
Abstract
A mounting body of evidence suggests that stress plays a major role in the injury progression after spinal cord injury (SCI). Injury activates the stress systems; this in turn may augment the generation of pro-inflammatory cytokines, stimulate pro-inflammatory immune cells, and alter the balance between the pro- and anti-inflammatory immune response. As a result, it is suggested that stress pathways may augment neuronal damage and loss after SCI. Considering these potential detrimental effects of stress after SCI, we hypothesized that inhibition of stress pathways immediately after SCI may offer protection from damage and improve recovery. To investigate the relevance of stress responses in SCI recovery, we investigated the effects of blocking three well-studied stress response axes in a mouse model of SCI. Propranolol, RU-486, and CP-99994 were administered to inhibit the sympathetic axis, the hypothalamus-pituitary-adrenal axis, and the neuropeptide axis, respectively. Surprisingly, assessing functional recovery by the Basso Mouse Scale revealed that RU-486 and CP-99994 did not affect functional outcome, indicating that these pathways are dispensable for neuroprotection or repair after SCI. Moreover, the beta-blocker propranolol worsened functional outcome in the mouse SCI model. In conclusion, immediate inhibition of three major stress axes has no beneficial effects on functional recovery after SCI in mice. These results suggest that injury-induced stress responses do not interfere with the healing process and hence, pharmacological targeting of stress responses is not a recommended treatment option for SCI. These findings are of great importance for other researchers to avoid unnecessary and potentially futile animal experiments.
Introduction
Spinal cord injury (SCI) causes substantial stress, not only on the molecular and cellular level, but also on the psychosocial level. 1 However, the role of stress mediators released in response to injury in the progression of damage and regeneration is unclear. Injury activates an acute pro-inflammatory neuro-endocrine stress response by stimulating neutrophil, macrophage, and lymphocyte trafficking. This ensures that the appropriate immune cells are present at the correct time and location to respond to the injury. 2 When this stress response is excessive or becomes chronic, all cells carrying the receptors for the various stress response mediators are continuously and often excessively stimulated.
In this study, we hypothesized that the inhibition of selected stress mediators may have beneficial effects after SCI by suppressing the detrimental influence of the three major stress axes on immune processes and neuronal survival (Fig. 1A, 1B). Most studies addressing the impact of stress on SCI suggest that stress may aggravate the damage via the following mechanisms: 1) augmenting the immune response via the generation of pro-inflammatory cytokines (e.g., tumor necrosis factor [TNF]-alpha and interleukin [IL]-1beta); 2) stimulating the activation, proliferation and trafficking of pro-inflammatory immune cells (e.g., microglia, dendritic cells, neutrophils, macrophages, and lymphocytes) 3 –5 ; and 3) disturbing the balance of innate and adaptive immune responses by suppressing the secretion of pro-inflammatory mediators such as interferon [IFN]-gamma, TNF-alpha, and IL-12, and stimulating the secretion of anti-inflammatory mediators like IL-4, IL-10, and IL-13 (Fig. 1B). 6 As a result, neuronal responses for example to glutamate, may be altered and increase excitotoxicity leading to neuronal loss. 3 Thus, instead of the normal immune protection and induction of regeneration, the excessive and unbalanced immune response leads to damage of cells and tissue that had been previously spared (i.e., secondary damage). Taken together, SCI-induced activation of the stress systems may have detrimental effects via a defective immune response that increases the secondary damage resulting in impaired functional recovery.
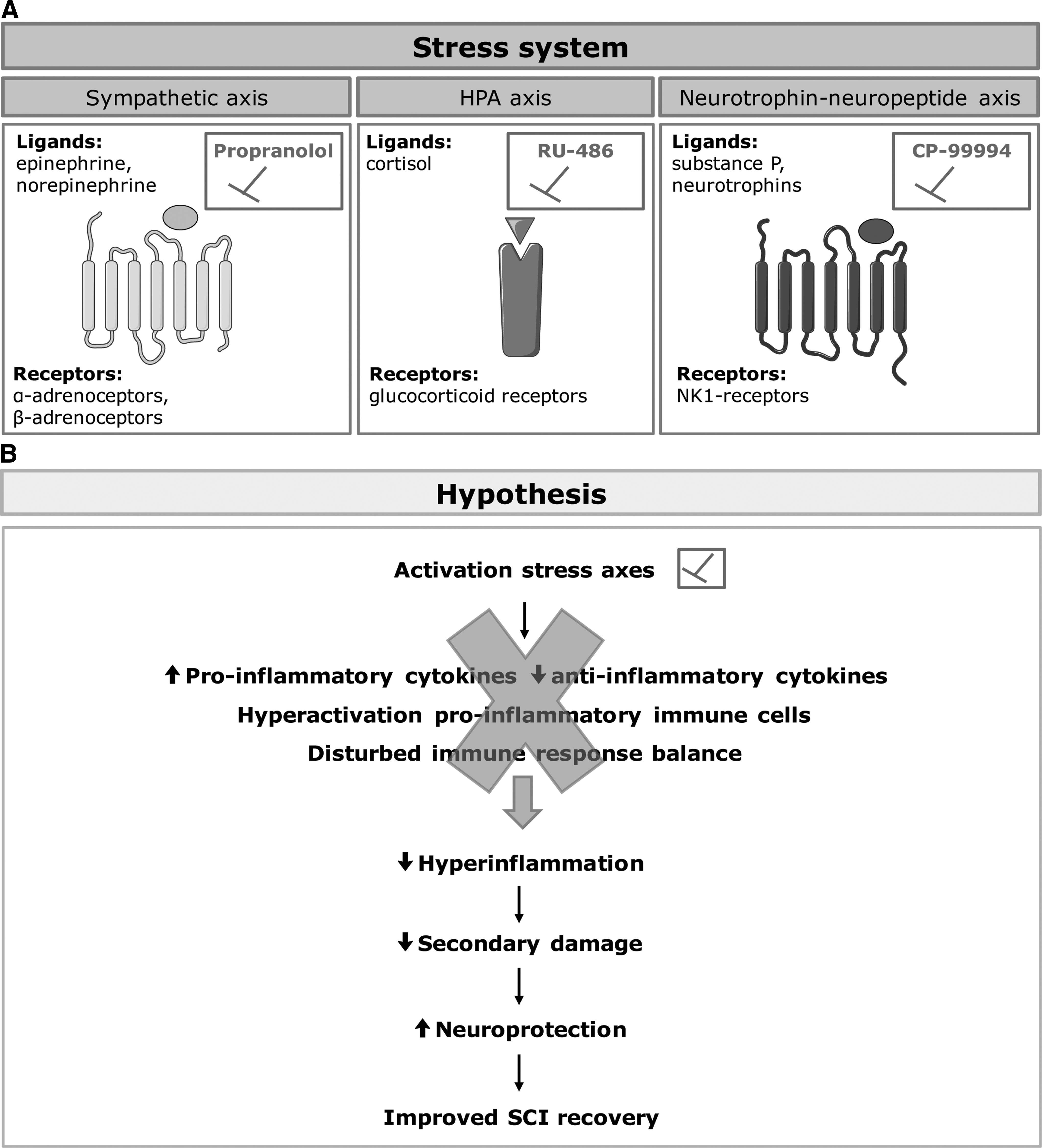
The stress system and its effects after spinal cord injury (SCI).
Consistent with our hypothesis, after traumatic brain injury (TBI), which has a comparable pathology to SCI, it has been shown that inhibition of the beta-adrenergic stress pathway reduces the inflammatory response by decreasing pro-inflammatory cytokines such as IL-1β, thereby improving motor performance. 7 In addition, cerebral glucose metabolism was significantly reduced after the induced TBI and propranolol attenuated this reduction. The rapid increase and extended decrease in cerebral glucose metabolism after TBI is associated with underlying cerebral cell death. In an abdominal incision mouse model of traumatic injury, stress pathway blockade attenuated the hyperinflammatory response such as TNF-α, IL-6, monocyte chemoattractant protein-1, and IFN-γ production by macrophages. 8 Similar results indicate reduced pro-inflammatory cytokine production by microglia after stress pathway inhibition following surgery. 3 In another study, stress hormones induced T-cell apoptosis, while chronic blockage of the beta-adrenergic pathway with propranolol ameliorated this post-traumatic immune suppression after high level SCI. 9
Therefore, modulating the stress response may be a promising strategy to reduce the hyper-inflammatory response 8,9 and secondary damage, thereby increasing neuroprotection, 7 and ultimately improving functional recovery after SCI (Fig. 1B).
Three stress response pathways have been described to mediate the effects of acute stress (Fig. 1A)
6
: The hypothalamus pituitary adrenal axis (HPA); The sympathetic nervous system axis (SA); The neurotrophin–neuropeptide axis (NNA);
To promote functional recovery after SCI, we have used the following inhibitors to block the above three axes: RU-486, a type II glucocorticoid receptor antagonist that blocks the binding of the natural ligand cortisol to target the HPA; propranolol, a general beta-adrenoceptor (β-AR) blocker to inhibit the beta-adrenergic pathway normally activated by epinephrine and nor-epinephrine secreted by the SA; and CP-99994, a neurokinin 1 (NK1) receptor antagonist to inhibit the NNA. Surprisingly, we found that the inhibition of three major stress pathways (HPA, SA, NNA) at the time of injury does not improve functional recovery. We also demonstrate an unexpected reduction in functional outcome in the mouse model after treatment with the beta-blocker propranolol, whereas stimulation of the beta-adrenergic pathway failed to improve functional recovery.
Methods
Experimental spinal cord injury
All in vivo experiments were performed using female C57BL/6j mice (9–10 weeks old; Janvier, France). They were housed in a conventional animal facility at Hasselt University under regular conditions (i.e., in a temperature-controlled room [20 ± 3°C] on a 12 h light-dark schedule and with food and water ad libitum). All experiments were approved by the local ethical committee of Hasselt University and were performed according to the guidelines described in Directive 2010/63/EU on the protection of animals used for scientific purposes.
A hemisection injury was performed as previously described. 10 Briefly, mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg, Ketalar; Pfizer, Elsene, Belgium) and xylazine (12 mg/kg, Rompun; Bayer, Diegem, Belgium). A partial laminectomy was performed at thoracic level 8 to expose the spinal cord. A bilateral hemisection injury was induced to the spinal cord by using iridectomy scissors to transect left and right dorsal funiculus, the dorsal horns and additionally the ventral funiculus. This “T-cut” hemisection results in a complete transection of the dorsomedial and ventral corticospinal tract and impairs several other descending and ascending tracts.
Muscles were then sutured and the back skin was closed with wound clips (Autoclip®; Clay-Adams Co., Inc.). Glucose solution (20%) was given after the operation to compensate for any blood loss during surgery. All mice were placed in a temperature-controlled chamber (33°C) until thermoregulation was established. Bladders were emptied manually until spontaneous return of the micturition reflex.
Locomotor recovery of the animals was determined by an investigator blinded to the experimental groups using the Basso Mouse Scale (BMS). 11 During the first week after injury, mice were scored daily. From the start of the second week until the end of the observation period (12–15 days post-injury), mice were examined every second day.
Initially, per experiment, 12 animals per group have been included. Animals were excluded from the study if the Basso Mouse Scale (BMS) was >1 at 0 days post-injury (dpi) or BMS = 1 at 10 dpi. None of the animal reached humane end-points.
Stress pathway modulator treatment
For CP-99994, propranolol, xamoterol, salbutamol, and clenbuterol first a stock solution has been prepared in Milli-Q (MQ) water. To dissolve propranolol, the pH has been adapted to 3. Next, the correct dilutions have been prepared in 0.9% NaCl prior to injection. For RU-486, a stock solution has been prepared in ethanol, after which it has been diluted in cyclodextrin prior to injection. Mice were injected intraperitoneally twice daily (morning and evening) with the stress pathway blockers (Table 1) starting 2 days before SCI and finishing 3 days post-injury. The vehicle control groups were treated with saline (0.9% NaCl), except for the RU-486 vehicle control group, which received cyclodextrin (Sigma-Aldrich) because RU-486 is not dissolvable in saline.
Overview of the Applied Stress Pathway Modulators
EtOH, ethyl alcohol; MQ, Milli-Q.
The same treatment protocol has been followed for the β-adrenoceptor (AR) agonists (Table 1). Salbutamol, clenbuterol, and xamoterol have been selected to stimulate the β-adrenergic pathway of the sympathetic nervous system, which also acts via α-ARs. We chose to focus on the β-adrenergic pathway because in the context of neuroprotection and SCI its role was not investigated thoroughly. For the α-adrenergic pathway, a lot of information was already available as reviewed by us. 12 Salbutamol was selected to stimulate the β-adrenergic pathway to investigate the effects on functional recovery after SCI, because it is a general β1 –and β2-AR agonist although with a higher selectivity for β2-ARs according to the datasheet. Next, xamoterol and clenbuterol have been used to determine the specific effects of β1- or β2-AR stimulation on functional recovery, because they exclusively act either on β1- or β2-Ars, respectively. Immunohistochemistry and quantitative image analysis are described in the Supplementary Methods.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5.01 software (GraphPad Software, Inc.). Data sets were analyzed for normal distribution using the D'Agostino-Pearson normality test. This test indicated that all data sets were not-normally distributed, except for the GFAP and Iba-1 expression data. Statistical differences between two groups were analyzed via the nonparametric Mann-Whitney U test and to compare multiple groups, a Kruskal Wallis test followed by a Dunn's Multiple comparison test was used. GFAP and Iba-1 expression were analyzed using regular two-way analysis of variance (ANOVA). Functional recovery in vivo was analyzed using a two-way ANOVA for repeated measurements, as well as a Bonferroni post hoc test for multiple comparisons, according to Basso and colleagues. 11 Data were presented as mean ± standard error of the mean. Differences were considered statistically significant when p < 0.05.
Results
Inhibition of the HPA does not affect functional recovery after SCI
To investigate the relevance of the HPA axis for recovery after SCI, SCI mice were treated with RU-486 during 6 days starting 2 days prior until 3 days after SCI to inhibit acute activation (Fig. 2A). The BMS curve shows a normal course with a mean score of 3 (i.e., plantar placing of the paw) for the vehicle group at the end of the observation period. 11 The BMS score of the mice treated with RU-486 reached the same level as the score of the vehicle mice. No differences have been detected between the BMS of the RU-486 treatment group and the vehicle group.
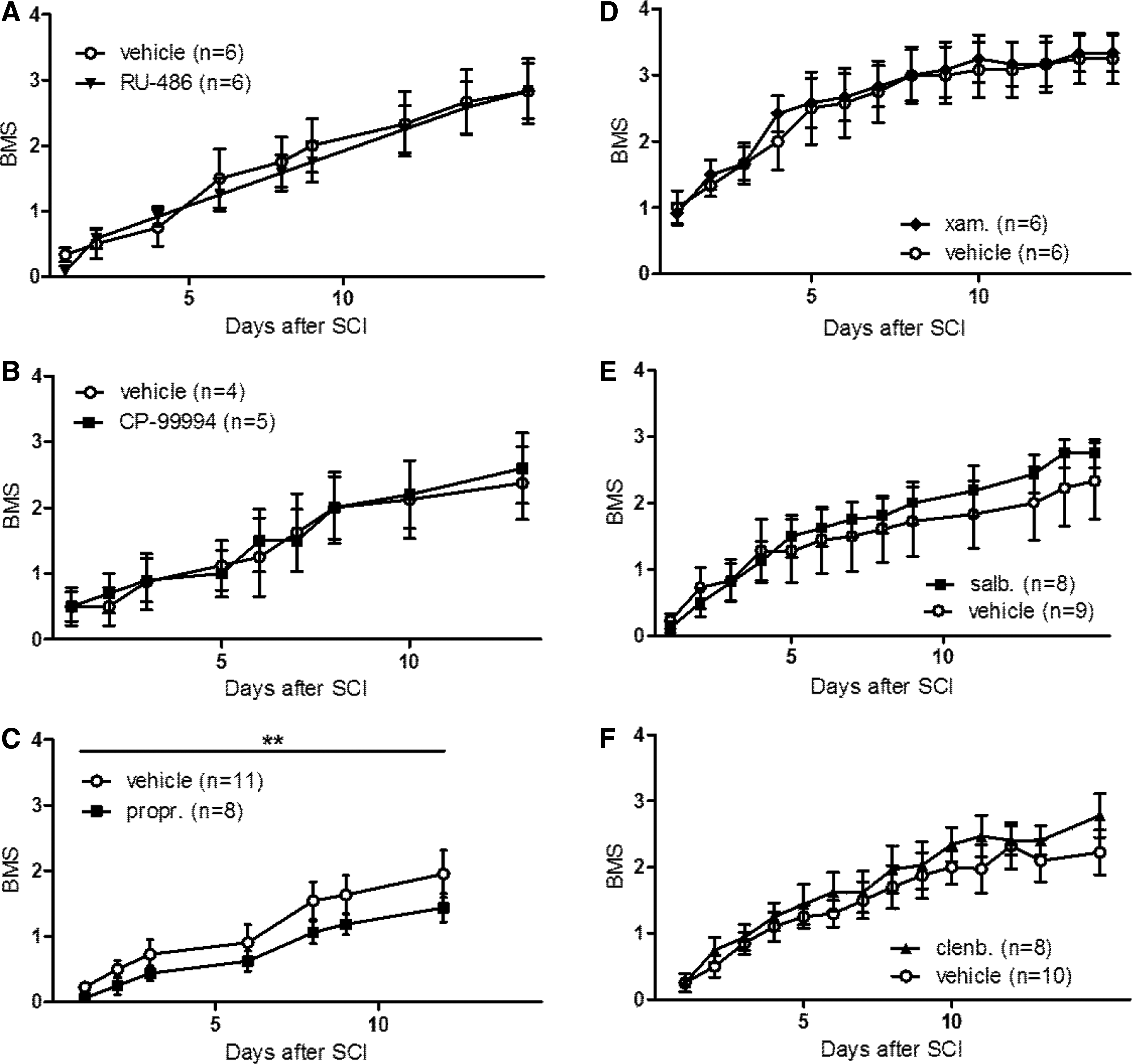
Inhibition of the adrenergic system (part of the sympathetic nervous system) with propranolol reduced functional outcome after spinal cord injury (SCI), whereas modulation of the other stress axes did not affect functional recovery.
Inhibition of the NNA does not affect functional recovery after SCI
To investigate the role of the NNA in functional recovery after SCI, mice have been acutely treated with CP-99994 (Fig, 2B). At the end of the observation period, the vehicle mice reached a BMS score of 2, meaning extensive ankle movement. The CP-99994 treatment group obtained this score as well. The BMS was not altered after CP-99994 treatment, compared with treatment with vehicle.
Inhibition of the beta-adrenergic pathway of the sympathetic axis worsens the functional outcome after SCI
The relevance of the sympathetic axis has been investigated by inhibiting the beta-adrenergic pathway with propranolol (Fig. 2C). BMS data indicated that propranolol slightly worsened functional recovery after SCI compared with the vehicle group (Fig. 2C; mean BMS score 1.4 vs. mean BMS score 2, respectively). Stress pathways are closely involved in secondary damage after SCI (e.g., excessive inflammation) resulting in neurodegeneration and reduced functional outcome. Therefore, we analyzed the perilesional presence of selected immune cells (CD4+ T-cells and macrophages/microglia) in addition to other important parameters that correlate with functional recovery (lesion size and astrogliosis; Supplementary Fig. S1). The decrease in functional outcome after propranolol treatment was accompanied with a decrease in the number of CD4+ T-cells (Supplementary Fig. S1D). Interestingly, the other neuro-immune parameters (lesion size, astrogliosis, and macrophage/microglia presence) were not influenced after treatment (Supplementary Fig. S1A-C).
β-adrenoceptor agonism with xamoterol, salbutamol, or clenbuterol does not improve functional recovery after SCI
The results described above suggest the importance of the beta-adrenergic pathway in functional recovery given that blocking it surprisingly reduces locomotion function. Therefore, we aimed to investigate the potential association between β-adrenoceptor (AR) activation and functional recovery after SCI in our T-cut hemisection mouse model, by stimulating the beta-adrenergic pathway. To this aim, mice were treated with either xamoterol (β1-AR agonist), salbutamol (β2-AR agonist), or clenbuterol (β2-AR agonist). Neither xamoterol, salbutamol, nor clenbuterol affected functional recovery after SCI, compared with the vehicle control, as no differences in the BMS were found (Fig. 2D-F).
Treatment with different stress pathway modulators increases the mortality rate after SCI
After RU-486, propranolol, salbutamol, and clenbuterol treatment more animals died compared with the vehicle control groups (Fig. 3), which indicates these mediators should be handled with care. However, the general health status of the animals has not been affected substantially, which is reflected by the BMS-results after RU-486, salbutamol, and clenbuterol treatment, as there was no change visible compared with controls animals (Fig. 2).
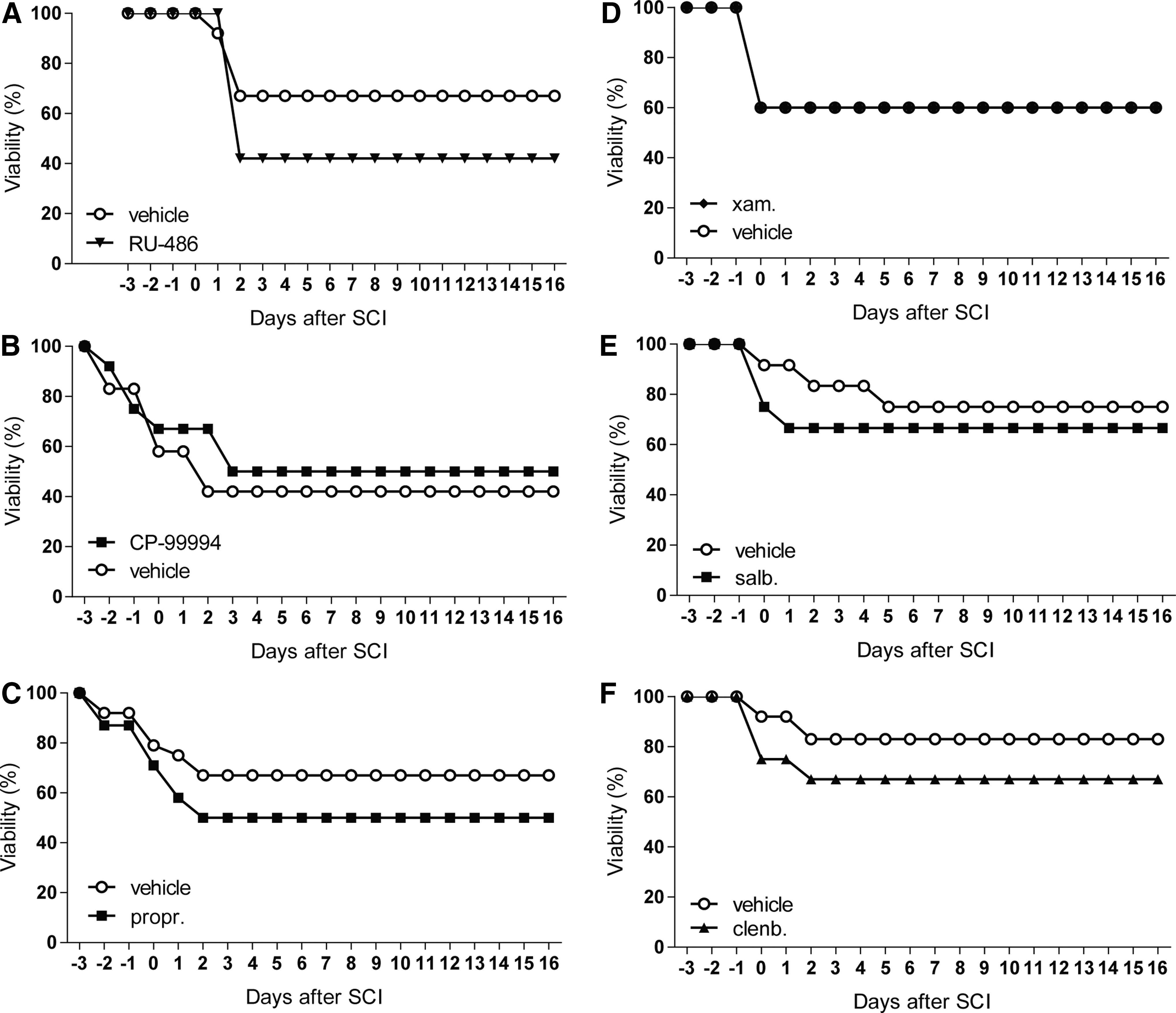
Treatment with different stress pathway modulators increases the mortality rate after SCI.
Discussion
In this study, we investigated the hypothesis that inhibition of either one of three key stress axes protects from neuronal and inflammatory damage and improves functional recovery after SCI in mice. Our results show that for the first time in a comparative study, the inhibition of the three major stress axes (HPA, SA, NNA) around the time of injury has no beneficial effect on functional recovery. Our unexpected negative results on the functional level (BMS score) are of great importance from a scientific and ethical aspect, as they will help to reduce research waste and avoid unnecessary animal experiments. Moreover, our results suggest, that beta-blockade, but not the interference with cortisol or substance P associated stress responses, may in fact harm the healing process. Our findings were especially surprising in the context of other traumatic central nervous system (CNS) disease models (e.g., aging, multiple sclerosis, and stroke), where blocking stress pathways has been shown to improve functional recovery. For example, after stroke, administration of the beta-blocker propranolol drastically reduced loss of function. A main reason for this may be the immunomodulatory effects of beta-blockers. 13 –16 Therefore, it appeared plausible that detrimental inflammation after SCI could be improved by sympathetic axis inhibition similar to other CNS diseases.
However, we are the first to report the lacking effects of sympathetic nervous system (SNS) inhibitors on functional recovery in SCI. Previous studies have reported beneficial effects of the beta-blocker butoxamine on the immune response after SCI. 9,17 SCI at thoracic level 3 (T3) leads to post-traumatic immune suppression by inducing splenocyte cell death, B-cell apoptosis, and suppressing antibody synthesis towards non-self-antigens. However, this has been shown to be reversible following treatment with the beta-blocker butoxamine, suggesting that excessive beta-adrenergic pathway signaling was responsible for the immune suppression. 9 Consistent with our study, injury at T9 did not induce post-traumatic immune suppression and modulating the beta-adrenergic pathway had no effects on the immune response. 17 We have induced SCI at T8 to avoid interference with the immune system in this manner. Considering the slight detrimental effect of propranolol on functional outcome, we hypothesized that stimulating the beta-adrenergic pathway may promote functional regeneration after SCI.
Beta-adrenoceptors (ARs) are abundantly expressed in the CNS. 18 Their expression on glial cells, astrocytes, and neurons is implicated in neuroprotection, neurotrophic and anti-inflammatory reactions both in vitro and in vivo. 19 –21 However, neither acute β1-AR agonism with xamoterol or β2-AR agonism with salbutamol or clenbuterol promoted functional recovery. The effects of β1-AR agonism in the context of SCI have not been previously investigated. Our results on the effects of β2-AR are in concurrence with other studies using rat and mouse transection SCI models. 22,23 In addition, others have investigated the effects of the β2-AR agonists in contusion SCI models and in combination with other experimental therapies, showing opposing results. Salbutamol treatment combined with functional electrical stimulation induced positive effects for spinal cord injured individuals. 24 Two studies showed no effects on locomotion recovery by using clenbuterol alone, although it may be a necessary co-factor to prevent muscle atrophy. 22,23 Zeman and colleagues investigated the effects of clenbuterol after SCI in a rat contusion model. 25 They showed that clenbuterol improves locomotion and reduces tissue loss, compared with vehicle control.
Differences between the various SCI animal models may explain these contradictory findings. For example, Zeman and colleagues 25 used a rat contusion injury model whereas we have used a T-cut hemisection mouse model in this study. In a contusion injury model, spared fibers may contribute to functional recovery. 26 –28 We use the T-cut hemisection in order to precisely transect the corticospinal tract allowing us to analyze true regeneration of the cut fibers and exclude the possibility of spared fibers. 28 Lesion severity may also play a big part, as positive effects are observed in contusion injury models with possible effects on spared fibers whereas no effects are often reported for complete or hemi-section injury models.
In addition to hyper-activation of the SNS, traumatic SCI activates the HPA axis, increasing levels of circulation glucocorticoids (GC), which bind to receptors on different CNS cell types, causing a multitude of effects. For many years, high iatrogenic doses of the GC receptor agonist methylprednisolone (MP) have been applied in the clinic within 8 h after SCI in order to suppress the immune system and decrease inflammation. However, there is a lot of debate about the administration of GC, and particularly MP, after SCI. First, if general immunosuppression is the aim, this should be undertaken with caution given that the system is already severely immune compromised after SCI which may result in serious infections or even early death. 5 Second, the clinical application of MP is based on a National Spinal Cord Injury Study (NASCIS) from the 1980s where patients treated with steroids within 8 h showed a statistically significant improvement in motor scores at 6 months and at 1 year after SCI. 29 However, a lot of methodological controversy regarding the NASCIS trials exists and the NASCIS III trial was unable to replicate the findings of NASCIS II, which identified the 8-h cut-off. Others also have questioned the extent of neurologic improvement. 29 In contrast to the NASCIS approach, which suppresses the immune system with high iatrogenic doses of MP, we systemically inhibited the binding of endogenous cortisol to its receptors in order to improve functional recovery—without effect. 30 Thus, manipulation of the HPA axis is not a promising approach to improve functional recovery shortly after SCI. Future studies may reveal more effects at later time-points after SCI.
Finally, inhibition of the neurotrophin neuropeptide axis also was inefficient to improve functional recovery after SCI. Our hypothesis was based on studies where substance P (SP) has been associated with increased blood–brain barrier (BBB) permeability and edema development following both TBI 31,32 and stroke, 33 while antagonism of the NK1 receptor reduced BBB permeability as well as edema, improving functional outcome after TBI and stroke. 31,32,34 Further, it has been demonstrated that SP stores are reduced following injury, indicative of SP release, whilst NK1 receptor immunoreactivity increased. 35 We have shown that NK1 receptor inhibition is ineffective after hemisection SCI in mice consistent with findings in a rat balloon compression SCI model. 36 In contrast, it has been reported that stimulation of the NNA increased functional recovery after compression and contusion SCI in mice and rats respectively. 37,38 Hence, manipulation of the NNA results in inconsistent findings.
Taken together our results show that the immunological changes after stress are complex and can be both pro- and anti-inflammatory, depending on the time point after SCI and the duration of the stress stimulus. Acute stress activates the SNS and provides a rapid redistribution of immune cells into target organs. 6 The HPA axis is both stimulated after acute and chronic stress. Acute stimulation induces a pro-inflammatory Th1 response whereas chronic stimulation leads to an anti-inflammatory Th2 response. 6 Activation of the NNA leads to the release of neuropeptides which then stimulate receptive immune cells in the CNS such as mast cells. 6 There are no data available on differences between acute and chronic activation of this axis. The outcome of the inflammatory response also will depend on which cell types are present and which receptors they express when the stress stimulus occurs. The receptors of the different stress pathways are expressed on many, if not all, immune cells in the CNS, 21 especially on macrophages and T cells. The outcome of the inflammatory response will depend on whether these cells have a pro- or anti-inflammatory phenotype. In our experiments, in vivo, propranolol decreased the number of CD4+ T cells after SCI. This reduction in helper T cells may dysregulate the action of CD8+ T cells causing a Th1-response and axonal loss, as shown in a mouse model for multiple sclerosis. 39,40 In contrast, others report promotion of differentiation towards Th2 cells after β-AR agonism. 40,41 Thus, β2-AR modulation on lymphocytes regulates the level of lymphocyte activity differentially, depending on the time of receptor engagement in relation to the activation and differentiation state of the cell, the molecular signaling pathway activated, and the cytokine microenvironment, which are all very variable in each stage of SCI (reviewed by Sanders). 42
In conclusion, our counterintuitive results suggest that injury-induced stress responses are so highly controlled by the body that inhibiting one of the stress axes has no major impact. When modulating one single axis, knock-on effects on the other axes cannot be excluded and alternative pathways may compensate for a blocked stress axis. 6,21 In addition, the receptors for the main stress pathway mediators are expressed on many, if not all, immune cells and CNS cells. 19 –21 Thus, this also may contribute to the contradictory results observed.
The number of studies on stress pathway modulation after SCI are limited and this may be due to publication bias. 43 For instance, only two papers outlining the effects of beta blocker treatment after SCI have been published to date. 9,17 This may suggest that either no effects or detrimental effects of β-AR agonism have been found but not published. Although negative results are difficult to publish, they provide extensive implications, high clinical relevance and can help avoid unnecessary experiments. Thus, their documentation is invaluable. The absence of beneficial effects after stress pathway modulation in the acute phase after SCI may question the use of stress axis modulators as an experimental strategy to improve functional recovery after SCI in future studies. It also may provide some reassurance to patients and their doctors that the high level of stress experienced at the time of injury does not appear to interfere with functional recovery down the line. The negative impact of propranolol suggests that beta-blockers should be used with great care in SCI patients. These data are of major importance to other researchers aiming to modulate the three main stress axes in an effort to promote functional recovery after SCI. Our findings are vital to help avoid scientific waste and potentially futile animal experiments.
Footnotes
Acknowledgments
The authors thank Dr. Leen Timmermans (Hasselt University) for excellent technical support.
Author Disclosure Statement
This study was supported in part by grants from ‘Fonds voor Wetenschappelijk Onderzoek – Vlaanderen’ (FWO) to SH (G.0389.12, G0A5813, G.0834.11N, G0A58113FWO), from FWO to NG (1.2.917.14N), and from “Agentschap voor Innovatie door Wetenschap en Technologie” (IWT) to SL (131230).
For the other authors, no competing financial interests exist.
Supplementary Material
Supplementary Methods
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.