
Abstract
Traumatic spinal cord injury (SCI) elicits a complex cascade of cellular and molecular inflammatory events. Although certain aspects of the inflammatory response are essential to wound healing and repair, post-SCI inflammation is, on balance, thought to be detrimental to recovery by causing “bystander damage” and the spread of pathology into spared but vulnerable regions of the spinal cord. Much of the research to date has therefore focused on understanding the inflammatory drivers of secondary tissue loss after SCI, to define therapeutic targets and positively modulate this response. Numerous experimental studies have demonstrated that modulation of the inflammatory response to SCI can indeed lead to significant neuroprotection and improved recovery. However, it is now also recognized that broadscale immunosuppression is not necessarily beneficial and may even carry the risk of contributing to the development of serious adverse events. Immune modulation rather than suppression is therefore now considered a more promising approach to target harmful post-traumatic inflammation following a major neurotraumatic event such as SCI. One promising immunomodulatory agent is intravenous immunoglobulin (IVIG), a plasma product that contains mostly immunoglobulin G (IgG) from thousands of healthy donors. IVIG is currently already widely used to treat a range of autoimmune diseases, but recent studies have found that it also holds great promise for treating acute neurological conditions, including SCI. This review provides an overview of the inflammatory response to SCI, immunomodulatory approaches that are currently in clinical trials, proposed mechanisms of action for IVIG therapy, and the putative relevance of these in the context of neurotraumatic events.
Introduction
Traumatic spinal cord injuries (SCIs) typically result from a sudden mechanical impact on the spine. This compromises the integrity of the spinal cord, inflicting immediate and irreversible damage due to compression, laceration, and/or shearing. Traffic accidents, falls, sports- and water-related injuries are the main causative factors of SCI worldwide. 1 The prevalence (total number of cases per population unit) and annual incidence (number of new cases per population unit per year) of SCI vary considerably across the globe but are among the highest in the United States, with a reported prevalence of 906 and >40 new cases annually per million population. 1 Specific to Australia, 300 to 400 new cases of traumatic SCI are reported each year, 2,3 with an estimated incidence rate of 21.0 to 32.3 cases per million population. 4 This incidence is below that of the United States but comparable to many other Western countries, particularly in Europe. 1 In the majority of studies from around the world, traumatic SCI has otherwise been shown to disproportionally affect the male population. 1 There is no reliable estimate of the global prevalence of SCI, but approximately 288,000 Americans and 15,000 Australians are currently living with this condition. 4,5
The cost of SCI is high, not just in terms of the physical and emotional impact on those who live with its debilitating consequences on a daily basis, but also for society at large. The vast majority of patients with traumatic SCI experience continuing medical complications that require specialized care and often result in their readmission to a hospital. 6 In Australia, the cumulative burden of SCI to the economy has been estimated as A$5 million per case of paraplegia (i.e., for individuals with lesions below the cervical spinal cord and preserved upper limb function) and A$9.5 million per case of tetraplegia (i.e., individuals with lesions involving the cervical spinal cord and partial or complete loss of function in both the upper and lower limbs). 6 When removing estimates for loss of productive life years and income, these figures are very similar to the high lifetime cost that has been reported for the United States. 5 With no proven effective treatment options for SCI currently available, 7,8 there is an urgent and unmet need to reduce its socioeconomic impact, and to bring hope for a better quality of life and return of lost function to those affected. 9
Many patients with SCI initially present with incomplete damage to the spinal cord, but destructive biological processes (the so-called secondary injury cascades) continue to inflict significant pathology in the weeks to months following the initial insult. 10 –12 Inflammation is a major feature of the acute phase response to SCI, 13,14 and neuroinflammatory changes persist well into the chronic phase of injury. 10,15 The temporal relationship between inflammation and progressive neuropathological changes at and around the site of SCI has been long recognized, and the inflammatory response itself is therefore seen as a prime target for reducing the secondary loss of neural tissue following insult.
It is increasingly recognized, however, that the nature of the neuroimmune response to central nervous system (CNS) injury is highly complex, in that it contains elements that can drive secondary damage as well as wound healing/repair, perhaps even doing both at different temporal stages of the injury process. 16 A global suppression of immune function will therefore not necessarily be beneficial to recovery. 16,17 This is further compounded by the fact that patients are already at high risk for developing potentially life-threatening infections, with pneumonia and septicemia being amongst the leading causes of death in SCI survivors in many countries. 5,18 –20
Much of the ongoing research effort is therefore focused on generating a better understanding of the neuroinflammatory response to SCI, and on defining discrete therapeutic targets or processes that can be modulated to improve outcomes. Here we provide an overview of the current understanding of the local inflammatory response to SCI (i.e., at the lesion site itself) and how this response may be modulated, focusing in particular on intravenous immunoglobulin G (IVIG) therapy as an emerging treatment option for acute neurological conditions such as SCI.
Development and Consequences of Inflammation in SCI
The neuroinflammatory response to SCI
Acute phase
The neuroimmune response to traumatic SCI begins immediately following the accident and involves a complex series of events at the lesion site that are partially overlapping but also temporally separated (Fig. 1). The sudden presence of necrotic cell debris and associated danger signals (DAMPs) trigger surviving CNS-resident cells to begin producing reactive oxygen species (ROS), complement factors, and cytokine products. Pro-inflammatory mediators (e.g., interferon gamma [IFN-γ], interleukin 1 alpha [IL-1α], IL-6, and tumor necrosis factor alpha [TNF-α]) can indeed be detected at CNS lesion sites within minutes of neural injury in both human patients and rodent models. 21 –25 The ensuing inflammatory response, in conjunction with the pathophysiological and biochemical changes that occur following the mechanical destruction of the blood–spinal cord barrier (BSCB), 26 including microhemorrhaging and the development of tissue edema and ischemia, further propagates the spread of damage into parts of the spinal cord that were originally spared but vulnerable to degeneration. 27
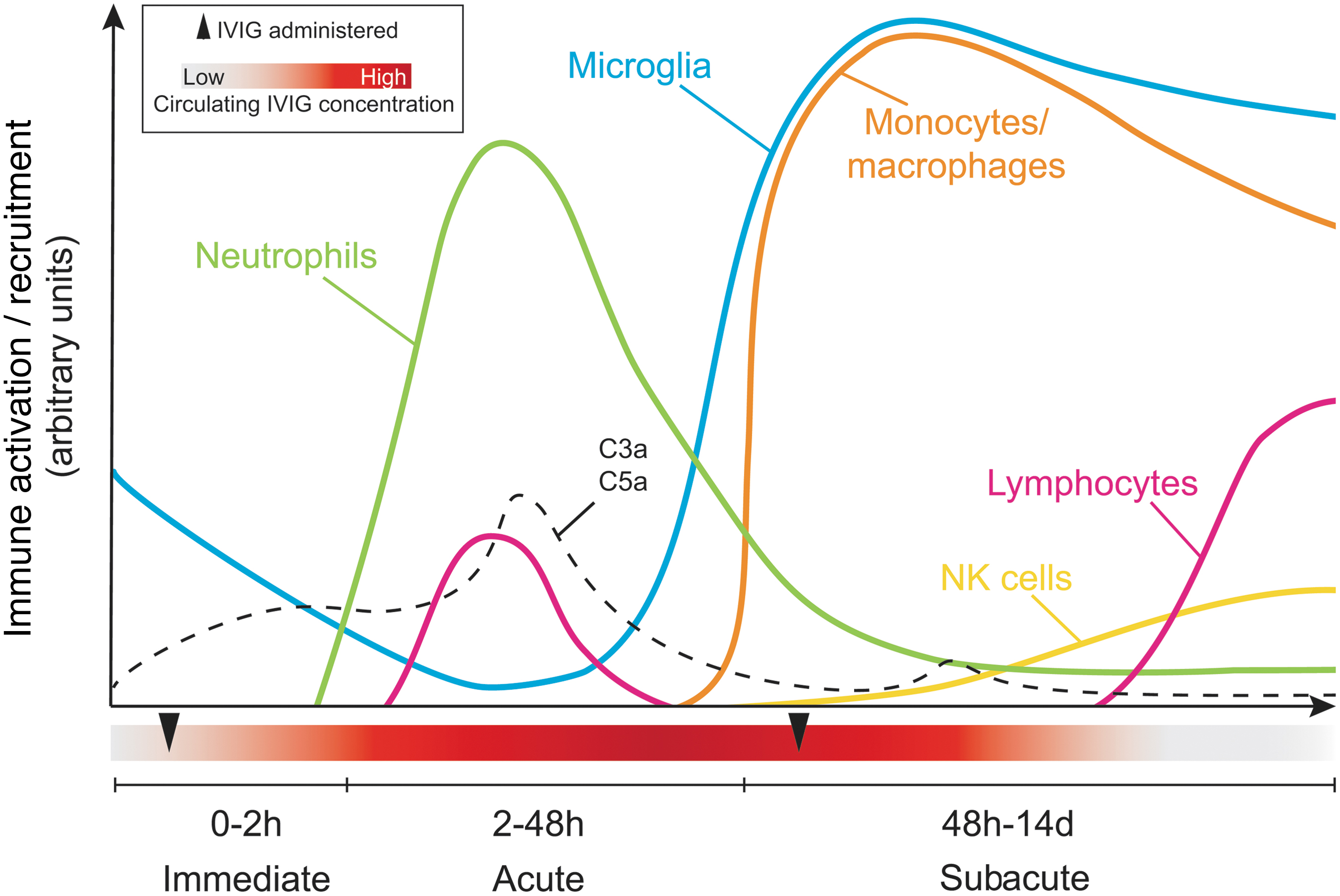
Overview of the inflammatory response and timing of immune cell recruitment during the acute, subacute, and intermediate stages of spinal cord injury. The “exposure window” for intravenous immunoglobulin (IVIG) therapy is also shown, with arrowheads indicating the timing of administration (1 h and 3 days post-injury [dpi]) to maintain therapeutic levels (red) during the first 7 dpi based on its known half-life in mice. Color image is available online.
The microglial population at the site of SCI, although traditionally proposed as being one of the first cellular responders, was recently shown by Bellver-Landete and colleagues to be significantly depleted immediately after injury due to death from both mechanical impact and apoptosis. 28 Remaining microglia change their shape, movements, and migration patterns in response to CNS injury, 29 and they begin to produce oxidative metabolites, ROS, and pro-inflammatory cytokines. 24 These cells directly associate with degenerating axons within minutes of injury but do not actively begin to phagocytose these axons until around 4 days post-injury (dpi). 24,28,30
The early hours and days post-injury are further characterized by profound complement system activation and the start of sequential leukocyte migration from the periphery/circulating blood into the injured neural parenchyma. 31 –33 It is interesting that, although the destruction of the BSCB would seemingly allow for unrestricted access/entry of circulating leukocytes into the injured segment of the spinal cord, recruitment of these cells remains a highly regulated process. 14,15 For example, although lymphocytes account for ∼80% of circulating leukocytes in the mouse, they do not really enter into the spinal cord in significant numbers until at least several weeks or months post-injury. 15 Instead, the early infiltrate is predominantly made up of myeloid cells, mostly neutrophils, that represent only a minority of all circulating cells but are the fastest peripheral responders to SCI and can be detected at the lesion site within 4 h of injury (Fig. 1). 34,35
Regulation of the acute peripheral response to CNS injury is highly complex, but some of our more recent work shows that chemotactic signals, such as CXCL1, are key triggering factors for the release of neutrophils from the bone marrow into the blood and the subsequent recruitment of these cells to the lesion site. 14 We further showed that this process is negatively regulated by the C3a/C3aR1 axis, which engages phosphatase and tensin homolog (PTEN) to counterbalance chemotactic signaling pathways driving neutrophil mobilization and recruitment. 14 Activated vascular endothelial surfaces within the injured segment of the spinal cord play an essential role in capturing circulating neutrophils, following which these cells are known to secrete matrix metalloproteinases (MMPs) to extravasate through the capillary walls and migrate toward sites of tissue injury and/or inflammation. 34 Neutrophil numbers in the injured spinal cord peak at 1 dpi, but these cells remain present at the lesion site for at least 42 dpi. 15,17
The recruitment of neutrophils is mostly viewed as detrimental to recovery from SCI, as well as wound healing in general. We recently showed that circulating neutrophil numbers in admission bloods from human patients were negatively correlated with outcomes at discharge. 14 Parallel experiments in mice with a contusive SCI showed that the extent of neutrophil presence at the lesion site was inversely correlated with neurological outcomes, whereas the depletion of these cells with an antibody against Ly6G significantly improved the recovery of locomotor function. 14 Interestingly, we also found that there is a small subset of B220mid B cells that enters into the lesioned spinal cord at 1 dpi, the presence of which depends on neutrophils. 14 Although questions remain around the precise identity of such cells, these results are particularly interesting given the new advances in our understanding of how neutrophils can either amplify or suppress B cell activity in other inflammatory conditions, including cancer and autoimmunity. 36
Some studies have postulated beneficial roles for neutrophils in augmenting SCI recovery, for example, through the expression of the secretory leukocyte protease inhibitor (SLP1). 37 In a model of peripheral nerve injury, neutrophil infiltration and associated cytokine/chemokine production were essential for the clearance of myelin debris. 38 Stirling and colleagues otherwise reported that neutrophil depletion in a contusion injury model increased lesion sizes and impaired neurological outcomes. 39 The Gr-1 antibody that was used in this study, however, not only depletes neutrophils but also inflammatory monocytes, and these results therefore need to be interpreted with some caution. It is nonetheless clear that the neutrophil response to SCI is too complex to be fully understood or appreciated when using blanket knockout or depletion approaches. Future research should therefore focus on probing the temporal and spatial dynamics of neutrophil recruitment in SCI, specifically their interactions with and modulation of other cell types to fully elucidate the complex mechanisms around the neutrophil response during wound healing processes. 40
Subacute phase
During the subacute phase of SCI, newly proliferated and recruited microglia begin to actively phagocytose necrotic cell debris and accumulate around the lesion epicenter. 24,28,30 The presence of microglia seems essential during the first week following SCI (but less so thereafter) as their depletion during this time with the colony stimulating factor-1 (CSF-1) inhibitor PLX5622 is associated with significantly worsened outcomes from SCI. 28,41 Consistent with a more beneficial role, early enhancement of microglial activation can reduce secondary pathology. 42
Circulating inflammatory monocytes also begin to be recruited during the subacute phase (Fig. 1). Adoptive transfer experiments showed that their recruitment picks up at around 3 dpi, and then peaks at 7 dpi. 43 Monocyte turnover at the lesion site appears rather high, at least during the subacute phase, 43 but infiltrating monocyte-derived macrophages remain prominently present at the site of SCI for many weeks to months post-injury. 15,43,44 The timing of monocyte recruitment appears delayed compared with non-neurological models of tissue injury. For example, monocytes are robustly recruited to the injured heart following myocardial infarction as early as 1 dpi, and their numbers quickly resolve to baseline by ∼16 dpi. 45 It is likely that factors specific to CNS injury may influence both the timing of monocyte recruitment and the chronic, non-resolving nature of this response.
Due to diversity in monocyte subsets and macrophage phenotypes, delineating the precise contribution of these cells to SCI pathology is complicated and only just beginning to be understood. 46 On one hand, some of the polarization states associated with recruited macrophages are thought to impede recovery by spreading secondary injury via the demyelination of axons, inhibition of regeneration, and by driving fibrotic scar tissue formation. 47 –49 On the other hand, activated macrophages can also secrete factors that support neuronal survival, axonal regrowth, and angiogenesis, and thus aid recovery. Phagocytically active macrophages are otherwise required for the clearance of cellular debris, which is an essential part of the successful wound healing process, although this has been linked to “bystander damage”. 47,50,51
There is general ongoing controversy within the literature based on rather non-discriminative depletion studies as to what the role of monocytes/monocyte-derived macrophages is in relation to recovery from SCI. Although many studies have shown that reduced myeloid cell infiltration into the injured spinal cord is typically associated with better outcomes, 43,49,52 others have reported that the depletion of circulating monocytes/macrophages results in significantly worse functional recovery and increased lesion size after contusive SCI; this phenotype was attenuated when blood monocyte numbers were restored. 44 Recent in vitro studies suggest that blood-derived macrophages may suppress microglial phagocytosis, 30 without reducing microglial proliferation and extension of processes. 53 Consistent with that, preventing the infiltration of circulating monocytes/macrophages into the lesion causes a subsequent increase in activated resident microglia and a worsened recovery. 53
Interestingly, some studies have suggested that monocyte/macrophage migration to the lesion site is regulated by a temporal T cell response to SCI that is essential for optimal outcomes. 54,55 Raposo and associates used a genetically modified mouse model lacking functional T helper 1 (Th1) cells (Tbx21-/- ) to elucidate the relationship between monocytes/macrophages, T cells, and their role in recovery from SCI. 55 Mice lacking Th1 cells had significantly fewer infiltrating myeloid cells within the lesion site at 7 dpi compared with wild-type SCI controls, and this was associated with worse functional outcomes. These differences were no longer observed at 13 dpi, suggesting that circulating Th1 cells are important for the beneficial recruitment of monocytes to the lesion site within the first week of SCI. 55 A separate study by this group suggested that the presence of CD4+ T cells within the choroid plexus is important for homeostatic immune surveillance, and that leukocytes, including T cells and CD11b+ myeloid cells, travel from the choroid plexus to the injured parenchyma following injury. 54 The trafficking of these cells was shown to be dependent on IFN-γ signaling as mice lacking the receptor for IFN-γ had a reduced number of T cells and CD11b+ myeloid cells within the CSF at 1 dpi, and at the lesion site itself by 7 dpi compared with controls; this was once again associated with a worsened locomotor recovery. 54
In summary, because of their divergent phenotypes, global inhibition of all monocyte/macrophage function in the injured cord will not be beneficial to recovery. 47 Going forward, a better appreciation of the diversity of these cells and modulating monocytes/macrophages accordingly, either by altering the timing of recruitment or which subset is being recruited, and/or by manipulating their polarization (i.e., functional activation state), may be more advantageous and will likely also resolve some of the controversy as to what role they play in SCI recovery. 15,47,53
Intermediate phase
The transition from the subacute to the intermediate phase of SCI (14 days to 6 months post-injury) is characterized by an increased presence of natural killer (NK) 14 and adaptive immune cells, 15,56 both of which have incompletely understood roles in both tissue remodeling and secondary damage after SCI (Fig. 1). The spatial and temporal role of B and T cells likely involves a complex interconnected network of cell and cytokine responses to injury. 36,54,55 CD3+ T cells are the first lymphocytes to be detected in significant numbers within the injured spinal cord at ∼14 dpi, and they remain present there for at least 180 dpi; the majority of these are CD4+ T cells. 15,17
Some evidence suggests that myelin-reactive T cells may confer neuroprotection by increasing neuronal survival. 57 Indeed, mice lacking T cells have reduced neuronal survival in response to CNS injury. 57,58 Other studies have, however, implicated T cells in tissue-destructive events after SCI. 59,60 Here, treatment of experimental SCI with a blocking antibody against the T cell chemoattractant, CXCL10, decreased the infiltration of T cells to the lesion site at 14 dpi, a phenomenon that was associated with improved functional recovery, reduced lesion size, and improved angiogenesis. 59,60 A more recent study found that the secretion of IFN-γ by a specific subset of T cells (γδ T cells) was detrimental to recovery from SCI, and specifically acted on macrophages to enhance pro-inflammatory responses. 61 Significantly more research is required to better understand the complex role of T cells following SCI, and how the presence of these cells shapes the overall inflammatory response and lesion site development.
The final major wave of peripheral immune cell infiltration is thought to consist of (a renewed) B cell recruitment that often surrounds blood vessels. These B cells form prominent follicle-like structures interspersed with T cells, macrophages, and microglia from around 28 dpi onward. They again remain detectable in the CNS well into the chronic phase of SCI. 56 The extent of B cell presence at the lesion site can vary substantially between animals but has been correlated with the emergence of self-reactive antibodies that recognize epitopes within protein homogenates of the spinal cord. 62 A putative pathogenic contribution of these to secondary inflammatory pathology has been deduced from adoptive transfer experiments, in which antibodies that were isolated from SCI mice were able to induce significant damage when injected into the neural parenchyma of naïve animals. Conversely, mice lacking B cells have a significantly improved recovery from SCI. 56
Whether the presence of these self-reactive antibodies precedes an autoimmune event or signifies a full-blown autoimmune disease remains unclear. Specifically, there is no evidence to date for B cell presence/accumulation in other areas of the CNS of these mice (i.e., away from the lesion site itself), and also no worsening of neurological performance is normally observed during the intermediate/chronic phase of SCI, that is, the period during which self-reactive antibodies emerge. An alternative explanation may therefore be that their presence is part of an adaptive mechanism for opsonization and debris clearance from the injury site. 63 An elevation in naturally occurring autoantibodies, which have well-established roles in tissue regeneration/repair, does indeed occur in response to SCI. 64,65 Any involvement of these in wound healing processes does not necessarily exclude unwanted and/or pathological consequences, as highlighted by a contribution of pre-existing natural IgM antibodies to secondary injury during the more acute phase of SCI. 66 In summary, although evidence is mounting, whether self-reactive antibodies are indeed pathogenic and a significant contributor to adverse post-SCI sequalae, including during the more chronic phase of injury, remains very much an area of debate.
Chronic phase
The impact of persistent neuroinflammatory changes during the chronic phase of SCI is much less understood. Most interventionist studies have focused on the first hours to days post-insult, when secondary injury reaches its peak and there is thus maximum potential for attenuating such changes by modulating the influx of immune cells and associated cytokine expression. These major waves of immune cell infiltration have, however, passed during the chronic phase of SCI and the lesion site itself has also undergone extensive remodeling by this time. 67 The glial scar now creates a well-defined border between the lesion core and the healthy tissue that surrounds it. 67 Infiltrating immune cells are mostly also spatially restricted to the lesion core itself rather than the spared tissue by this stage. Neutrophils, large granular macrophages, and B cell clusters, interspersed with T cells, can be detected here for many months. 15,62,68 Significant metabolic dysfunction, characterized by increased oxidative stress and reduced production of lipid metabolites, as well as increased production of pro-inflammatory mediators is also observed within the spinal cord throughout the chronic phase of injury. 69 Without a clear functional readout, however, it is difficult to appreciate the true contribution of these changes to outcomes.
Few studies have probed the mechanisms driving the chronicity or non-resolving nature of the inflammatory response to SCI. Greenhalgh and coworkers have suggested that chronic microglial activation and inflammation is suppressed by communication with infiltrating monocytes/macrophages after SCI. 53 Prevention of this communication was associated with a worsened functional recovery, suggesting that although the initial response of microglia to injury may be beneficial, 28 their long-term activation can become detrimental. 53 Another study investigated the use of the anti-inflammatory drug, licofelone, in improving recovery from chronic SCI. 69 Rats were treated with licofelone daily for 1 month, beginning at 8 months post-SCI. Although some improvements in metabolic functions were observed, this late-stage treatment with licofelone had no effect on locomotor recovery. 69
In summary, understanding the impact of persistent inflammation during the more chronic phase of SCI is challenging, complicated in particular by the fact that locomotor recovery from SCI typically plateaus well prior to the chronic phase of injury. 70,71 This makes it particularly problematic to delineate the roles of specific immune cell types to ongoing neurological dysfunction and pathology during the more chronic phase of SCI, and there is thus an urgent need for better readouts to assess this.
Targeting inflammation in acute SCI: immunosuppression versus immunomodulation
Until recently, the gold standard treatment across the globe for targeting inflammation in acute SCI has been the broad-spectrum immunosuppressant methylprednisolone (MPSS), in conjunction with surgical decompression of the spinal cord where required. 8 MPSS is a glucocorticoid therapy that binds to nuclear receptors and acts to suppress the expression of pro-inflammatory cytokine genes. Early pre-clinical studies indicated that MPSS could provide neuroprotection from SCI, mostly in rodent models, as evident from a reduction in neurofilament breakdown, increased neuronal excitation, and improved blood flow to the lesion site. 72 These studies formed the basis for multiple large-scale clinical trials (National Acute Spinal Cord Injury Studies [NASCIS]), the outcome of which suggested that MPSS infusion within 8 h of SCI improved both motor and sensory function in patients with SCI compared to those treated with an opiate-based therapy (naloxone) or placebo. 73 –75
The validity of these results has, however, been questioned, 76 with a Canadian multi-center spinal cord injury registry finding no difference in recovery of motor scores between patients treated with or without steroid therapy, even when corrected for age, neurological injury level, and injury severity. 77 In fact, MPSS treatment following acute SCI was significantly associated with a higher rate of total complications, including an increased risk of death. 78 The use of MPSS in treating acute SCI has therefore become controversial among clinicians. Adverse effects of MPSS administration following SCI have also been reported in pre-clinical studies. Specifically, Cabrera-Aldana and colleagues showed increased tissue edema and plasma extravasation into the injured parenchyma of the spinal cord with MPSS administration. 79 In rodent models of SCI, MPSS treatment did reduce immune cell infiltration into the lesion site, 80 but treatment also negated the effects of an anti-CD11b therapy, 81 which was previously shown to improve SCI outcomes, 82 suggesting methylprednisolone treatment may prevent other immunomodulatory therapeutics from working through mechanisms that are yet to be elucidated.
As our appreciation grows for how SCI can also disrupt the neuroimmune interface, additional reasons have emerged as to why broad-acting immunosuppressants such as steroid treatment may not be optimal and/or even carry risk. Specifically, SCI-IDS (spinal cord injury-immune deficiency syndrome) is observed in both human patients and animal models of SCI. This phenomenon is hallmarked by a significant reduction in circulating leukocyte numbers within the first few days of injury 83,84 and increased infection susceptibility. 85 SCI-IDS is already accompanied by systemic increases in glucocorticoid levels due to disrupted innervation and outflow to the adrenal glands, particularly in high-level SCI. 86 Steroid treatment should be carefully considered in light of that, with recent studies indeed showing that broad immunosuppression following SCI can be accompanied by harmful side effects, including respiratory failure, hemorrhage, and a higher incidence of infection in an already at-risk population. 8,78,87 There is thus a pressing need for the further development and testing of therapies that more selectively modulate aspects of the inflammatory response to traumatic SCI, rather than universally suppressing these. A number of such agents are currently being trialled in human patients, and a brief overview of these is provided below.
A new era for modulating inflammation in acute SCI: bench to bedside (and back)
Minocycline
The antibiotic minocycline has shown promise in both experimental 88 –90 and clinical studies. 91 Minocycline manipulates the inflammatory response by inhibiting phosphorylation of p38 MAPK, thereby preventing/reducing the expression of pro-inflammatory chemokine and cytokine genes. It has also been shown to have direct neuroprotective effects. 92 In rodent models of SCI, minocycline augmented functional recovery, with reduced inflammation and improved histological outcomes. It was also shown to be superior to MPPS treatment in SCI rats. 89 As minocycline is an antibiotic, whether there is a beneficial influence of this treatment on the gut microbiota, or any SCI-induced changes therein, 93 remains undetermined.
Results of a randomized Phase 2 clinical trial into the use of minocycline for acute SCI were published in 2012. 91 Patients were administered intravenous minocycline (n = 27) or placebo (n = 25) for 7 days following acute SCI. Those with an injury to the thoracic spinal cord showed no improvement in recovery. On the other hand, patients with an injury to the cervical spinal cord that was classed as “motor incomplete” (i.e., those with some degree of voluntary movement remaining below the lesion level on admission) showed a significant improvement of 14 points on the American Spinal Cord Association (ASIA) Impairment Scale (AIS) functional motor scoring system. 91 Analysis of CSF biomarkers found that minocycline treatment did not alter levels of IL-1β, MMP-9, CCL2, CXCL10, NCAM, or nitric oxide oxidation products (NOx), but did attenuate heme oxygenase-1 (HO-1) levels at 7 dpi. 94 A double-blinded, randomized, multi-center Phase 3 follow-up trial is now underway (Minocycline in Acute Spinal Cord Injury [MASC], NCT01828203).
G-CSF
Phase 1/2a 95 and 2b 96 clinical trials have also confirmed the safety and feasibility of granulocyte-colony stimulating factor (G-CSF) treatment in patients with acute SCI (within 48 h of injury). Results from a recent double-blind, placebo-controlled, randomized Phase 2/3 clinical trial (n = 60 patients) further reported significant functional improvements and a reduced neurological severity of SCI (measured by AIS grade) in association with G-CSF treatment (300 μg) for individuals who were motor-incomplete and between 1 and 6 months post-injury. 97 These results follow positive outcomes in animal models 98 –100 and earlier, more exploratory clinical studies with this hematopoietic growth factor. 95,96
G-CSF is well known for its mobilizing effects on hematopoietic stem/progenitor cells and granulocytes from the bone marrow; it also can promote the survival, differentiation, and proliferation of both neutrophil progenitors and mature neutrophils. 101 Consistent with this, G-CSF treatment following SCI increases recruitment of bone-marrow-derived cells to the injured site. 99 It is presently unclear as to whether increases in preservation of white matter, 99 improved BSCB integrity, and enhanced angiogenesis 98 following G-CSF therapy are the direct result of its immunomodulatory effects, or perhaps more so a putative involvement of G-CSF in pro-regenerative mechanisms, for example, angiogenesis and/or neural precursor activation. 102 Addressing these knowledge gaps may aid in the development of a better understanding of its use and therapeutic window in SCI.
Intravenous immunoglobulin
IVIG is another emerging immunomodulatory strategy for acute neurological conditions, including neurotraumatic events. IVIG is a plasma product comprising mostly immunoglobulin G (IgG) from the blood of thousands of healthy donors, 103 and it was originally developed as an antibody replacement therapy in primary immunodeficiency disorders. 104 In the 1980s, high-dose IVIG therapy was also found to increase platelet numbers in patients with idiopathic thrombocytopenic purpura (ITP), 105 paving the way for its use as an immunomodulatory therapy. High-dose IVIG therapy is now widely used to treat a range of autoimmune and inflammatory conditions, including ITP, Kawasaki's syndrome, and arthritis 104 as well as neurological autoimmune diseases such as Guillain-Barré syndrome 106,107 because of its potent immunomodulatory effects and limited side effects.
Recent research from our laboratory (and also from other labs; see below) has shown that high-dose IVIG is a very promising therapeutic for acute SCI, where it successfully modulates inflammation and improves recovery. 108 A Phase 1/2a clinical trial is now underway exploring the feasibility, safety, and efficacy of IVIG therapy in human patients with acute SCI (Australian Clinical Trials, ACTRN12616001385437). However, the mechanism behind IVIG's neuroprotective effects in SCI is unknown. 109,110 The sections below summarize current literature on the experimental use of IVIG in animal models of acute neurological insults, before focusing on the current knowledge surrounding IVIG's possible mechanism(s) of action in inflammatory disease, and how these may be of relevance to SCI.
IVIG in Acute SCI
Current knowledge about IVIG in neurotrauma
Multiple pre-clinical studies have now demonstrated that IVIG treatment can improve outcomes from acquired CNS injury in a range of neurological conditions. 108,111 –115 In traumatic brain injury (TBI), IVIG-treated mice showed improved neurobehavioral outcomes in rotarod and Morris water maze tasks compared with vehicle-treated controls. 111 This improvement was associated with a significant reduction in brain edema and neuronal degeneration, both acutely (1 dpi) and chronically (33 dpi). High-dose IVIG (2 g/kg) also significantly reduced mortality rates, neurological impairment, and infarct volumes in mice with middle cerebral artery occlusion, a common model of stroke. 114,116 Similar results were obtained in neonatal hypoxic-ischemic injury where IVIG reduced infarct size, attenuated neurological deficits, and improved mortality rates. 115
Using a mouse model of contusive SCI, our own previous work showed that IVIG therapy in the clinical dose range (0.5–2 g/kg body weight) resulted in improved locomotive recovery, as well as increased myelin and axon preservation, decreased astrogliosis and a 30–40% reduction in lesion size. 108 Doses below 0.5 g/kg IVIG were found not to be effective in this study. We also showed that albumin treatment was not able to recapitulate the IVIG effect, indicating a level of specificity in that it did not just emanate from protein loading itself. 108 The effectiveness of IVIG in acute SCI is consistent with the outcome of independent studies that used purified human IgG (i.e., the main ingredient of IVIG) in both a high-level (vertebral level C7–T1) clip compression model of SCI 112,113 and a low-level (T9) contusion injury in rats. 117
IVIG has been demonstrated to enter into the neural parenchyma within hours of injury, but only under conditions where the blood–brain barrier (BBB)/BSCB is compromised. 108,114 In SCI, IVIG localizes to neurons, astrocytes, oligodendrocytes, and microglia/macrophages, 108 as well as to pericytes and blood vessels. 113 The significance of these interactions remains unclear. IVIG treatment has otherwise been associated with acute reductions in the presence of immune cells in both brain injury and SCI models (F4/80+ microglia/macrophages and polymorphonuclear cells, respectively). 111 –113 Our own previous work has shown a reduced presence of CD68+ microglia/macrophages at and around the lesion site with IVIG therapy during the more chronic phase of injury (35 dpi). 108 It should be noted, however, that these previous studies did not distinguish between CNS-resident microglia and infiltrating peripheral monocytes/macrophage subsets. More research is therefore required to determine if and how IVIG influences the recruitment and activation state of specific peripheral myeloid cell subsets.
Possible Mechanisms for IVIG in CNS Injury
Overview of immunomodulatory effects of IVIG in neurological disease
Despite IVIG's well-established benefits in a variety of neurological conditions, its mechanism of action remains poorly understood. Because IVIG consists of pooled antibodies from thousands of donors, preparations include a diverse repertoire of antibodies specific against millions of unique antigens, and these can therefore have a wide variety of effects depending on the disease in question. The immunomodulatory properties of IVIG in autoimmune disease, including neurological conditions such as demyelinating polyneuropathy and Guillain-Barré syndrome, have been summarized extensively elsewhere. 104,106 Although there are likely some shared mechanisms as to how IVIG acts in these conditions compared to acquired CNS injuries, the inflammatory pathology that occurs during the acute phase of SCI is not traditionally considered to be driven by autoimmune processes. Hence, IVIG's therapeutic efficacy in acute CNS insults is therefore more likely mediated via a modulation of innate rather than adaptive immune components.
IVIG's potential mechanisms are generally split between those that are mediated via the F(ab)’2 portion of the IgG molecule, which is responsible for antigen recognition, and those that link to the constant Fc domain, that is, the portion of the antibody that binds Fc receptors. 104 On the one hand, F(ab)’2-dependent mechanisms in neurological disease are thought to include the possible blockade of cell surface receptors, the neutralization of cytokines and autoantibodies by antibodies present in IVIG preparations, as well as the scavenging of complement. On the other hand, Fc-dependent mechanisms include the regulation of Fc receptor expression, saturation of the neonatal Fc receptor (FcRn), blockade of activating Fc receptors, and a modulation of T cell activity. 104,106,118 IVIG has also been shown to modulate the expression of several cytokines and chemokines in models of neurological injury, which could involve both F(ab)’2 and Fc-dependent signaling cascades. 118 The remainder of this review will discuss in more detail proposed IVIG mechanisms that may be applicable to acute SCI, based on our current knowledge of the factors that drive secondary inflammatory pathology.
Modulation of inflammation via the variable F(ab’)2 region
Autoimmunity
Circulating self-reactive antibodies have been detected in chronic rodent models of SCI 56 as well as in human patients with SCI (>12 months post-injury). 119 However, these are unlikely targets for acute IVIG therapy as the temporal relationship between the emergence of these autoantibodies and the window of IVIG administration does not match. Naturally occurring autoantibodies, which are germline encoded and produced by B1 cells, may play a role in acute SCI, 65,66 but whether IVIG modulates their action remains to be determined. Another outstanding question is whether early IVIG therapy has any impact on the development of autoimmunity during the more chronic phase of SCI and/or whether delayed IVIG therapy can reveal a pathological role for autoantibodies at this stage of the injury process.
Fas/FasL signaling
Another possible F(ab’)2-dependent mechanism may involve the neutralization of the cell death mediator Fas (also known as CD95) by IVIG. In a study investigating Lyell's syndrome, a disease in which active Fas ligand (FasL or CD95L) binds Fas present on keratinocytes to induce apoptosis, IVIG was able to completely inhibit FasL-induced cell death both in vitro and in human patients. This effect was due to IVIG's blockade of Fas, rather than FasL, as protection only occurred when the cells were pre-treated with IVIG; incubation of soluble FasL with IVIG did not attenuate cell death, thus suggesting that IVIG contains antibodies specific for Fas. 120,121
Putative modulation of the Fas-FasL pathway by IVIG is directly relevant to SCI, 122 as knockout mice lacking Fas have shown reduced apoptosis at the lesion site, attenuated glial scar formation, and an improved locomotor recovery from SCI. 123 The translational significance of these findings is further evident from post-mortem studies, which showed an increase in Fas- and FasL-positive neurons and glial cells in spinal cord tissue from human patients. Interestingly, these SCI-associated changes in expression were only observed during the acute but not chronic phase of injury, suggesting this apoptotic pathway exerts most of its detrimental effects early after injury. 123 It is therefore entirely plausible that at least some of the benefits that are conferred by IVIG therapy during the acute phase of SCI result from the neutralization of Fas, thereby attenuating secondary cell death. Somewhat counterintuitively perhaps, agonistic anti-Fas antibodies have also been detected within IVIG preparations. 121 How these may impact on neural cells within the injured spinal cord following BSCB compromise remains unclear, but they could beneficially influence outcomes by inducing apoptosis of circulating leukocytes, including neutrophils. 124
Consistent with that, IVIG therapy has been associated with a reduced presence of polymorphonuclear cells within the lesion site at 1 dpi, 112,113,117 although it should be noted that IVIG-induced apoptosis has so far only been observed in human leukocytes rather than in rodent models. 121,124 Reduced recruitment could also be explained by IVIG's ability to regulate the expression of adhesion molecules and cytokines/chemokines involved in leukocyte trafficking. IVIG has indeed been shown to downregulate the expression of integrins on leukocyte cell surfaces in an ischemia-reperfusion injury model, preventing the adhesion, rolling, and subsequent extravasation of these cells into the damaged area. 125 These findings are contrasted, however, by observations in experimental stroke where IVIG actually increased leukocyte and platelet trafficking to the injury site, causing them to form aggregates within the cerebral vasculature. 126
Complement scavenging
Non-antigen-binding regions of the F(ab’)2 fragment of IgG have been shown to bind and neutralize the complement activation products C3a and C5a, both in vitro 127 and in vivo, 128 thereby preventing complement-mediated tissue damage. Several studies, including our own, have reported that IVIG is able to attenuate complement in various models of acquired CNS injury. 108,114,115 For SCI, we have demonstrated that IVIG therapy reduces the overall levels of complement activation products, C5a and C3b, within the damaged segment of the spinal cord. 108 IVIG treatment similarly suppresses the rise in C3b levels that would otherwise occur within the infarct area following experimental stroke 114 and hypoxic-ischemic insults to the neonatal brain. 115 Immunoprecipitation analysis found that human IgG did directly bind to mouse C3b, lending further support to the hypothesis that IVIG may act here by neutralizing complement activation products in a manner similar to that observed in non-neurological injuries. Intriguingly, IVIG also appeared able to attenuate the oxygen deprivation-induced production of C3 itself by neurons in primary cultures, suggesting that IVIG may act not only by scavenging complement activation products already generated/secreted, but also by reducing the local production of these. 114
Modulation of inflammation via the constant Fc region
As alluded to earlier, therapeutic efficacy has also been ascribed to the Fc fragment, that is, the portion of the IgG molecule that normally binds to Fcγ receptors (FcγRs) present on most leukocytes as well as many resident CNS cells (Fig. 2). The FcγR family consists of multiple activating receptors that typically induce a phagocytic response to degrade pathogens or other opsonised targets, and one inhibitory receptor that signals to dampen and/or balance effector cell responses. 104 The amount and types of FcγRs expressed differ between immune cell types, and it is the combination of which FcγRs are expressed by a given cell that determines what immunoglobulin isotypes they respond to and how. All myeloid cells express some combination of activating FcγRs, as do various innate lymphoid cells without a classical antigen receptor (e.g., NK cells), while key cells of the adaptive immune system (i.e., T and B cells) do not. 129 The inhibitory FcγRIIb receptor is again expressed on all myeloid cells, as well as B cells, but not NK cells or resting T cells. 130 Within the CNS, there is some controversy as to the expression and function of FcγRs in neurons, but messenger RNA (mRNA) for all FcγRs receptors has been detected in vitro in neuronal cultures. 131 Microglia, astrocytes, and oligodendrocyte precursor cells also express FcγRs and can upregulate these under disease or injured conditions (Fig. 2). 131
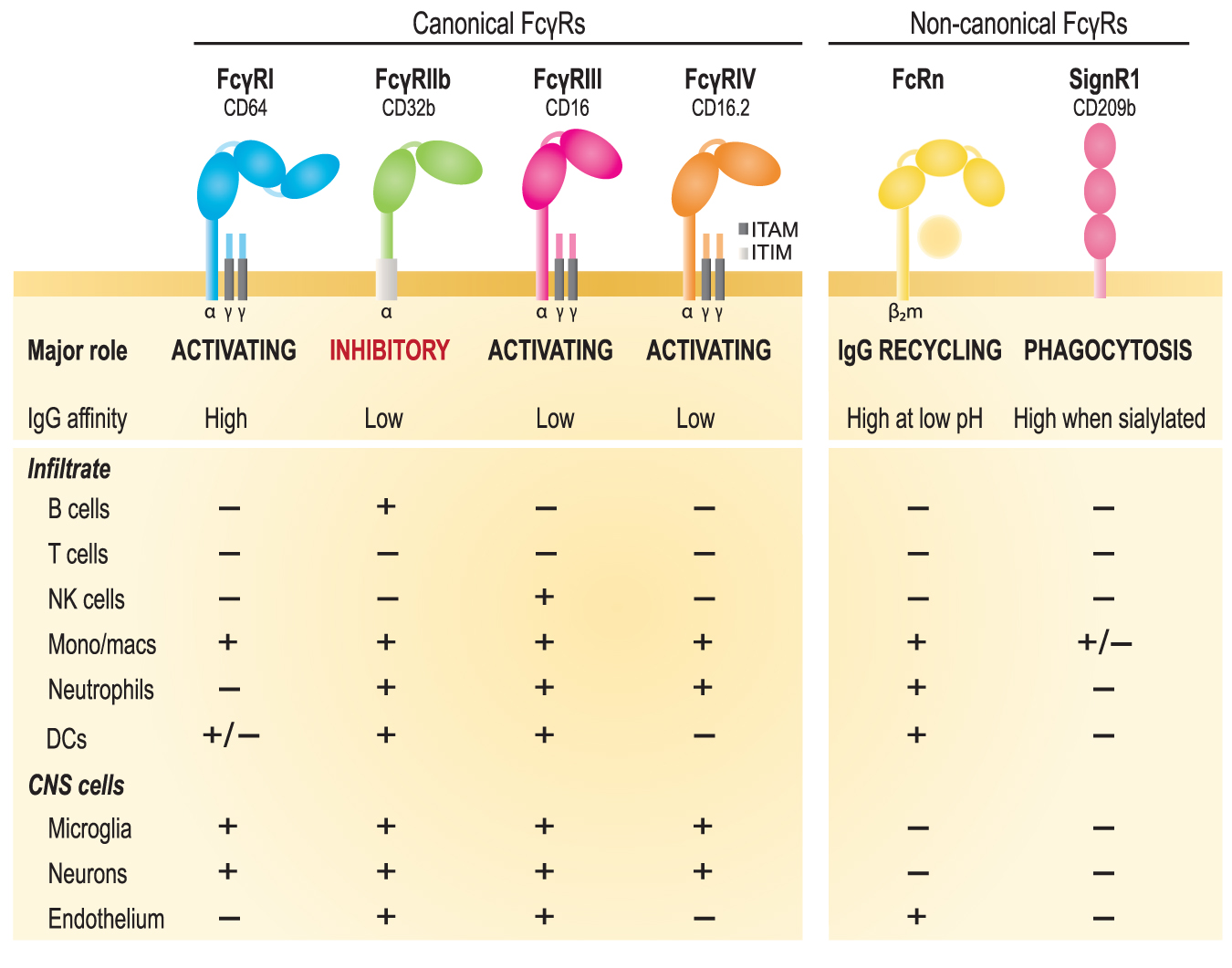
Overview of the murine family of canonical and non-canonical Fcγ receptors (FcγRs). The function, affinity for monomeric immunoglobulin G (IgG), and distribution of these receptors on relevant cell types is indicated. The canonical family of FcγRs comprises three activating receptors for IgG (FcγRI/CD64, FcγRIII/CD16, and FcγRIV/CD16.2) and one inhibitory receptor (FcγRIIb/CD32). Activating FcγRs have an ITAM sequence for signal transduction that is associated with the accessory γ chains; the inhibitory receptor contains an ITIM sequence. Non-canonical IgG receptors include the neonatal Fc receptor (FcRn), which is an MHC class 1 molecule, and DC-SIGN-related protein 1 (SIGN-R1), which is a glycoform-specific FcγR. NK, natural killer; DCs, dendritic cells. Color image is available online.
Evidence for a key involvement of FcγRs in IVIG's mechanism of action has come from studies where treatment with just the Fc fragment was equally effective in modifying disease course in a range of non-neurological autoimmune conditions, including ITP, 132,133 K/BxN arthritis, 134 –136 and nephrotoxic nephritis models. 137 Although certainly not considered an autoimmune condition at the outset, there is some evidence within the literature for a contribution of FcγRs to secondary inflammatory pathology in acute CNS injury. Specifically, mice that lack the common γ-chain, and thus have no functional FcγRs, are protected from experimental stroke as well as SCI. 56,138
Several Fc/FcγR-dependent mechanisms via which IVIG may exert its immunomodulatory effects have been proposed. 104 Certainly, within the context of antibody-mediated autoimmune disease, saturation of FcRns with high-dose IVIG therapy may reduce the half-life of circulating endogenous IgG, in particular pathogenic autoantibodies. 104 High-dose IVIG may also saturate activating FcγRs, essentially blocking off access and preventing effector cells from being activated by opsonised materials coated with endogenous IgG. Despite comprising mostly non-complexed IgG, IVIG does indeed bind to activating receptors on human cells that are low-affinity for monomeric Ig. 139 –141 However, the “FcγR saturation concept” has never been fully proven, and the focus of research has consequently shifted to other mechanisms via which IVIG may act. 142
Another possibility is therefore that IVIG may modulate dendritic cell (DC) activity via the DC immunoreceptor, which is an ITIM-linked c-type lectin Fc receptor present on the surface of these cells. In allergic airways disease, IVIG-treated DCs induced regulatory T cell (Treg) differentiation and suppressed the inflammatory Th1 response. 143 Interestingly, adoptive transfer of DCs that were primed ex vivo with IVIG and then transferred into mice with ITP was equally effective in modifying disease course as IVIG treatment itself. 144 IVIG has further been shown to reduce the expression of the activating receptor FcγRIII in human patients with ITP, and to also lower type 1 interferon responses. 145 The putative significance of these effects in the context of SCI requires further investigation. 146
One of the most favored theories as to how IVIG modulates innate immune effector mechanisms is that it may act via the inhibitory FcγRIIb. Supporting evidence for this comes from observations that the presence of FcγRIIb is critically required for IVIG's immunomodulatory effects in multiple models of immune complex-mediated disease, 104 and also that IVIG increases the expression of this inhibitory FcγRIIb on macrophages across multiple models. 133 –135,137,141 The latter is proposed to lower the ratio between activating (A) and inhibitory (I) FcγRs on the surface of these cells, thereby changing their threshold for activation and/or the way in which they respond to inflammatory stimuli.
Although there is consensus that presence of FcγRIIb is typically required for IVIG therapy to be effective, there is still much controversy as to if and how IVIG upregulates FcγRIIb expression. The prevailing hypothesis is that an IVIG-dependent upregulation of FcγRIIb, along with the anti-inflammatory effects that result from this, are dependent on the glycosylation state of the IgG molecules present within IVIG preparations. 104 The presence of a sialic acid residue at the N-linked glycosylation site Asn297 within the Fc portion of IgG in particular is thought to be essential for IVIG's disease-modifying effects. 135,143,147,148 Consistent with that, a loss of effect has been reported for de-glycosylated IVIG in peripheral autoimmune disease, whereas sialic-acid-enriched preparations showed a 10-fold increase in therapeutic efficacy and a concomitant increase in FcγRIIb expression by effector macrophages. 135
As sialylated IgGs only make up a very small fraction of the total amount of Ig that is present in IVIG preparations (approximately 5%), this could explain why IVIG often needs to be administered at relatively high doses for it to be effective, including in SCI. 108 Consistent with that, studies by Anthony and colleagues have shown that sialic-acid enriched Fc fragments from IVIG preparations were effective at a much lower dose than normal IVIG and acted via a mechanism that involved the type 2 c-type lectin Fc receptor SIGN-R1 (Fig. 2). 147,148 However, many other studies have questioned this need for sialylation, 136,149,150 and also whether IVIG upregulates FcγRIIb on macrophages. 141 Nevertheless, as the benefits of acute IVIG therapy in acquired CNS injury are now firmly established through independent studies across a range of models, further investigation is warranted to better understand whether IVIG acts here in an F(ab’)2- and/or an Fc-dependent mechanism, and also whether there is any role for sialylation therein.
Modulation of cytokines/chemokines by IVIG
In addition to the above-listed F(ab’)2- and Fc-dependent mechanisms, there is also substantial evidence within the literature that IVIG changes the cytokine response, either directly or indirectly, in the context of SCI. In a clip compression model of SCI, administration of hIgG (0.4g/kg; IV) significantly reduced the intraparenchymal concentration of pro-inflammatory cytokines IL-6, IL-1β, and MCP-1 (monocyte chemoattractant protein-1) at 4 h post-injury. 112 A follow-up study from the same group showed, however, that this phenomenon is effectively reversed by 24 h post-injury. Specifically, Chio and associates used a proteome profiler array to measure cytokine levels in the injured spinal cord, and reported significantly higher levels of IL-10, CX3CL1, IL-1β, and TNF-α in hIgG-treated rats. 113 A similar trend was observed in the blood, with significant increases in IL-8, MIP-1α, CCL2, IL-5, and VEGF (vascular endothelial growth factor) levels in association with hIgG treatment. These effects were specific to IVIG and not observed with MPSS, 113 highlighting the fact that IVIG acts differently than classical immunosuppressive agents.
The above-detailed results are nonetheless somewhat surprising, or perhaps counterintuitive even, given the general consensus views of IVIG as an anti-inflammatory, and that pro-inflammatory signals during the acute phase are often considered detrimental to recovery from SCI. Early in vitro studies looking at the effect of IVIG on blood cell cultures also found, however, that the effects of IVIG on cytokine production can change with time. The synthesis of various cytokines, such as IL-2, TNF-β, GM-CSF, IL-3, IL-4, IL-5, and IL-10, were all suppressed during the first 48 h of stimulation, but by 72 and 96 h after treatment this effect was reversed and many of these factors became upregulated. 151 In the context of neurological disease, IFN-γ levels were found to be increased in the plasma of patients with epilepsy who received IVIG therapy, from as early as 20 min and for at least up to 3 days post-treatment; plasma IL-6 levels were also increased following IVIG treatment. 152 Collectively, these findings highlight the immunomodulatory rather than immunosuppressive nature of IVIG therapy, and also our still limited understanding of how inflammation in general links to recovery. The outstanding challenge now lies in understanding how these changes beneficially influence the inflammatory response to acute neurological insults.
Conclusion
The overall profile of the inflammatory response to SCI in terms of the various immune cell subsets present at the lesion site has now been quite well documented. How these cells interact to influence lesion site development and outcomes remains much less understood. The scientific consensus has been that post-SCI inflammation is, on balance, harmful to recovery and therapeutic interventions have therefore mostly been focused on suppressing this response. For successful translation, therapeutic modulation of the inflammatory response to SCI is unlikely going to be as simple as depleting one type of cell or overexpressing one type of cytokine. It is indeed becoming increasingly clear that, for the field to advance, it is essential to create a more holistic understanding of how the inflammatory networks of cellular and molecular responses work together.
As for translation, and having the advantage of being an immunomodulatory rather than an immunosuppressant, there is sufficient evidence to support the exploratory use of IVIG in acute SCI clinically. As discussed, the multitude of ways via which IVIG can beneficially modulate the immune response here include both F(ab)’2 and Fc-dependent mechanisms, as well as a temporal regulation of cytokine expression, both at the lesion site and in the periphery. In addition to exploring its potential as a treatment option for acute SCI in humans, we can also continue to take advantage of IVIG (as well as other promising immunomodulatory agents) in pre-clinical studies by using it as a tool to dissect the effector mechanisms and signaling pathways that positively modulate the inflammatory response to SCI and improve neurological outcomes.
Footnotes
Funding Information
E.R.G. is supported by a Research Training Program Scholarship (Australian Government). Work in the laboratory of M.J.R. is supported by SpinalCure Australia (020759), the Wings for Life Spinal Cord Research Foundation (WFL-AU-19/17), and the National Health and Medical Research Council of Australia (1060538 and 1163835).
Author Disclosure Statement
No competing financial interests exist.