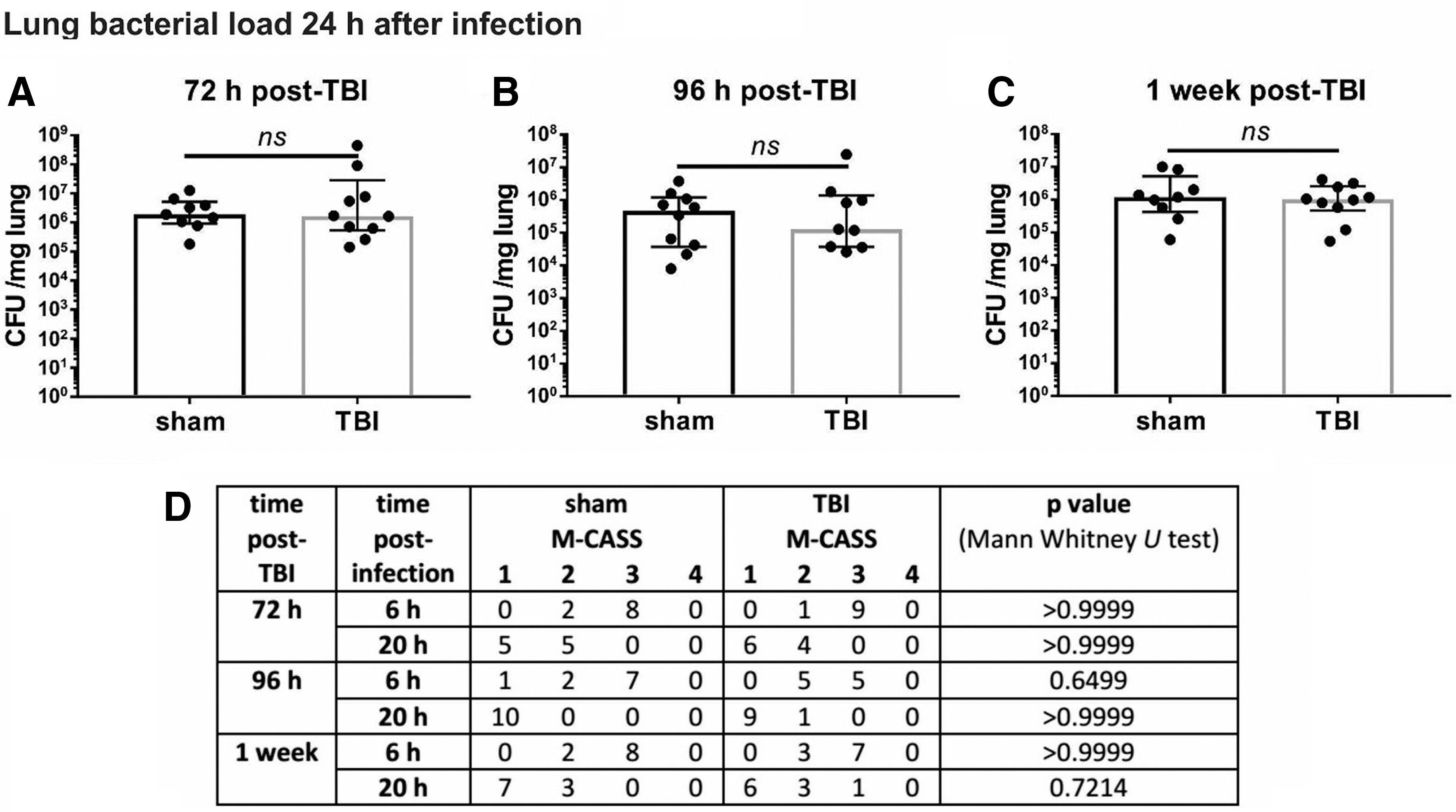
Traumatic brain injury (TBI) + pneumonia: TBI does not affect clearance of bacteria from the lung, or mouse clinical assessment score for sepsis (M-CASS) scores for up to one week. In comparison with the sham procedure, TBI did not adversely affect bacterial load of the right lung 72 h (n = 9–10) (
TXA does not affect the lung bacterial load, but induces changes in the cellular immune response to TBI in infected mice
Next we assessed the effect of TXA treatment in the TBI + pneumonia cohort. The TXA did not influence the bacterial load in lungs of traumatized mice at the 72 h (Fig. 5Ai) and one week time points (Fig. 5Aii). Moreover, TXA did not affect the M-CASS scores 6 h and 24 h after infection in TBI animals (Fig. 5B). In comparison with vehicle administered mice, however, animals treated with TXA displayed increased numbers of splenic macrophages 72 h post-TBI relative to sham controls (sham-vehicle controls for TBI-vehicle mice, and sham-TXA level for TBI-TXA mice) (Fig. 5Ci). Also, the proportion of CTLA4-expressing cells within the Treg population (Fig. 5Cii) was reduced by TXA. At one week post-TBI, TXA reduced the numbers of monocytes and pDC relative to sham (Fig. 5D).
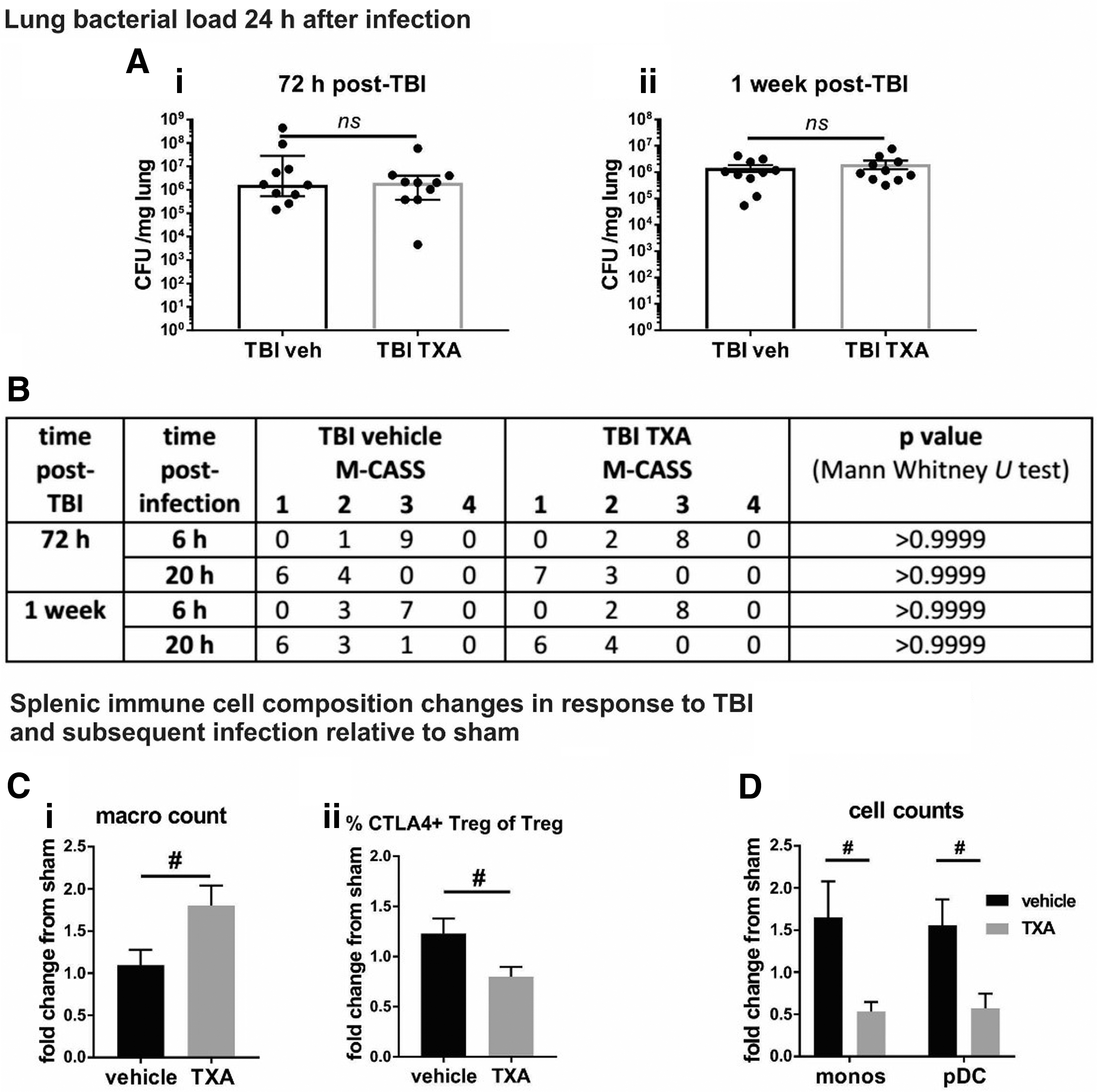
Effect of tranexamic acid (TXA) on traumatic brain injury (TBI) + pneumonia: TXA does not change the capacity of mice to clear bacteria from the lung, but induces significant alterations in the splenic immune response after TBI + pneumonia. (
TXA does not induce changes to the splenic cellular immune profile in response to TBI in non-infected mice
We next assessed whether these immunological changes were related to infection itself or TXA alone by assessing the immune response in non-infected mice treated with TXA/vehicle. In contrast to infected mice, TXA did not significantly influence splenic macrophage counts (Fig. 6Ai) or the proportion of CTLA4-expressing Treg (Fig. 6Aii) relative to sham 72 h post-TBI. Similarly, one week after TBI, no difference was detected in TXA-treated animals in monocytes and pDC counts (Fig. 6B) relative to sham. Thus, it is evident that TXA specifically influences the immune response after TBI + pneumonia.
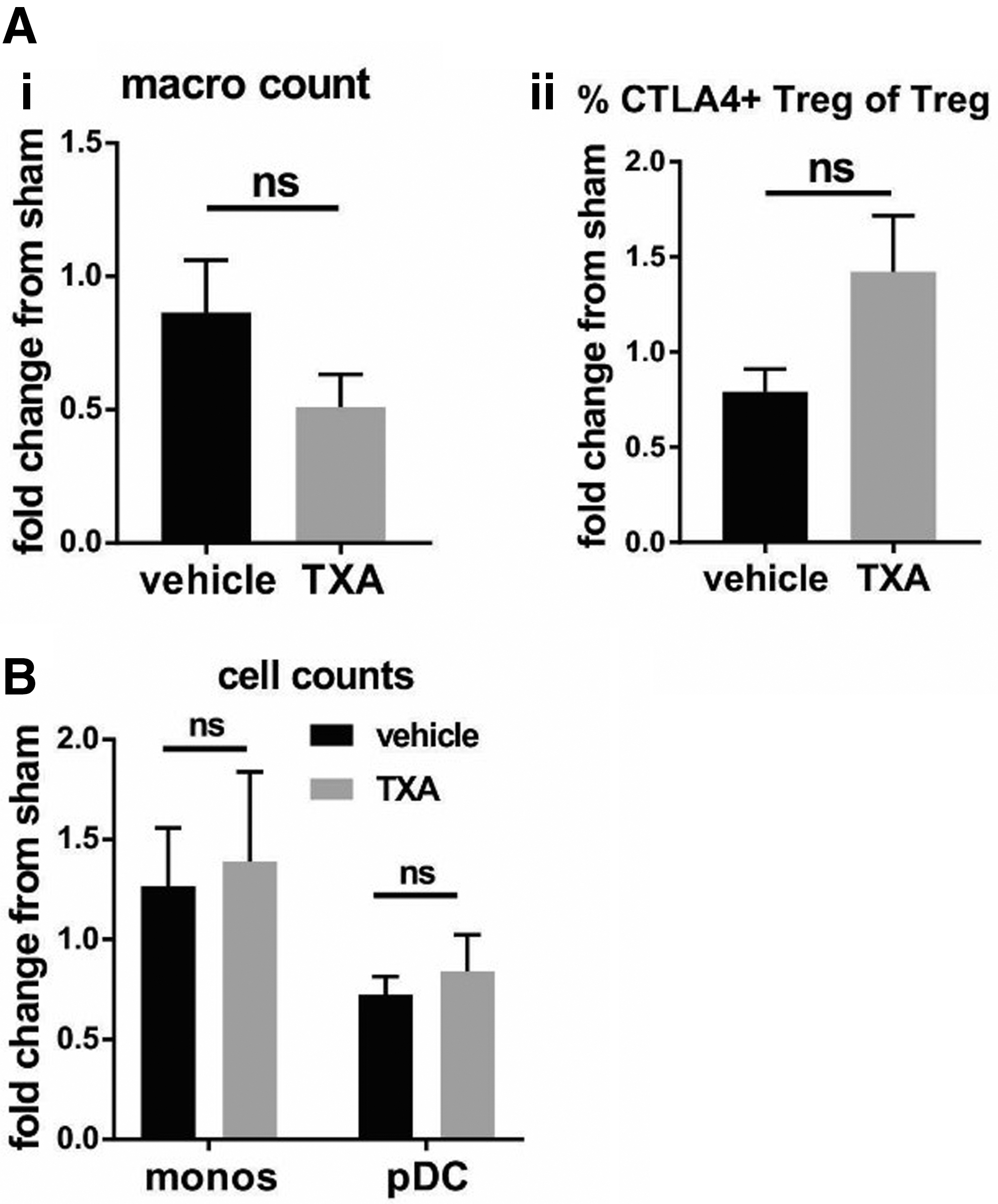
Differential effects of tranexamic acid (TXA) after traumatic brain injury (TBI) alone: TXA does not alter the splenic immune profile when administered to mice subjected to TBI alone. (
Restriction of TXA effects on the immune response to TBI + pneumonia can be explained by the difference in the plasmin generating potential between infected and non-infected mice
We next assessed plasma levels of the PAP complex (representing plasmin formation) and D-dimer (representing plasmin-mediated fibrinolytic activity) to determine whether the effects of TXA on the immune response were because of suppression of plasmin generation and activity (Fig. 7). We found that infection alone (i.e., without a previous TBI) resulted in a significant reduction in plasma PAP complex levels (Fig. 7Ai). In stark contrast, levels of PAP complexes were increased significantly in mice subjected to infection but with a previous TBI, whereas TBI alone had no effect. Interestingly, the selective increase in PAP levels in the TBI + infection group was reversed fully by TXA administration.
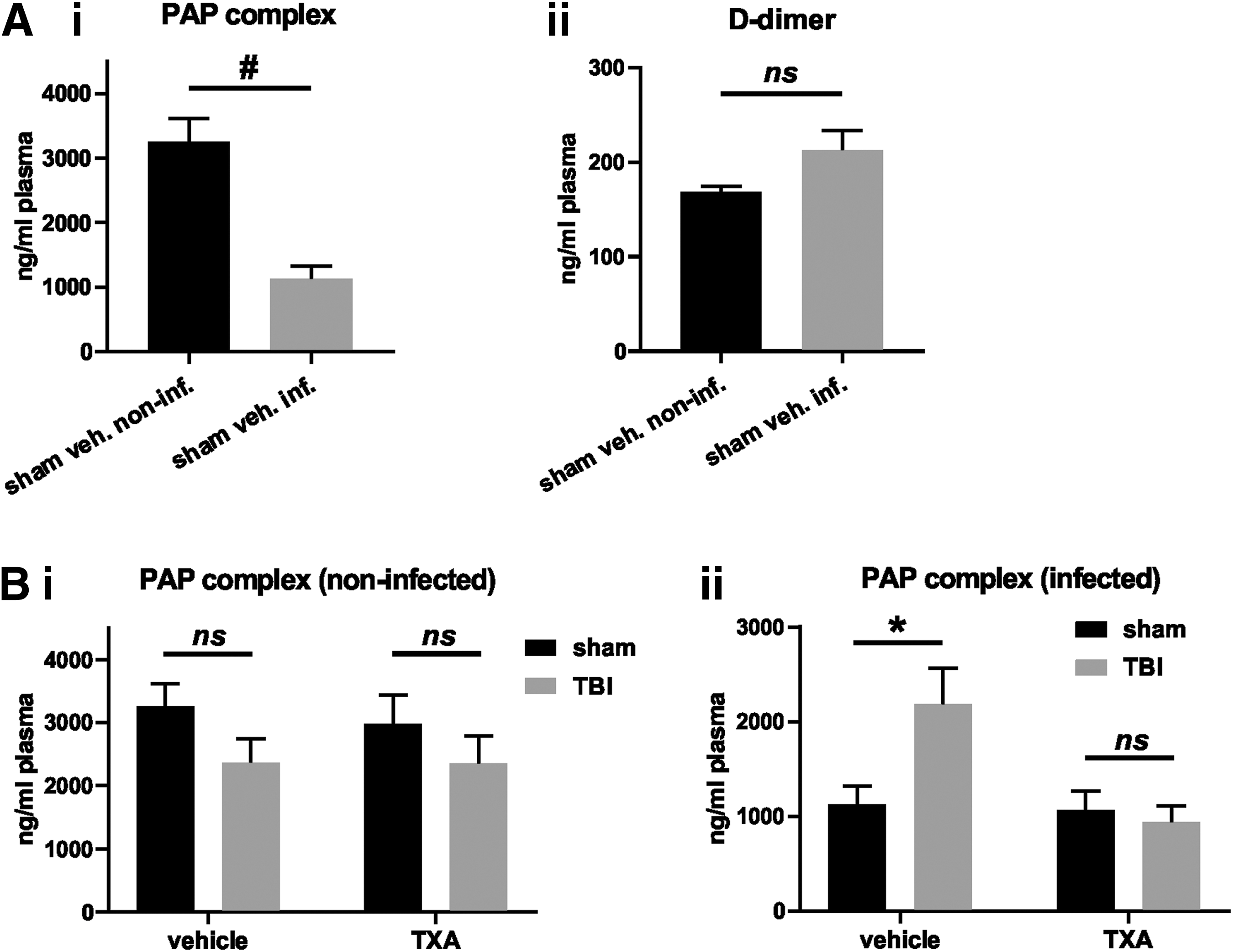
Infection reduces plasma levels of the plasmin-antiplasmin (PAP) complex, but not D-dimer at the 72 h time point: Effects of tranexamic acid (TXA) on PAP levels in infected mice. Plasma levels of the PAP complex and D-dimer were assessed by enzyme-linked immunosorbent assay at the 72 h time point. (
Discussion
TXA has been implemented in resuscitation guidelines around the globe for major trauma, 23 yet guidelines for its use in isolated TBI are still missing. While clinical trials addressing this important issue are under way, 7,8 an improved understanding of the off-target effects of TXA is necessary to interpret clinical outcome data correctly and conclusively and to facilitate optimal patient selection for this treatment.
Based on previous studies suggesting immune-modulatory properties of TXA, 24 we tested the effects of TXA on the immune response after TBI. As a lysine analogue, TXA is also likely also to exert effects independent of plasmin formation inhibition. Given the broad and well-characterized implication of plasmin in inflammation and the adaptive immune response, it is reasonable to assume that plasmin also regulates the immune response after TBI, and these effects may be inhibited by TXA. 11
Notably, lysine-dependent plasmin formation is not restricted to fibrin clots. Plasminogen receptors on various leukocytes, endothelial cells, and other components of the vasculature provide a cofactor for plasmin generation by t-PA as well. 25 In addition, misfolded proteins formed during late-stage cell death and crosslinked via disulfide bonds also expose lysine residues, a process coined “nucleocytoplasmic coagulation.” 26,27
Given that infection is regarded as a major contributor to morbidity and mortality in TBI, 3 we were interested specifically in the effects of TXA on the systemic immune response and the capacity to fight infection. In addition to ventilator-, line- and device-related origins, bacterial migration from the gut into the circulation has been suggested as an important cause of infection in patients with TBI. 28
A recent publication described changes in colon morphology with an increase in paracellular permeability on day 28 post-TBI. 29 Nevertheless, even though endogenous infection from the intestinal tract has been described in other models of brain injury, such as the middle cerebral artery occlusion (ischemic stroke) model, 15 we have shown here that in our CCI model of TBI, this does not occur, most likely because the degree of injury is not sufficiently severe. We therefore established a novel model of post-CCI TBI pneumonia (a common TBI-related complication 3,14 ) to study the functional consequences of TBI on the systemic immune response, and hence to evaluate potential effects of TXA treatment on the capacity to fight infection.
We first established a pneumonia model that could be applied later to our CCI model of TBI. The S. aureus infection induced profound lymphopenia measured 24 h after infection, which is in line with previous reports of lymphopenia in cases of S. aureus bacteremia. 30,31 We also observed spleen atrophy with changes in the frequencies of several splenic cell populations and a downregulation of activation markers (CD80, CD86, and MHCII) on central antigen-presenting cells. Downregulation of CD86 and MHCII has been described previously for human monocytes cultured with S. aureus. 32 To our knowledge, this study provides the first detailed flow cytometric characterization of the immune response to murine pneumonia induced with the S. aureus strain ATCC25923.
To assess the role of plasmin on infection alone, we compared plg-/- mice, which are unable to generate plasmin, with their plg+/+ littermate controls. In our study, genetic deletion of plasminogen did not affect bacterial clearance from the lungs, which was surprising given the profound baseline differences in numbers of various leukocyte subsets in the spleen, their expression of activation markers, and plasma cytokine levels between plg-/- and plg+/+ mice. These differences likely can be attributed to the ubiquitous deposition of fibrin, which cannot be removed in the absence of plasmin, inducing a chronic inflammatory response in plg-/- animals. 33
Another explanation would be a compensatory increase in leukocyte numbers in response to their reduced function, as indicated by previous reports. For instance, plg-/- mice displayed reduced migration of monocytes to the peritoneum in a peritonitis model. 34 Macrophages from plg-/- mice have also been described to have reduced phagocytic capacity. 35 Moreover, in a model of S. aureus arthritis, plg-/- mice completely lacked the ability to clear bacteria from the knee joint, in stark contrast to their plg+/+ littermates. 36
The time points for evaluation of immune response and infection were selected to span the periods post-TBI that are most likely to present immune dysfunction. Impaired cellular immunity has been described to occur within a few hours, but persists up to several weeks after brain injury, 14 and a peak in hospital-acquired infections can be observed 5–11 days after neurotrauma. 3 Neither TBI itself nor acute plasmin inhibition with TXA after TBI significantly affected bacterial clearance in the lungs, however. Future studies will have to harness more severe models of TBI in which post-traumatic immunosuppression naturally occurs to further investigate whether TXA can modulate this phenomenon.
Indeed, we did not detect a profound systemic immune response after induction of TBI. This is in contrast to a recent article by Ritzel and associates 37 that impressively demonstrated immune changes consistent with immunosuppression using the same TBI model. 37 Differences in the anesthetic procedure (avertin vs. isofluorane 37 ), the age of mice (8 weeks vs. 10–12 weeks 37 ), a higher impactor velocity (5 m/sec vs. 6 m/sec 37 ), and a wider impactor tip (2.5 mm vs. 3.5 mm37), however, indicate that their settings for TBI were more severe, and this may explain why their model induced spontaneous immunosuppression.
Also the closed head injury model/weight drop model might be a useful alternative for this research question given the more diffuse impact compared with the focal TBI in the CCI model. 17 In fact, a recent study also described immune-modulatory effects (decreased splenic cell count, a reduced B cell count, and increased naive CD4+ T cell counts) of TXA 24 h after TBI using the weight drop model, and the authors also highlighted the need to assess whether TXA impacts on post-TBI immunosuppression and infection risk. 38
The S. aureus strain we used did not disseminate, as evidenced by the absence of bacterial colonies in cultured spleen lysates. This allowed us to study specifically the immune response against post-TBI infection rather than confounding effects on the pathogen's ability to disseminate, a process which is also influenced by the plasminogen activation system. 12
Different bacterial strains and species can be tested in future experiments because bacterial dissemination and specific interaction of bacteria with the plasminogen activation system 11 will certainly influence the net effect of this treatment. Notably, TXA previously has been demonstrated to enhance rather than decrease bacterial burden in mouse models of S. aureus sepsis and septic arthritis. 39 In contrast, plasmin is also detrimental in S. aureus mediated sepsis, yet protective in low grade S. aureus infection 40 and S. aureus mediated arthritis, 36 indicating that the circumstances and location of the infection determine the impact of plasmin, and therefore a potential effect of TXA. Accordingly, whether TXA is administered prophylactically or instead given after the onset of infection also needs to be considered.
Nevertheless, despite the lack of improvement of bacterial clearance in the lung, TXA did alter the immune response post-TBI and pneumonia. Relative to sham animals, TXA increased splenic macrophage counts and reduced the proportion of Tregs expressing the potent immunosuppressive surface marker CTLA4 72 h post-TBI–pneumonia, consistent with immune-enhancing effects. 41,42 In contrast, reduced splenic monocytes and pDC at the one week time point could be interpreted as potentially immunosuppressive changes. Nonetheless, all of these observations confirm the notion of immune-modulatory properties of TXA.
Contrary to infected mice, in non-infected animals, TXA did not alter the described immune parameters, indicating that the subsequent infection is essential for TXA to induce changes to the immune response post-TBI. A possible explanation for this would be an actual increase in plasmin generating potential in infected animals that contributes to the immune response in TBI + pneumonia and allows TXA to modulate immune changes by TBI to a greater extent.
This is supported by the fact that the majority of the affected leukocyte subsets (macrophages, monocytes, DC) are known for their expression of plasminogen receptors and plasmin responsiveness. 11 Notably, plasmin generation induced by staphylokinase, a plasminogen activator produced by many S. aureus strains, has been shown to be inhibited by TXA. 43 Staphylokinase, however, has also been demonstrated to have limited activity on murine compared with human plasminogen. 44
In addition, lysine-dependent effects independent of plasmin generation have to be considered because TXA, a lysine analogue, 9 would potentially compete with any lysine residues for their binding partners. Given the role of lysine in connective tissue protein structures and crosslinking, 45 epigenetics, 46 and fatty acid metabolism, 47 TXA might interfere with all of these processes. The involvement of these processes in wound repair and infection might well allow TXA to modulate further any additional immune changes TBI induces on top of the infection.
Interestingly, in our model, infection significantly reduced PAP levels, indicating attenuated plasmin generation, while D-dimer levels were not significantly affected. While it is known that plasmin formation can occur independently of fibrin formation as shown by Gall and coworkers 58 (where elevated PAP levels were seen in some trauma patients without TEG-based evidence of hyperfibrinolysis), it is important to bear in mind the potential influence of the biological half-life of these circulating proteins (PAP half-life 12 h vs. D-dimer half-life 12 h). 49
Nonetheless, to the best of our knowledge, this study is the first to report how TBI can influence the host plasminogen activating system to a subsequent infection. One interpretation of this finding is that the reduction in PAP levels (reflecting reduced plasmin activity) after infection is a host protective response, possibly to reduce the immunosuppressive impact of plasmin. This protective effect, however, is lost in mice previously subjected to TBI by some as yet to be identified mechanism.
The TXA treatment of TBI/infected mice, however, restores this protective mechanism, by reducing plasmin that in turn promotes immune activation (i.e., as reflected by increases in macrophages and reduction of the CTLA4-expressing proportion of Treg cells), again consistent with the notion of an immunosuppressive potential of plasmin. 13 While this is one likely scenario, additional experiments are required to confirm this and also how other components of the plasminogen activating cascade are modulated under these conditions.
It is also interesting to note findings from a rat model of hemorrhagic shock, where TXA prolonged survival despite the fact that the rats did not exhibit increased fibrinolysis in response to the hemorrhage. 48 The authors suggested that the survival benefit of TXA was unrelated to its antifibrinolytic properties, perhaps similar to the survival benefit of TXA reported in the CRASH-2 trial. Although the presence of acute traumatic coagulopathy and the degree of fibrinolysis has not been evaluated functionally in this study, it has been shown that the majority (about 2/3) of severely injured patients actually present to the emergency department with a shutdown of fibrinolysis, 49 making the antifibrinolytic effects of this agent redundant. Hence, reduced mortality might in fact be from effects independent of fibrinolysis, such as the modulation of the immune response. Whether TXA indeed has the potential to reduce infection post-TBI still needs to be assessed.
Tissue injury without significant blood loss has been linked to reduced fibrinolytic activity, referred to as “fibrinolytic shutdown,” which like hyperfibrinolysis has been associated with increased mortality in some, but not in other reports. 50 –54 Further, TBI has been linked to a procoagulant state later after injury, because of platelet activation at the site of the lesion independent of systemic coagulopathy in both humans and rodents. 55,56
Viscoelastic whole blood testing, nowadays frequently used in surgery and trauma care, may not fully reflect the extent of local fibrinolysis at the site of the lesion, as indicated in a study on brain tumor-induced changes in coagulation. 57 Mechanistically, it has been suggested that an “occult” hyperfibrinolytic state is present locally even in conditions in which fibrinolysis shutdown is detected by viscoelastic tests in whole blood, which is mediated by the cell-bound plasminogen receptor S100A10. 58 However, others disagree with this notion, given that patients with fibrinolysis shutdown, according to viscoelastic tests in this study, had increased rates of thromboembolic events and did not die early from bleeding. The potential phenotypes of fibrinolysis in trauma patients remain a topic of controversy and discussion.
It is also important to mention that in this study, TXA treatment was started 20 min after TBI, yet maintained over three days. We chose this administration regimen because we were interested in the effect of prolonged suppression of fibrinolysis on immune competence, given our hypothesis that plasmin, generated on the leukocyte surface, attenuates their ability to mount an allogeneic immune response. 13 However, this potentially comes at the price of enhanced plasmin generation via urokinase. In a report by Hijazi and colleagues, 59 it was demonstrated that fibrinolysis in the cerebrospinal fluid of rats is inhibited by TXA early after TBI while t-PA levels are high. 59 The u-PA levels were reported to increase at later time points, however, and plasmin levels could be increased by TXA (referred to as the “TXA-paradox” 60 ).
Hence, while TXA might reduce intracranial hemorrhage if administered early, it could, in fact, promote intracranial hemorrhage if administered later (8 h post-TBI) because of the presence of u-PA. This is consistent with the findings of the CRASH-2 trial demonstrating a survival benefit of TXA administration within 3 h of injury, yet increased mortality from bleeding if the drug is given after 3 h 5 and might, in fact, provide a mechanistic explanation for this observation.
Summary
The CCI model of TBI does not induce translocation of bacteria from the gut microbiota to the lung, liver, or mesenteric lymph node. We determined the ideal dose for a S. aureus (ATCC25923) bacterial challenge as 1 × 107 CFU for a sublethal pneumonia that induces clinical sickness and a profound immune response in C57Bl/6 mice. CCI-TBI does not affect bacterial clearance from the lung. TXA significantly alters the immune response in mice undergoing trauma and infection, despite the fact that it had no effect on bacterial clearance from the lung. However, the TXA has no influence on systemic immune parameters in response to TBI in uninfected mice. This can potentially be explained by increased plasmin generation in response to TBI exclusively in animals subjected to subsequent infection, which is counteracted by TXA.
Conclusion
Our data confirm the immune-modulatory properties of TXA, yet this effect was specific to mice subjected to both TBI and pneumonia infection. Hence, inhibition of plasmin formation (or perhaps other lysine-dependent processes) is of greater importance to the immune response post-TBI, if a subsequent infection occurs. Further investigations are warranted to assess the true potential of TXA in reducing immunosuppression after TBI and hence in improving the host defence against invading pathogens.
Footnotes
Acknowledgments
We want to acknowledge AMREP Flow Cytometry Core Facility for technical assistance with the flow cytometry experiments. We would also like to thank Debbie Ramsey and David Spiteri from Baker IDI for their assistance in the infection experiments. Moreover, we want to acknowledge Tom Chung, PhD, from Jomar Life Research and Crux Biolab for performing the multiplex cytokine assays and Roger Lijnen, PhD (KU Leuven, Centre for Molecular and Vascular Biology), for providing reagents and protocol for the PAP ELISA. This investigation was funded by NHMRC. RLM is a NHMRC Principal Research Fellow. MS is the recipient of an Alzheimers Australia Dementia Research Foundation grant.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.