
Abstract
Patients with chronic traumatic brain injury (TBI) requiring long-term, permanent care suffer a myriad of clinical symptoms (i.e., impaired cognition, fatigue, and other conditions) that persist for years beyond the acute brain injury. In addition to these comorbid clinical symptoms, chronic TBI patients exhibit altered amino acid and hormonal profiles with distinct cytokine patterns suggesting chronic inflammation. This metabolic link suggests a role of the gut-brain axis in chronic TBI. Thus, we utilized a two-site trial to investigate the role of the gut-brain axis in comorbidities of chronic TBI. The fecal microbiome profile of 22 moderate/severe TBI patients residing in permanent care facilities in Texas and California was compared to 18 healthy age-matched control subjects working within the participating facilities. Each fecal microbiome was characterized by 16S(V4) ribosomal RNA (rRNA) gene sequencing and metagenomic genome sequencing approaches followed by confirmatory full 16S rRNA gene sequencing or focused tuf gene speciation and specific quantitative polymerase chain reaction evaluation of selected genera or species. The average chronic TBI patient fecal microbiome structure was significantly different compared to the control cohort, and these differences persisted after group stratification analysis to identify any unexpected confounders. Notably, the fecal microbiome of the chronic TBI cohort had absent or reduced Prevotella spp. and Bacteroidies spp. Conversely, bacteria in the Ruminococcaceae family were higher in abundance in TBI compared to control profiles. Previously reported hypoaminoacidemia, including significantly reduced levels of l-tryptophan, l-sarcosine, ß-alanine, and alanine, positively correlated with the reduced levels of Prevotella spp. in the TBI cohort samples compared to controls. Although the sequelae of gut-brain axis disruption after TBI is not fully understood, characterizing TBI-related alterations in the fecal microbiome may provide biomarkers and therapeutic targets to address patient morbidity.
Introduction
Traumatic brain injury (TBI) and its accompanying chronic morbidities affect more than 2.5 million individuals annually in the United States. 1 Concern about the long-term effects of brain injury has grown recently with increased publicity of sport-related chronic traumatic encephalopathy 2 and military blast-related injury. 3 TBI is a complex neurological insult that can lead to chronic, progressive deterioration of patient health and increased morbidity. During convalescence, TBI-associated pathologies can advance to full disability after significant periods of relative health and normalcy. In the case of chronic TBI, where severity of the injury often requires residence in a long-term care facility, clinical symptoms are often complex and varied ranging from cognitive deficiencies to fatigue. In this patient population, supportive treatment is standard of care and additional understanding is needed to open new therapeutic avenues.
The sequelae of chronic TBI are not well understood. Aside from the recognized pituitary dysfunction that occurs in some patients, the cause(s) of the myriad of other long-term clinical symptoms and their mediators are uncertain. In our previous work, chronic TBI patients were found to exhibit abnormal metabolic responses and altered relationships between circulating amino acids, cytokines, and hormones. 2 These findings are consistent with reports that the gut-brain axis may be linked to neuropathologies. 4 –7
Notably, the rapidly expanding study of interactions between the gastrointestinal (GI) tract, immune system, and brain has led to a number of discoveries suggesting that the GI microbiome is a major influence on each of the systems. 8 –10 Further, recent next-generation sequencing (NGS) analyses have associated altered GI microbiomes with a growing list of neuropathologies, including Parkinson's disease, autism, Guillain-Barre syndrome, anxiety, and depression. 11 The GI microbiome is one of the most diverse communities of bacteria in the human body, with estimates of up to 1000 distinct species included in typical communities. 12,13 Current understanding is largely based on fecal analyses by NGS of the variable regions of the bacterial 16S ribosomal RNA (rRNA) gene or metagenomic analyses. 13 The large data sets created from various cohorts have clearly established that this community of bacteria contributes to basic physiology, immunity and inflammation. More recently, dysbiosis has been associated with altered cognition, behavior, and even mood disorders. 4 –6
Mild TBI is associated with alterations in gut metabolism soon after injury with direct impacts upon the intestinal mucosa, including loss of tight junctions, that contribute to increased intestinal permeability, inflammation, and malabsorption. 14,15 Patients with mild TBI also can develop sequelae years after injury, presenting with profound fatigue and altered cognition. Such mucosal alterations likely foster dysbiotic conditions to which the intestinal microbiota adapt, ultimately establishing an altered bacterial population that propagates the sequelae.
In the current study, we investigated whether the fecal microbiome is altered in chronic, moderate-severe TBI patients in permanent care facilities compared to healthy control subjects working at the facilities. We hypothesized that if TBI-induced dysbiosis indeed disrupts the gut-brain axis, the fecal microbiome will provide biomarkers and therapeutic targets to address additional sequelae observed in chronic TBI patients.
Methods
Ethics statement
All patients provided informed consent before blood drawing or collection of a fecal sample was attempted. All procedures were approved by the associated institutional review boards contracted by the Tideway facility at the Transitional Learning Center (TLC; Galveston, TX) and Centre for Neuro Skills (CNS; Bakersfield, CA). Samples were assigned a unique study number to minimize exposure of personal information.
Patients
TBI patients were targeted for recruitment in the study based on the long-term effects of their brain injury and not based on an initial trauma score (i.e., Glasgow Coma Scale). The effects of the previous brain trauma of enrolled TBI subjects were severe enough that they were incapable of independent or semi-independent living because of cognitive and physical issues and required permanent long-term residential care. A total of 22 participants were enrolled from both the Tideway TLC facility in Galveston, Texas and the CNS facility in Bakersfield, California to provide fecal samples. Eighteen individuals were recruited to the control cohort composed primarily of facility workers who shared both environment and some meals with the TBI patients. TBI participants residing in the Tideway TLC facility ate meals prepared at an on-site cafeteria. Participants residing at the CNS facility had access to their own kitchen and prepared dietician-approved meals independently or with assistance. None of the participants in this study were vegetarian or on a regimented pre-biotic, probiotic, or nutritional supplement plan.
After informed consent was obtained, participants were fed a standardized meal and blood was collected 90 min later for serum amino acid analysis. Participants were provided fecal sampling kits and were instructed on sampling technique. Fecal samples were collected using a commercial kit (Omnigene-GUT; DNA Genotek Inc., Ottawa, Ontario, Canada) as close to ingestion of the study meal as possible. Samples from control participants were self-collected, whereas TBI participants were assisted by center staff. Demographics and details of each cohort are provided in Table 1.
Study Cohort Demographics
Average values for each subgroup are shown in bold text with (SDEV) indicated. Biopool (BP) assignments for the second round of Ion Torrent–based NGS testing are indicated. Several fecal DNA samples were of insufficient quality for molecular evaluations and were not tested (NT) in the biopools, but were recovered for subsequent qPCR assays.
Bakersfield controls were significantly younger (p < 0.05) than any other subgroup, but the overall average age of controls and participants with TBI was not significantly different. There were no differences in average height or weight among the subgroups or main cohorts; however, the controls from Bakersfield had a significantly higher average BMI than the Galvestonians with TBI (p = 0.0087) and the combined TBI cohort (p = 0.03).
TBI, traumatic brain injury; SD, standard deviation; M, male; F, female; N, no; Y, yes; BMI, body mass index; N/A, not applicable; SDEV, standard deviation; NGS, next-generation sequencing; qPCR, quantitative polymerase chain reaction.
Microbiome analysis and DNA preparation
Preparation of DNA
Fecal samples were shipped to the Baylor College of Medicine's (Houston, TX) Alkek Center for Metagenomics and Microbiome Research facility on dry ice after being frozen and stored at −80°C in the stabilizing chemical mixture provided in the DNA Genotek OMNIgene GUT kit (DNA Genotek). After an initial thaw of the material, cell lysing and DNA extraction were performed using the QIAGEN processing protocol (described in detail for Illumina NGS below; QIAGEN GmbH, Hilden, Germany) recommended by the National Institutes of Health (NIH)–Human Microbiome Project. 16,17 At The University of Texas Medical Branch (UTMB), a second DNA extraction was performed on the original fecal material (described in detail below for Ion Torrent full 16S rRNA gene NGS or subsequent quantitative polymerase chain reaction [qPCR] analyses). For both approaches, DNA quality was assessed for total bacterial content and human genomic material by qPCR.
16S rRNA gene sequencing and compositional analysis
16S rRNA gene sequencing methods were adapted from the methods developed by the NIH–Human Microbiome Project. 16,17 Bacterial DNA was extracted using the PowerMag Microbiome DNA isolation kit (QIAGEN), following the manufacturer's instructions. The V4 region of the 16S rRNA gene was amplified by PCR and sequenced using barcoded Illumina adapter-containing primers 515F and 806R 18 on the MiSeq platform (Illumina, Inc., San Diego, CA), using the 2 × 250 base pair (bp) paired-end read protocol yielding pair-end reads that overlap almost completely. Sequencing read pairs were demultiplexed based on the unique molecular barcodes, and reads were merged using USEARCH (v7.0.1090). 19 Merging allowed zero mismatches with a minimum overlap of 50 bases, and merged reads were trimmed at the first base with a Q ≤ 5. In addition, a quality filter was applied to the resulting merged reads and those containing above 0.05% expected errors were discarded. Sequences were step-wise clustered into operational taxonomic units (OTUs) at a similarity cut-off value of 97% using the UPARSE algorithm. 20 Chimeras were removed using USEARCH (v7.0.1090). OTUs were determined by mapping the centroids to the SILVA database 21 containing only the 16S rRNA gene V4 region to determine taxonomies. A custom script constructed a rarefied OTU table (rarefaction was performed at only one sequence depth) from the output files generated in the previous two steps for downstream analyses. We utilized multiple quality-control measures, including the use of non-template controls, at the microbial DNA extraction, 16S rRNA gene amplification, and amplicon sequencing processes. Resulting OTU tables were rarified to 5953 reads per sample.
Metagenomic shotgun sequencing and analysis
Individual libraries constructed from each sample were sequenced using the 2 × 100 bp paired-end read protocol on the HiSeq platform (Illumina). The process of quality filtering, trimming, and demultiplexing was performed using a pipeline developed at the Baylor College of Medicine that uses a number of publicly available tools, such as Casava (v1.8.3; Illumina) for the generation of fastqs, Trim Galore and cutadapt 22 for adapter and quality trimming, and PRINSEQ 23 for sample demultiplexing. Additionally, Bowtie 2 (v2.2.1) 24 was used to map reads to custom databases for bacteria, viruses, human, and vectors and remove non-bacterial reads from the data set. For bacterial reads, the highest identity match was chosen. If there were multiple top hits, the lowest common ancestor was determined, but these reads did not contribute to the analysis. Reads whose genomic coordinates overlapped with known Kyoto Encyclopedia of Genes and Genomes (KEGG) orthologs (KOs) were tabulated.
Coding sequences from reference genomes that have not been specifically annotated by KEGG were aligned to all known KOs. Any coding sequence that had >70% identity and >70% query coverage to a known KO was assigned to that KO. This process, in effect, created links between new genomes and the KEGG database. KEGG modules (M numbers) were calculated stepwise and determined to be complete if 65% of the reaction steps were present per detected species and for the metagenome as a whole. Pathways were constructed for each taxa and metagenome by calculating the minimum set through MinPath 25 resulting from the gene orthologs present. The number of reads matching a KEGG module was averaged across the TBI or control cohorts and compared (unpaired Student's t-test) to identify modules that were significantly over- or under-represented by the TBI microbiome profiles.
Ion torrent 16S rRNA gene NGS and qPCR evaluations
To confirm the 16S rRNA gene NGS, the second preparation of DNA was analyzed by Ion Torrent 16S NGS and qPCR at UTMB for selected organisms. This second DNA preparation addressed differences in recovered DNA species created by different kits as well as the age of the previously extracted material. For the second extraction, after thawing the original material preserved in the OmniGene kit solution, suspended fecal matter was diluted 1:1 in RNeasy PowerMicrobiome kit-provided lysis solution, incubated at 55°C for 30 min (QIAGEN, Germantown, MD), and then subjected to bead beating utilizing 0.1-mm glass bead-based (QIAGEN) homogenization (5 min at 30 Hz) in a TissueLyser LT (QIAGEN). Clarified liquid fractions (13,000g for 1 min) were mixed with internal reflection spectroscopy solution (QIAGEN) and incubated at 4°C for 5 min before final extraction of DNA using a MagNA Pure 96 automated nucleic acid extraction DNA and viral NA small-volume kit (Roche Applied Science, Indianapolis, IN).
Ion Torrent fecal microbiome sequencing was carried out using a fusion-PCR method. Briefly, fusion primers were designed in accord with the manufacturer's guidelines (Ion Amplification Library Preparation–Fusion Method; Life Technologies, Carlsbad, CA) using Ion Xpress Barcodes linked to 16S gene primer pairs targeting five overlapping contigs that covered hypervariable regions 1–8 sequencing over 90% of the gene. 26 DNA from 36 individual fecal samples were stratified into 10 biopools created by equal mixing of individual material that then were used as templates for creation of fusion 16S libraries. Stratification was accomplished for healthy control samples based solely upon body mass index (BMI; above or below 30), creating two biopools (Table 1).
TBI samples were stratified by location, BMI, and whether residence time was above or below the median time participants lived in either the Bakersfield (average of 232 months) or Tideway (average of 241 months) facility, creating an additional eight biopools (Table 1). Fusion PCR libraries, generated in a c1000 thermocycler (Bio-Rad Laboratories, Hercules, CA), were purified using QIAquick spin-columns (QIAGEN) and quantified using a spectrophotometer (Bio-Rad) before being diluted and then sequenced on an Ion Torrent Personal Genome Machine using 400-bp read kits together with 316 size chips, following the manufacturer's instructions (Life Technologies).
One TBI biopool (BP; #3) failed to meet minimal quality metrics and was not analyzed further. Sequencing reads from the other nine libraries were filtered for quality and binned according to Ion Xpress barcodes using Ion Torrent Suite software (v5.0.5). Filtered sequencing reads in FASTQ format were normalized using the FASTQ groomer tool function in the web-based Galaxy software. 27 Next, each barcoded read was trimmed to remove primer sequences and filtered to the expected size before being compared to the SILVA 16S rRNA gene database using Bowtie 2 software. 24,28 Curated reads showed >98% alignment to the database and ranged from 8E5 to 1.2E6 sequences to establish genera-level hit rates. Where multiple calls to the same genera were made, the number of hits was added accordingly. These numbers were then converted to the percentage of the total to give an overall relative proportion for each biopool.
Ion Torrent sequencing of the tuf gene for Bacteroides and Prevotella genera also was performed through fusion-primer design using Ion Express barcodes. Two sets of primers covering conserved areas flanking the hypervariable regions were designed based on multiple sequence alignment of 13 Bacteroides and 14 Prevotella species sequences obtained from Genbank. Each primer pair was designed to sequence both DNA strands of the specified gene region (primer pair 1, F: CAAACCGCATGTWAAYRTTGGTAC, R: CCRTCCATCTGDGCAGCACC; primer pair 2, F: CGTACTTCTBGCHCGTCAGGT, R: ACCTGTAGCHACNGTACCACG), producing coverage of approximately 50% of the tuf gene. Data processing and analysis of sequence reads were performed as described above. To identify specific species, NGS-derived tuf sequences were compared against a customized database of reference sequences of Bacteroides and Prevotella species sequences obtained from Genbank.
qPCR evaluation
Based on the sequence data from both approaches and calculated community profiles, qPCR primers were designed using Oligo Architect (MilliporeSigma; Burlington, MA) or obtained from the literature (Supplementary Table S1) to quantify organisms or genera of interest. These assays were evaluated in silico for specificity to their particular genera/species and determined to be specific through amplification of a single PCR product from a biopool of the NGS-evaluated control and TBI fecal DNAs. PCR products were subsequently cloned and Sanger sequenced, authenticating specificity. These cloned amplimers were then used to create high-resolution melt temperatures that served as a further confirmation of identity after SYBR green-based, real-time amplification.
Each 25-μL qPCR was carried out on each fecal DNA sample using: 12.5 μL of iQ SYBR green supermix™ (Bio-Rad), 1 μL of each forward and reverse (5 μM) primer, 9.5 μL of nuclease-free water and 1 μL of of DNA template. qPCR was completed in a c1000 thermocycler equipped with a CFX™ reaction module (Model info; Bio-Rad). By this method, all but two of the fecal samples (controls A19 and A20 failed to meet minimum quality assessments) were successfully evaluated. Fluorescent signal data were collected at the end of each extension step. Starting quantity values were extrapolated from standard curves of plasmids harboring the PCR targets run in parallel for each target.
Amino acid analysis
Post-prandial plasma amino acid levels were assessed for both TBI and control subjects after consuming a standardized meal as previously described. 2 Briefly, after overnight fasting, subjects consumed a standardized mixed-macronutrient breakfast providing 762 kcal composed of 40.3 g of protein, 34 g of fat, and 74 g of carbohydrate (26 g of sugar). Approximately 90 min after completing the meal, 5 mL of blood was drawn from subjects by venipuncture. Fresh blood was separated by centrifugation at 3000 RPM for 20 min and serum fractions were stored at −80°C (or on dry ice while in transit from Bakersfield to Galveston) until analysis. Serum amino acid concentrations were determined after protein precipitation using a Hitachi L8800 amino acid analyzer (Hitachi, Tokyo, Japan), according to manufacturer guidelines.
Statistical analysis
Bacterial diversity was assessed by calculating the number of observed OTUs and the Shannon diversity index (SDI) based on 16S rRNA gene compositional analysis. The number of observed OTUs measures bacterial richness in a sample, whereas the SDI measures both richness and evenness. Between sample diversity, or resemblance, was assessed with weighted and unweighted UniFrac metrics and plotted using principal coordinate analysis (PCoA) ordination. 29 To examine the contribution of different taxa to diversity and community composition, the relative abundances of taxa at the OTU, genus, and phylum level were calculated. Diversity data were analyzed using R software (R Foundation for Statistical Computing, Vienna, Austria). 30 Differences between alpha diversity in TBI versus controls were assessed with Wilcoxon rank-sum tests. Differences in beta diversity (UniFrac distance) were assessed using permutational multi-variate analysis of variance (PERMANOVA). All p values were adjusted for multiple comparisons with the false discovery rate algorithm. 31 MixOmics 32 was implemented in R (version 3.3; R Foundation for Statistical Computing) 30 to determine the correlations between bacterial relative abundance and the concentration of selected amino acids. 33 MixOmics used sparse partial least squares regression and was performed in canonical mode with least absolute shrinkage and selection operator (LASSO) penalization.
Statistical analyses of the qPCR data were performed using Excel™ (Microsoft Corp., Redmond, WA) or Prism (v7.0e; GraphPad Software Inc., La Jolla, CA) software packages. Clustering analysis was completed using Morpheus web-based software (Broad Institute, Cambridge, MA). For comparisons of the qPCR absolute abundance data, statistical significance was determined using multiple t-tests by the Holm-Sidak method (Prism; GraphPad Software). Each qPCR target was analyzed individually, without assumption of a consistent standard deviation (SD). A p value of <0.05 was considered significant.
Results
Study cohort
Fecal samples were collected from a cohort of patients with chronic, moderate-to-severe TBI residing in permanent care facilities located in Galveston, Texas and Bakersfield, California (n = 22) and from healthy control subjects (n = 18). The control cohort was enrolled from the care facilities in an effort to limit potential confounding factors, given that they shared both environment and ate some of the same meals with the TBI patients. Demographics of the cohorts are summarized in Table 1. In general, patients from the Texas cohort were minimally medicated whereas a more-aggressive drug therapy approach was utilized in the California cohort. All but one of the TBI samples were collected from participants who had not taken any recent antibiotic treatments. Similarly, only one of the controls was actively taking amoxicillin at the time of fecal sampling. Most of the other medications were oral dietary supplements (e.g., fish oil) or medications related to mood disorders (e.g., Zoloft).
Individuals in the Galveston program resided in private or semiprivate rooms within one building. Meals were planned and supervised by a dietician and plated for the individual. When the individuals went on outings, they were free to eat ad lib. Individuals in the Bakersfield program lived in one- or two-bedroom apartments within a large apartment complex. They followed a meal-planning menu supervised by a dietician and went to the grocery store to purchase their own food. Meals were prepared by that individual with help, as needed, and they were also free to eat ad lib when on outings.
There were no significant differences between the two main cohorts (overall TBI vs. overall control) regarding average age, height, weight, or BMI (multiple t-test, p > 0.05). Comparisons of the average time post-TBI injury revealed no differences between the two sites nor were there differences between a facility cohort and the overall average (p > 0.99). Considering each facility subgroup, the Bakersfield controls were significantly younger (p < 0.05) than any other subgroup, but the overall average age of controls and participants with TBI was not significantly different. There were no differences in average height or weight among the subgroups; however, the control cohort from Bakersfield had a significantly higher average BMI than the Galvestonians with TBI (p = 0.0087) and the combined TBI cohort (p = 0.03). Because weight/BMI differences may be significant contributors to the intestinal microbiome profile, we completed all analyses of 16S gene sequencing data and qPCR outcomes both by comparing the overall TBI versus overall control and by comparing site-specific control versus site-specific TBI results. This approach also addressed any potential impact for differences in drug therapy regimens between the facilities.
Shifted fecal microbiomes were identified in chronic TBI cohorts compared to controls
Fecal DNA from each person in the cohort was initially evaluated for quality before subsequent molecular evaluations. Four samples failed to meet quality metrics for amplifiable DNA (16S gene and human targets) and were excluded from the NGS analyses (Table 1; not tested [NT]). The remaining DNA samples were subjected to 16S rRNA gene V4 sequencing. Inclusion of all available samples from both collection sites illustrated that the microbiome profiles were significantly different between TBI and controls, both by weighted UniFrac (Fig. 1A; p = 0.002) and unweighted UniFrac (Fig. 1B; p = 0.005) PCoA analysis. UniFrac is a distance metric that incorporates phylogenetic relatedness of taxa in the analysis. The weighted UniFrac metric considered relative abundance of bacterial OTUs, showing that the overall structure of the microbial communities was different between groups (Fig. 1A). The unweighted UniFrac metric evaluated only the presence or absence of bacterial OTUs and demonstrated that the community structure also was significantly different between TBI and controls (Fig. 1B).
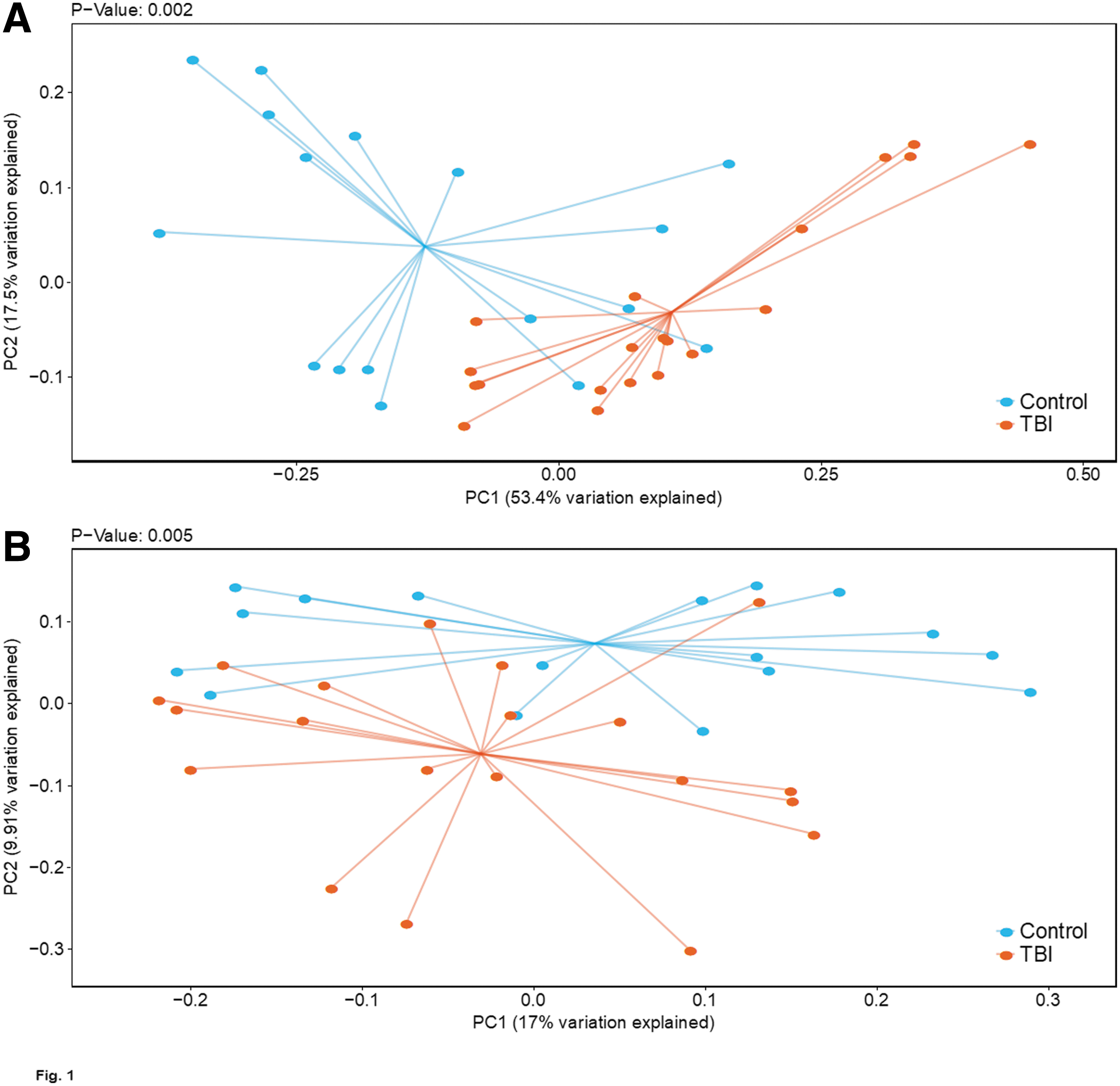
Comparison of fecal microbiome community structure of TBI and control cohorts determined by 16S V4 NGS. Weighted (
To evaluate whether the observed differences were maintained when only a single location was compared, we reran the weighted UniFrac PCoA analysis between TBI and matched controls from each site, and confirmed that significant differences between TBI and controls persisted in both the Galveston (p = 0.002) and Bakersfield (p = 0.006) cohorts (Supplementary Fig. S1A,B). Notably, the overall relative abundance of bacterial genera from samples collected at each site was comparable, and no single genera was found to be significantly different between locations (Supplementary Fig. S1A,B), supporting a lack of geographical bias and reducing concerns about the impact of other confounders. We next compared alpha diversity and bacterial relative abundance between the overall TBI and control groups. The number of observed OTUs was not significantly different between TBI and controls (p = 0.21), but the SDI (richness and evenness) was significantly higher in TBI cohorts compared to controls (p = 0.008; Fig. 2).
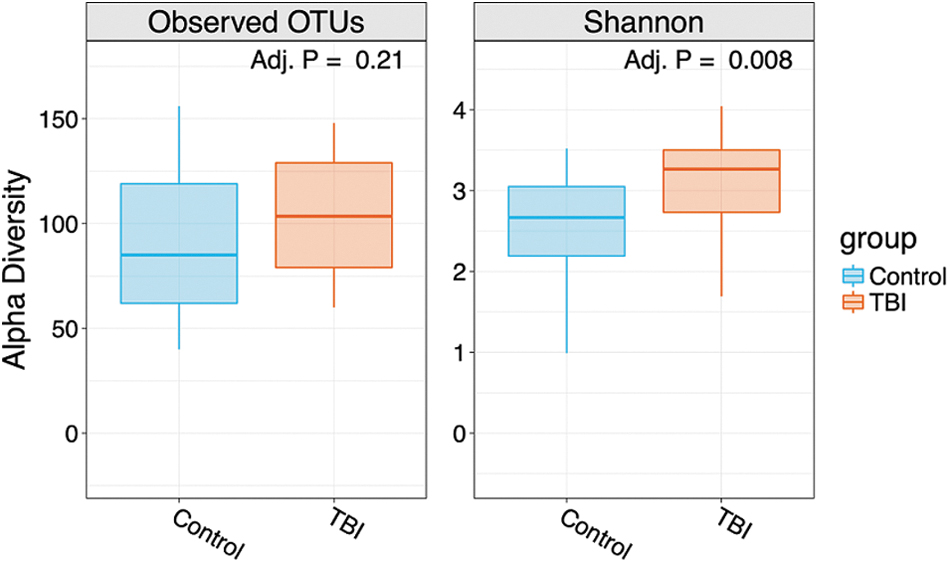
Alpha and Shannon diversity index (SDI) comparisons between TBI and control cohort community profiles. Overall numbers of OTUs were not statistically different (p = 0.21); as shown by alpha diversity, calculations were completed using R software (R Foundation for Statistical Computing, Vienna, Austria). 30 The SDI was significantly higher in TBI cohorts compared to controls (p = 0.008). Alpha-diversity differences between TBI and control data sets were assessed by Wilcoxon rank-sum tests. Evaluation of beta diversity (UniFrac distance) was completed by PERMANOVA. All p values were adjusted for multiple comparisons with the FDR algorithm. 31 Adj. P, adjusted p value; FDR, false discovery rate; OTUs, operational taxonomic units; PERMANOVA, permutational multi-variate analysis of variance; TBI, traumatic brain injury.
The data revealed that there were significant shifts, even at the phyla level, when the average TBI community structure was compared to the control cohort. As expected, the most abundant bacterial phyla were Bacteroidetes and Firmicutes, with the relative abundance of Bacteroidetes significantly higher in controls (p = 0.002) and the relative abundance of Firmicutes significantly higher in TBI (p = 0.002; Fig. 3A). Also, the relative abundance of Actinobacteria (p = 0.002) and Verrucomicrobia (p = 0.038) were significantly higher in TBI compared to controls (Fig. 3A). At the family level, Prevotellaceae (phylum Bacteroidetes) was significantly more abundant in controls (p = 0.03), and two unclassified genera of Firmicutes from the family Ruminococcaceae were significantly higher in TBI (p < 0.001; Fig. 3B). The relative abundance of the three significant bacterial families was indistinguishable between the two collection locations (data not shown).
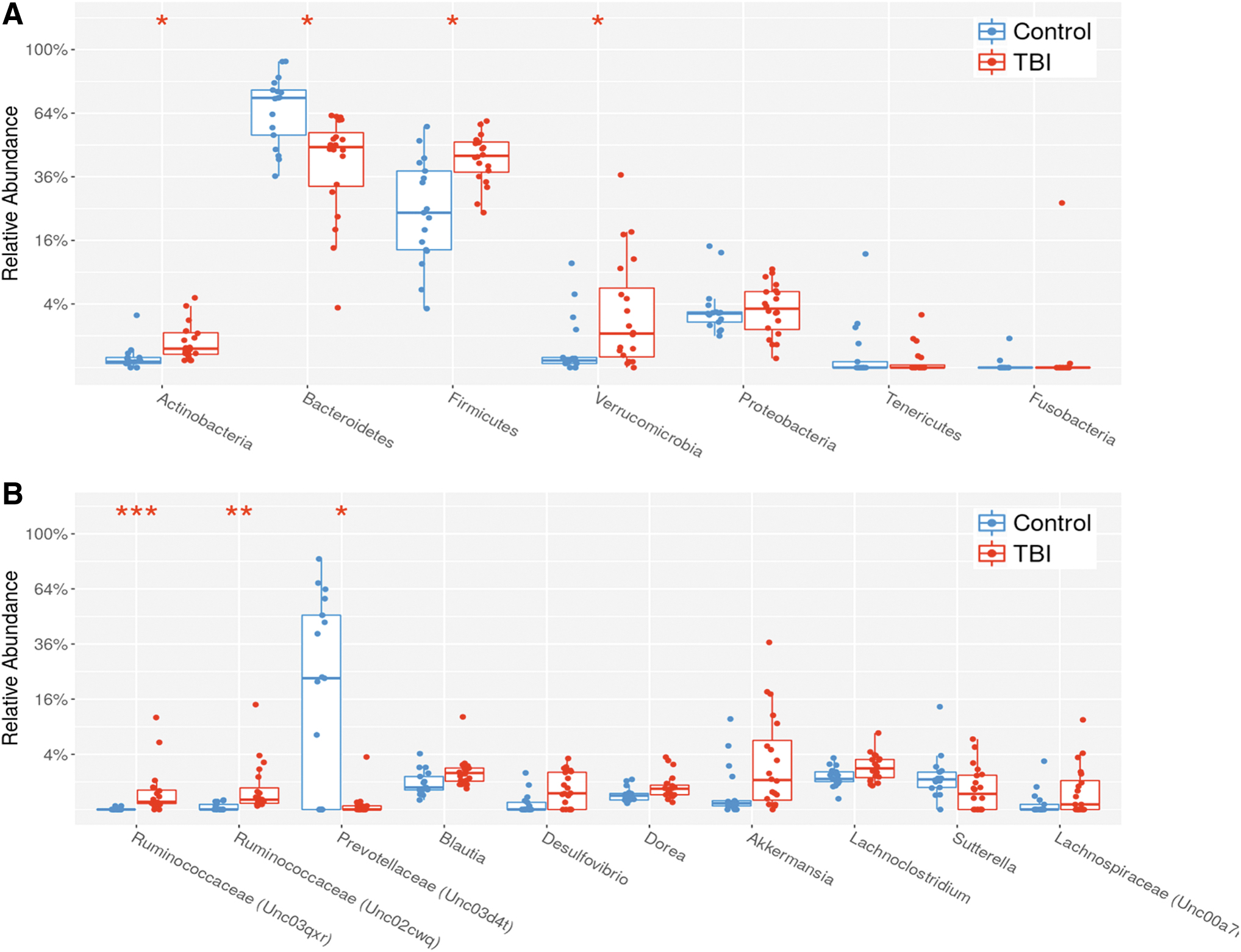
Comparison of the average relative abundance of indicated OTUs detected in TBI and control fecal bacterial communities. The plots illustrate individual levels for each indicated target (scatter plot) as well as the mean, standard deviations, and 95% confidence intervals (box and whiskers). (
Because 16S V4 rRNA gene sequencing data are limited to family/genus-level analyses, we performed metagenomic shotgun sequencing and analyzed the resulting data at the genus and species levels. These analyses corroborated the 16S rRNA gene evaluations, confirming that Prevotella was significantly more abundant in controls (p < 0.001), and Ruminiclostridium trended toward a greater abundance in TBI (p = 0.063; Supplementary Fig. S2). Within the metagenomic data, at the species level, Prevotella stercorea was significantly higher in controls (p < 0.001; Supplementary Fig. S2). However, qPCR assays, described below, targeting two distinct P. stercorea genes, showed that most samples were negative for this species. Because this organism was found in only a few samples from both controls and TBI, we concluded that other Prevotella species were likely missed by the NGS analyses.
To rigorously confirm the differences identified through Illumina 16S V4 and metagenomic comparisons, we also used a novel NGS approach that utilized Ion Torrent sequencing of five overlapping contigs created by amplification of the bacterial 16S rRNA gene. 34 This method produced bidirectional sequences from >90% of the bacterial 16S rRNA gene, including all of the variable regions, but had lower throughput relative to Illumina NGS. As a result, we created 10 biopools of fecal DNA (equivalent bacterial genomic contributions of the indicated samples mixed into a single DNA pool), by stratifying the cohorts by BMI, and by time, post-TBI (Table 1). Coincidentally, this also led to some Bakersfield- and Galveston-specific biopools, allowing for more-direct comparisons to confirm a lack of collection-site effects. One of the libraries (representing biopool 3) failed to meet minimal quality criteria and was excluded.
Results from the nine successful biopools (two control and seven TBI) corroborated much of the Illumina 16S rRNA V4 and metagenomic data, with some exceptions. At the genus level, the control samples had higher average relative abundance of Prevotella (88-fold increase over TBI sample average), Clostridium (5.7-fold), and Faecalibacterium (2-fold; Supplementary Table S2). Relative to the average control abundance, TBI samples had higher levels of Akkermansia (21-fold), Anaerotruncus (2-fold), uncultured Christensenellaceae (5.7-fold), Clostridium (5-fold), Collinsella (3-fold), Desulfovibrio (4-fold), Flavonifractor (4-fold), Odoribacter (16-fold), Parabacteroides (2.3-fold), and Streptococcus (8.8-fold; Supplementary Table S2). Comparisons between biopools also indicated no obvious differences associated with collection-site or BMI differences.
Interrogation of NGS 16S rRNA gene sequences against databases can mask differences in individual organisms that may share closely related sequences. To better differentiate specific organisms in fecal samples and address the P. stercorea outcome noted above, we developed and completed Ion Torrent NGS of the bacterial tuf gene for the Bacteroides, Parabacteroides, and Prevotella genera, because of the similarity of their 16S rRNA gene sequences and their differential abundance between control and TBI samples. This approach identified sequence matches to over 70 species within these genera and further confirmed that Prevotella spp. and Bacteroides spp. were generally more abundant in healthy controls compared to TBI, with notable exceptions to this generalization (Supplementary Table S3).
The tuf gene results revealed that P. copri was the most abundant Prevotella species in the controls (73% of tuf gene sequences representing a 54-fold greater abundance than measured in TBI samples), followed by the previously identified P. stercorea (4.6% of the control detections and 69-fold more abundant than in TBI samples). Bacteroides species that were more abundant in controls included B. plebius (3.7% of detections and 19-fold greater abundance) and B. massilensis (2.6% and 7-fold). However, this analysis identified specific Bacteroides species that were more abundant in TBI samples, including B. uniformis (32% of detections and 17-fold higher abundance), B. stercoris (19.6% of detections and 13-fold higher abundance), B. dorei (6.3% of detections and 8-fold more abundant), B. pectinophilus (2.4% and 3.6-fold), and B. vulgatus (6.5% and 2-fold). Finally, this analysis revealed species that were proportionally rare, but were detected in either controls or TBI samples only (Supplementary Table S3).
The consistency in the shifted TBI community structure was further confirmed by clustering analysis of qPCR data that provided absolute abundance (data are summarized in Fig. 4 and Table 2). These data were produced from a novel qPCR array we created based on the combined 16S rRNA and tuf gene NGS data as well as information from the literature. The array allowed simultaneous quantification of 94 bacterial (genus or species level), fungal, and viral targets as well as quantification of both total 16S and human glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Supplementary Table S1). Using this array, we evaluated every DNA sample with sufficient quality, allowing direct comparisons of the average absolute abundance profiles in TBI and control samples for these specific targets. This approach also provided accurate detection rates for each target across the samples. By this measure, there were no significant differences in average control 16S rRNA gene or human GAPDH copy numbers between TBI and control cohorts (Table 2).
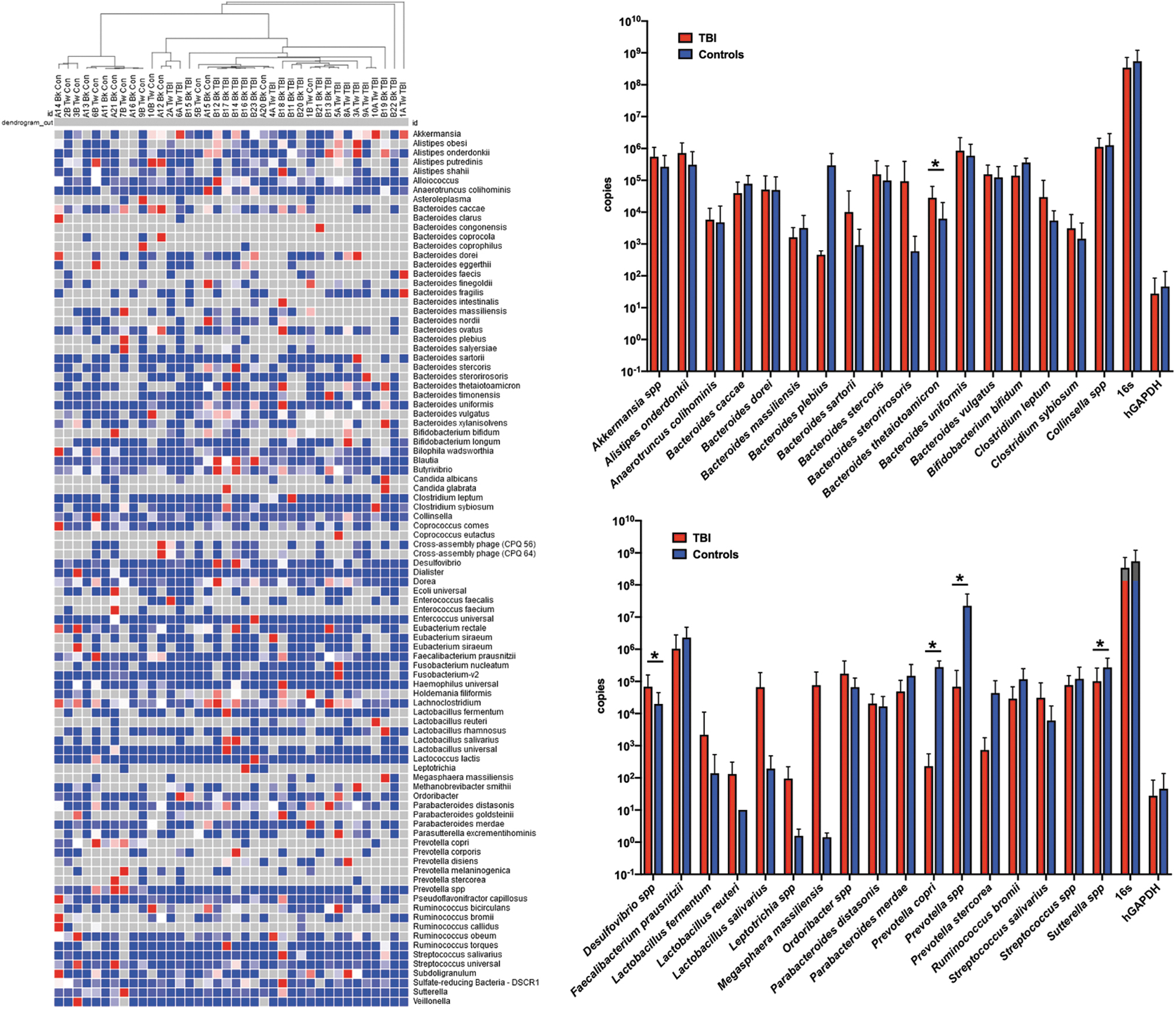
Significant differences in absolute abundance established by customized fecal microbiome qPCR array. Using a novel 96-target qPCR array developed for this study, absolute abundance for 94 selected bacterial targets and two controls (16S rRNA gene and human GAPDH gene), supported clustering analysis using Morpheus web-based software (Broad Institute, Cambridge, MA), was completed on individual TBI and control profiles as shown in the left panel. Statistically significant differences in the average qPCR absolute abundance data were determined using multiple t-tests by the Holm-Sidak method (GraphPad Prism, v.7.0e; GraphPad Software Inc., La Jolla, CA). Right panels show the average abundance and standard error for each selected target as indicated in the Results section. No differences were observed in 16S abundance or human genomic DNA content. Significant differences are indicated by the asterisks. Detailed data are provided in Table 2. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; qPCR, quantitative polymerase chain reaction; rRNA, ribosomal RNA; TBI, traumatic brain injury.
Absolute Abundance of Selected Bacterial Targets Based on qPCR Analyses
qPCR, quantitative polymerase chain reaction; TBI, traumatic brain injury; SD, standard deviation; NA, not applicable; hGAPDH, human glyceraldehyde 3-phosphate dehydrogenase.
Clustering analysis of the absolute abundance data showed remarkable grouping of controls and TBI, with some exceptions (e.g., 1BTW control and 2ATW and 6ATW TBI; Fig. 4A). There were no obvious characteristics in the available metadata (Table 1) that explained the unexpected clustering of these samples. From the 94 microbiome targets, the average abundance of targets identified as different by one of the previous methods, or found to be significantly different between the cohorts (multiple t-test by the Holm-Sidak method, p < 0.05), or to have >10-fold differences between the cohorts were plotted (Fig. 4, right panels) and tabulated (Table 2).
PCR results indicated Prevotella spp. were detected in 90% of the samples in each cohort with a 318-fold increase in absolute abundance in the control samples (p = 0.009; Table 2). There were no significant differences in the Lactobacillus spp. Levels, but L. fermentum and L. salivarius were substantially higher in titer in TBI samples (average fold change of 15.6 and 341, respectively; Fig. 4, right panels; Supplementary Table S4). Bacterial targets that had significantly higher absolute abundance in TBI fecal samples and were commonly found in both cohorts included Bacteroides sartorii (11-fold), B. sterorirosoris (158-fold), Clostridium leptum (5.5-fold), and Streptococcus salivarius (5.1-fold; Table 2). Two additional targets that were rarely detected, but were substantially higher, in TBI samples were Leptotrichia spp. (61-fold) and Megasphaera massiliensis (53,000-fold). Bacteroides plebius (630-fold) and Prevotella stercorea (59-fold) were also rarely detected across both cohorts, but when present, were substantially higher in the control samples.
Correlations between specific microbes and amino acid levels and biosynthesis potential
We previously reported that one of the long-term impacts of moderate/severe TBI was altered amino acid levels post-meal. 2 Such alterations could lead to or be caused by altered mucosal microenvironments contributing to the creation and/or maintenance of a shifted microbiome. Subjects in both cohorts were asked to fast and then were provided a standardized meal followed by a blood draw 90 min after the meal was consumed. Consistent with the previous report, TBI patients had significant reductions in concentration of a number of amino acids, as shown in Table 3. These data were correlated with the metagenomic data to reveal a positive correlation between relative abundance of Prevotella spp. and all amino acids.
Serum Amino Acid Concentrations in TBI and Control Cohorts after a Standardized Meal
Values shown are concentration in uM except for the last two rows that show the ratio of the indicated amino acids for each cohort. p value was calculated by a Student's t-test, with p < 0.05 considered significant as indicated by bolded text.
TBI, traumatic brain injury; Avg, average; SDEV, standard deviation; LNAA, large, neutral amino acid; BCAA, branched-chain amino acid.
In contrast, relative abundance of the two unclassified Ruminococcaceae spp. was generally negatively correlated with relative levels of the amino acids. Tryptophan/BCAA (branched-chain amino acid) ratio, tryptophan/LNAA (large, neutral amino acid) ratio, l-tryptophan, ß-alanine, and alanine showed the strongest positive correlations with Prevotella (Fig. 5). Using the one minus Pearson correlation algorithm for the absolute quantities (Fig. 4), we eliminated those samples that failed to group with the proper cohort, producing a data set with those samples that clustered exclusively to control or TBI cohorts (most similar to the average group profile). Similar statistical differences were identified for both l-tryptophan (control, 1.67 vs. TBI, 1.54; p = 0.025) and also for l-sarcosine (control, 1.66 vs TBI, 1.00; p = 0.029) by unpaired t-testing, corroborating the metagenomic correlations.
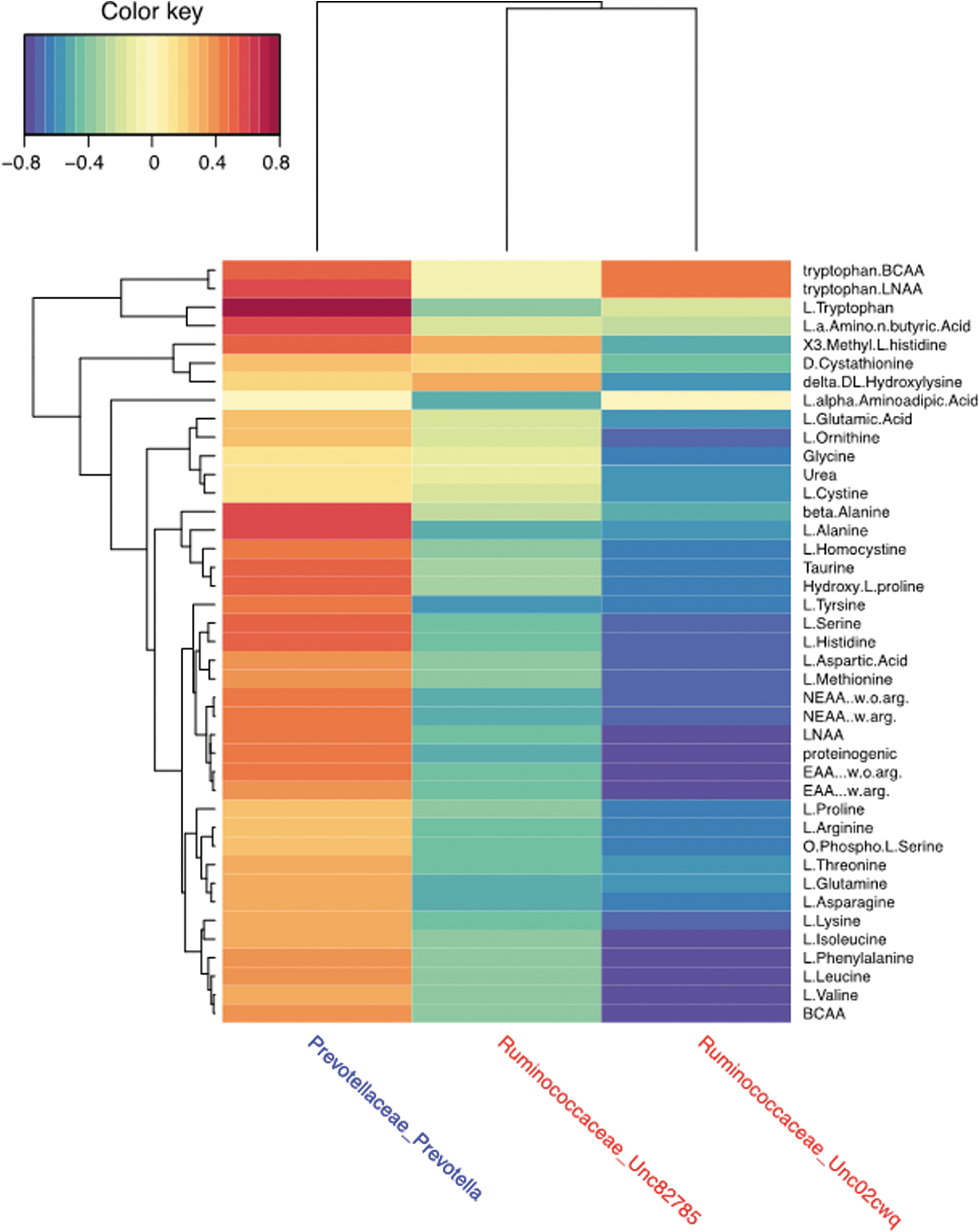
Heatmap associating amino acid concentrations with bacterial relative abundance. Using MixOmics
32
implemented in R software (R Foundation for Statistical Computing, Vienna, Austria),
30
correlations between the bacterial relative abundance and concentration of selected amino acids were completed using sparse partial least-squares regression performed in canonical mode with LASSO penalization. The results for two Ruminococcaceae spp and Prevotella spp are presented as a heatmap. BCAA, branched-chain amino acid; LASSO, least absolute shrinkage and selection operator; LNAA, large, neutral amino acid; NEAA, non-essential amino acid;
Metagenomic data from individual samples also were used to identify KEGG modules related to amino acid metabolism and fatty acid biosynthesis (summarized in Table 4). The results showed that the Akkermansia, Bacteroides, and Streptococcus genera, as well as a number of unclassified metagenomes that were more abundant in TBI samples, carried significantly higher levels of amino acid biosynthesis capabilities relative to control samples (Table 4). Although not significant, the presence of Roseburia contributed to overabundance of amino acid biosynthetic pathways in TBI samples. Bacteroides levels were of mixed results between the cohorts, similar to the NGS and PCR results, showing that specific species were associated with TBI or controls. At the Bacteroides genus level, control communities carried significantly higher levels of biosynthetic machinery for isoleucine, leucine, lysine, and serine (Table 4).
KEGG Pathway Modules Identified as Significantly Over- or Under-Represented in TBI Fecal Samples Compared to the Control Cohort
KEGG, Kyoto Encyclopedia of Genes and Genomes; TBI, traumatic brain injury; AVG, average; SEM, standard error of the mean; PRPP, phosphoribosyl pyrophosphate; DAP, diaminopimelate.
Considering the compiled KEGG module data, metabolic machinery for cysteine, histidine, isoleucine, leucine, lysine, methionine, ornithine, proline, serine, tryptophan, tyrosine, and valine were identified as more abundant in the TBI fecal communities compared to controls. Because of the potent signaling and neurological functions of short-chain fatty acids, we also noted that Akkermansia, Roseburia, and unclassified metagenomes also significantly increased the machinery for initiation and elongation of fatty acids (Table 4).
Discussion
Patients with chronic TBI requiring long-term, permanent care suffer a complex and varied set of clinical symptoms and comorbidities that persist for many years beyond the acute brain injury, including hypoaminoacidemia and altered pituitary function. The results from our novel, two-site, clinical investigation unequivocally illustrates that chronic TBI patients exhibit different fecal microbiome community structures compared to controls. These differences persisted and became more defined after rigorous secondary targeted analyses in combination with group stratification and confound removal. The absence or reduction of Prevotella spp. and Bacteroidies spp., and higher abundance of Ruminococcaceae spp. in chronic TBI compared to controls, offers immediate therapeutic targets for further investigation in minimizing patient morbidity.
Notably, data from this study are an extension of our previous findings of altered amino acid levels and modified inflammation 2 to now include altered bacterial communities. These shifts in community structure are likely created and maintained by, and also contribute to, an altered intestinal mucosa that creates a selective microenvironment in a perpetuating cycle. More focused work on the state of the GI mucosa in TBI patients will be required.
Decreased post-meal amino acid levels of the TBI patients in our cohorts support the idea that the intestinal microenvironment also would have altered amino acid metabolism. Unknown mechanisms may be selecting for bacteria that carry necessary machinery to compensate for amino acid deficits. Varied abundance of P. copri and B. vulgatus have previously been associated with increased serum amino acid concentrations, especially the branch chain amino acids. 35 Our findings also indicated that P. copri was the most abundant Prevotella in the control cohort, but this species was significantly reduced or absent in the participants with TBI. However, B. vulgatus was relatively uncommon in our cohorts and, when present, was more abundant in the TBI samples.
Bacteria in the Ruminococcaceae family have been previously associated with amino acid deficient microenvironments 36 and were higher in fecal samples from TBI patients in our cohort. KEGG analyses confirmed that the bacterial shifts occurring in TBI patients encoded higher levels of necessary machinery for biosynthesis of several amino acids. These community shifts also may contribute to altered inflammation that was detected 2 and, through production of different metabolites including short-chain fatty acids, would further disrupt normal function of the autonomic nerves (e.g., vagal), altering intestinal contractility. 37 –39 Reduced gut motility also disturbs microflora balance and promotes small intestinal bacterial overgrowth, contributing to dysbiosis. 40 Our current study supports this process based on the combined results in these two geographically distinct cohorts and emphasizes the need for careful cellular and molecular analyses of the TBI intestinal mucosa.
These first-round studies were completed in individuals that suffered moderate-to-severe TBI leading to disabilities requiring full-time supportive care. To address the impact of residence in a care facility, we recruited from two distinct communities that offered distinct lifestyles and diets. Even in our small TBI cohorts from the two locations, we identified significant changes in specific organisms that were supported and confirmed through multiple NGS approaches and subsequent qPCR analyses. Consistent with published reports, the fecal microbiome profiles we obtained from our controls were typical of healthy communities with greater diversity than was observed in the TBI samples. 12,13 Stratification of the data using participant metadata did not reveal any obvious confounder beyond TBI. Importantly, the lack of distinction between the two geographical sites, where different food, environment, and even medication approaches were present, added confidence in the observed differences between the control and TBI cohorts.
Our rigorous approaches, each confirming the last, have identified a number of potential biomarker organisms in fecal samples with altered abundance, supporting the development of targeted qPCR panels for less-expensive, higher-throughput analyses of larger cohorts that are now being performed. Completion of these analyses are critical to understanding the biological mechanisms behind clinical sequelae of chronic debilitating symptoms commonly associated with a range of injury from severe to mild TBI.
Consistent with other chronic inflammatory conditions, including obesity, the fecal microbiome of chronic TBI patients demonstrated higher abundance of Firmicutes and decreased abundance of Bacteriodetes compared to controls. Within the Bacteroidetes phylum, TBI fecal communities showed a loss of Prevotella spp. compared to controls. Despite the development and utilization of a tuf gene-targeted NGS approach, we were unable to account for all of the detected Prevotella sequences at the species level. By several approaches, it was clear that Prevotella spp. were very common in both cohorts (over 90% of the individual samples were positive), but the P. copri and P. stercorea species that showed significantly lower abundance in TBI samples were not as common. Additional analyses will be required to account for the substantial proportion of Prevotella spp. that were not fully identified. Interestingly, expansion of P. copri and P. stercorea in microbiome communities has been associated with localized systemic disease, including periodontitis, rheumatoid arthritis, bacterial vaginosis, and other chronic inflammatory conditions. 41
Mechanistically, overly abundant Prevotella spp. have been linked to function of T-helper type 17 cells that can be directly causative for types of inflammation. 42 Studies of individuals with non-alcoholic fatty liver disease have suggested that Prevotella spp. is also significantly reduced relative to controls. 43 This same study also identified that the Alistipes genus was significantly reduced whereas Anaerobacter and Streptococcus genera were increased. The group concluded that such community changes led to alterations in microvilli and intestinal barrier integrity (caused by reductions of tight junctions), consistent with impacts on GI function. 43
Similar disruption of intestinal integrity has been described for TBI outcomes, but the mechanisms remain unknown. After TBI in animal models and in some clinical studies, expression of proteins associated with tight junctions, including zonula occludens-1 and occludin, are significantly decreased as are anatomical aspects of the GI tract. 11 Short-term evaluations of the intestinal microbiome after TBI showed quick changes in the community profiles in experimental animals. Our findings are the first to examine and report the long-term outcomes of TBI on human intestinal communities, including impacts on amino acid levels and chronic inflammation. 2
Consistent with the reported short-term impact of TBI, we observed a significant increase in abundance of Ruminococcaceae spp. in TBI patients that were, on average, 20 years post-injury. Although total Bacteroides spp. was higher in the controls, thorough tuf gene analysis and subsequent species-specific PCR approaches clarified that some specific Bacteroides species were more abundant in TBI fecal communities. Specifically, three very commonly detected Bacteroides genus members, B. uniformis (detected in all fecal samples), B. stercoris, and B. thetaiotoamicron (both found in more than half of all samples) were more abundant in TBI by both tuf gene and qPCR analyses. In a recent study, rats exposed to silver nanoparticles showed altered behavior in elevated mazes that was associated with gut microbiota profile changes, including B. uniformis. 44 Unclassified Bacteroides abundance was also increased in patients with myalgic encephalomyelitis/chronic fatigue syndrome without inflammatory bowel syndrome, where patients were evaluated for severity of pain, fatigue, and reduced motivation. 45
Finally, PCR analyses also revealed that Sutterella spp., that were detected in every sample, were significantly less abundant in TBI samples than controls. Recent work with microbiomes transplanted from patients with multiple sclerosis (MS) to mice confirmed that decreased Sutterella spp. was associated with MS. 46 In previous studies, higher Sutterella spp. was associated with reduced development of autoimmune encephalomyelitis in mice 47 and better outcomes for individuals with inflammatory bowel disease. 48 However, a recent study involving human fecal microbiome transplant (FMT) treating ulcerative colitis indicated that enrichment of Sutterella wadsworthensis was associated with poor treatment outcomes. 49 In this same study, Roseburia inulivorans was positively associated with successful FMT treatment in concert with increased short-chain fatty acid synthesis. 49
Utilizing the reconstruction of metabolic pathways from metagenomic data, relative abundance of specific metabolic pathways was evaluated based on the composition of fecal microbiota communities. These data illustrated the selection of bacterial communities that had greater capability of biosynthesis of selected amino acids, consistent with reduced available amino acids detected in our previous study. 2 In addition to critical essential amino acids, sequences encoding synthesis machinery for a number of non-essential amino acids were significantly more abundant in TBI samples. Fewer KEGG modules related to fatty acid biosynthesis were significantly associated, but it is notable that the Akkermansia spp. and Roseburia spp. also led to increased fatty acid biosynthesis capability in the TBI communities.
The data generated in this novel, two-site human clinical study involving chronic TBI and control subjects support an altered intestinal mucosa lacking available amino acids and fatty acids, leading to enrichment of bacterial types that carry the necessary metabolic machinery to address this deficit. Although post-prandial nutritional absorption influences growth hormone (GH) secretion, the nutrients responsible for regulating this secretion are not clear. 50 The enterocytes lining the small intestinal tract have rapid turnover and are highly metabolic, scavenging a large proportion of dietary amino acids. This suggests that the chronic and long-lasting effects in these TBI patients are not quickly overcome and can lead to comorbidities long after the initial injury. The hypoaminoacidemia observed in TBI patients could help to explain the subsequent reduction in GH function and other pituitary issues observed in many TBI patients.
Together, the results support the conclusion that the injury-based disruption of intestinal metabolism in TBI patients, in addition to alterations in nutrient utilization by the microbiota, likely contributed to the altered amino acid profiles we observed in TBI patients. 2 The cascade of sequelae may require multiple treatment methods, including FMT. Additional research will be required with larger cohorts of chronic TBI to confirm the mechanism(s) connecting the original injury to an altered intestinal mucosa that would select for shifted or even dysbiotic microbiomes.
Finally, an important goal of these studies was to identify novel, therapeutically relevant biomarkers that can be utilized to treat symptomatic, chronic TBI patients and offering clinically meaningful treatment options for TBI-related comorbidities. Given the complexity of the impact on both the brain, central nervous system, immune, metabolic, inflammatory, pituitary, and intestinal microbiome, this study has taken that necessary first step, yielding both potential therapeutic and mechanistic insights into TBI. Notably, our results lend themselves to the next step of supplementation or replacement of the dysbiotic intestinal community by FMT. Although laudable, more-extensive studies of the fecal and intestinal profiles from individuals with and without TBI and its associated comorbidities, in addition to supporting animal and ex vivo models, are needed.
This article is published in this Journal issue along with two other articles highlighting the etiology of persistent symptoms after TBI. One of these is an original research article finding that GH replacement therapy altered brain morphometry and functional connectivity and reduced fatigue and related symptoms in mild TBI patients. 51 The other article is a letter to the editor that describes a unified complex of symptoms that persists long term in a subset of TBI patients, including profound fatigue and altered cognition; we have dubbed this symptom complex brain-injury–associated fatigue and altered cognition. 52 Together, these three articles characterize chronic symptoms that plague many patients after TBI and provide evidence to further explore therapeutic treatments to relieve those symptoms.
Footnotes
Acknowledgments
The authors thank the participants in the study as well as the care providers who assisted. The authors also acknowledge the efforts of the Baylor Alkek Center and the UTMB assay development services division for thorough molecular analyses of the fecal samples. In addition, the authors acknowledge Dr. Don Powell for conversations inspiring the concepts guiding this research.
Author Contributions
R.U., W.D., B.M., and M.S.M. contributed to the conception and design of the study. J.S., C.S., M.M., L.K., and B.M. contributed to data acquisition. R.P., N.A., W.D., A.M., K.R., C.D., and E.D. contributed to data analysis, and R.U., C.S., W.D., T.W., and M.S.M. contributed to further data interpretation. R.U., R.P., C.S., and M.S.M. contributed to writing the manuscript, and N.A., K.R., W.D., C.D., E.D., J.S., C.S., M.M., L.K., B.M., A.M., and T.W. contributed to additional editing of the manuscript. All authors have reviewed the submitted manuscript.
Funding Information
This study was funded by the Moody Endowment (funding M.S.M.) and conducted with the support of TideWay, part of the Transitional Learning Center (Galveston, TX) and the Centre for Neuroskills (Bakersfield, CA).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.