
Abstract
Gastrointestinal dysfunction is a common peripheral organ complication after traumatic brain injury (TBI), yet the underlying mechanism remains unknown. TBI has been demonstrated to cause gut microbiota dysbiosis in animal models, although the impacts of gut microbiota dysbiosis on gastrointestinal dysfunction were not examined. Bile acids are key metabolites between gut microbiota and host interactions. Therefore, the aim of this study was to investigate the mechanistic links between them by detecting the alterations of gut microbiota and bile acid profile after TBI. For that, we established TBI in mice using a lateral fluid percussion injury model. Gut microbiota was examined by 16S rRNA sequencing, and bile acids were profiled by ultra-performance liquid chromatography-tandem mass spectrometry. Our results showed that TBI caused intestinal inflammation and gut barrier impairment. Alterations of gut microbiota and bile acid profile were observed. The diversity of gut microbiota experienced a time dependent change from 1 h to 7 days post-injury. Levels of bile acids in feces and plasma were decreased after TBI, and the decrease was more significant in secondary bile acids, which may contribute to intestinal inflammation. Specific bacterial taxa such as Staphylococcus and Lachnospiraceae that may contribute to the bile acid metabolic changes were identifed. In conclusion, our study suggested that TBI-induced gut microbiota dysbiosis may contribute to gastrointestinal dysfunction via altering bile acid profile. Gut microbiota may be a potential treatment target for TBI-induced gastrointestinal dysfunction.
Introduction
Traumatic brain injury (TBI) is a worldwide public health problem and is the leading cause of death in young adults. 1,2 Each year, TBI causes a large number of deaths, and the survivors also experience a spectrum of physical, psychiatric, emotional, and cognitive disabilities. Therefore, TBI poses a heavy burden on individuals, families, and society. It is known that TBI is a complex injury involving the primary injury that occurred at the time of trauma and a secondary injury developed hours to months following trauma. 3 The secondary brain injury is mediated by several biochemical cascades, which are not only detrimental to the brain, but also can have significant negative effects on various organ systems, such as the respiratory system, cardiovascular system, and gastrointestinal system. 4 There is considerable evidence demonstrating that TBI can cause gastrointestinal dysfunction, which is characterized by gut motility abnormality, inflammation, and gut barrier impairment. 4,5 Gastrointestinal dysfunction will further lead to feeding intolerance, translocation of bacteria, and infections, which will prolong hospitalization periods and increase the mortality rate of TBI patients. 6,7 To investigate the underlying mechanisms of TBI- induced gastrointestinal dysfunction, a number of studies have been performed in this area and revealed that increased level of pro-inflammatory factors, mucosal barrier disruption, and decreased mucosal blood flow were associated with TBI- induced gastrointestinal dysfunction. 8 –10 Yet, the exact mechanism is not fully understood.
The microbiota are defined as the collective genomes of the microorganisms that reside in the human body. 11 The gastrointestinal tract is colonized by trillions of microorganisms, including >1000 different species of bacteria, known as gut microbiota. 12,13 These intestinal microbes regulate many aspects of host physiology and pathological process and are crucial to human health. 14 In recent years, there is increasing evidence that indicates a link between gut microbiota and the brain via the “gut–brain axis”. 11,15,16 Disruption of gut microbiota has been found to be related to several brain function disorders including Parkinson's disease, mood disorders, and autism. 17 –19 On the contrary, brain function disorders can in turn alter gut microbial composition. 16
Recently, several studies showed that TBI can lead to alterations of gut microbiota. 20 –22 Houlden and coworkers found that TBI altered the cecal microbiota via altered autonomic activity and mucoprotein production in mice. 21 Nicholson and coworkers reported that mild TBI induced gut microbiome changes in the first 7 days after TBI in rats, 20 and Treangen and coworkers demonstrated alterations of gut microbiota at 24 h after TBI in mice. 22 Although these works are important in this field, some limitations exist. The previous studies only provided preliminary descriptions of gut microbiota changes after TBI, without probing their subsequent effects. Therefore, further studies are needed to investigate the impacts of TBI- induced gut microbiota alterations. For example, it remains uninvestigated whether gut microbiota dysbiosis is involved in the pathogenesis of TBI-induced gastrointestinal dysfunction. It is known that microbial metabolites including bile acid (BA), short chain fatty acids, and tryptophan are key media between gut microbiota and host interactions. These bacterial metabolites can act locally in the intestine or cause systemic reactions after accumulating up to certain concentrations in the serum. 23 Among the various bacterial metabolites, BA is considered to be closely related to intestinal function. BAs are small molecules synthesized from cholesterol in the liver and then released to intestinal tract. Alterations in the composition of gut microbiota and BA profile in inflammatory bowel disease (IBD) patients have been described in a number of studies, and decreased level of specific secondary BAs will contribute to intestinal inflammation. 24,25 These findings are suggestive that gut microbiota dysbiosis after TBI may contribute to gastrointestinal dysfunction via altering the BA metabolic profile. Therefore, in the present study, we aimed to examine the alterations of gut microbiota and BA metabolism profile after TBI, and to investigate the associations between specific bacterial taxa and metabolic changes.
Methods
Protocols and animals
The protocol of brain injury establishment and gut microbiota sequencing has been described in our previous studies. 26,27 Male C57BL/6J mice (5–6 weeks old, 20–25 g) were obtained from the Experimental Animal Center of Zhejiang University School of Medicine. The mice were maintained on a 12-h light–dark cycle, and food and water were provided ad libitum. All mice were fed with the same diet throughout the study. All experiments were approved by the Experimental Animal Ethics Committee of Zhejiang University. All efforts were made to minimize animal pain and discomfort.
Experimental model of TBI
Mice were assigned randomly to the sham and TBI group. In TBI group, brain injury was induced with a lateral fluid percussion injury (LFPI) device. Briefly, after the animal received anesthesia, the scalp was incised sagittally. A craniotomy (3 mm in diameter) was trephined into the skull over the right parietal cortex. A plastic injury cannula was placed over the craniotomy and cemented to the skull using dental acrylic. The injury cannula was then connected to the LFPI device (VCU Biomedical Engineering, Richmond, VA). Moderate severity of TBI (1.6–1.8 atm) was induced by rapid injection of saline into the closed cranial cavity. After inducing the brain injury, the plastic cannula was removed and the incision was sutured. A temperature-controlled heating pad was used to maintain rectal temperature at 37°C during surgery and for 30 min after TBI. Mice in the sham group were subjected to the same procedure except for LFPI.
Microbiome sequencing
Fecal samples (n = 5) were collected from sham and at 1 h, 6 h, 24 h, 3 days, and 7 days after TBI using sampling tubes provided by GENEWIZ (Genewiz Bio-Tech Co.Ltd.; Suzhou, China). The fresh fecal samples were stored at −80°C until further processing. DNA was purified from all fecal samples using the E.Z.N.A. ®Stool DNA Kit (D4015, Omega, Inc., USA). Genomic DNA was then quantified and the V3-V4 variable region of the 16S rRNA genes amplified with custom-designed primers (Forward: CCTACGGRRBGCASCAGKVRVGAAT, Reverse: GGACTACNVGGGTWTCTAATCC ). The 5' ends of the primers were tagged with specific barcodes per sample and sequencing universal primers. Polymerase chain reaction (PCR) amplification was performed in a total volume of 25 μL reaction mixture containing 25 ng of template DNA, 12.5 μL PCR Premix, 2.5 μL of each primer, and PCR-grade water to adjust the volume. The PCR conditions to amplify the prokaryotic 16S fragments consisted of an initial denaturation at 98°C for 30 sec, 32 cycles of denaturation at 98°C for 10 sec, annealing at 54°C for 30 sec, extension at 72°C for 45 sec, and then final extension at 72°C for 10 min. The PCR products were confirmed with 2% agarose gel electrophoresis. Throughout the DNA extraction process, ultrapure water was used to exclude the possibility of false-positive PCR results as a negative control. The PCR products were purified by AMPure XT beads (Beckman Coulter Genomics, Danvers, MA, USA) and quantified by Qubit (Invitrogen, USA). The amplicon pools were prepared for sequencing and the size and quantity of the amplicon library were assessed on Agilent 2100 Bioanalyzer (Agilent, USA) and with the Library Quantification Kit for Illumina (Kapa Biosciences, Woburn, MA, USA), respectively. The libraries were sequenced on NovaSeq PE250 platform.
Enzyme-linked immunosorbent assay (ELISA) for inflammatory factors in intestinal tissue
Inflammatory factors including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, and IL-10 were detected by ELISA. The experiments were performed using specific ELISA kits (MultiSciences Biotech, Hangzhou, Zhejiang, China) for the factors according to the instructions of manufacturer. Briefly, the wells were firstly coated with capture antibody and then the remaining protein-binding sites were blocked. Then samples were added and incubated with monoclonal antibodies and secondary antibodies, respectively. For detection, horse radish peroxidase (HRP) was used and optical density (OD) values were measured. Concentration values were converted from OD values after standard curve was established.
Quantitative real-time reverse transcription-PCR (qRT-PCR)
Intestinal tissue were harvested from sham and TBI mice and pre-treated, then total RNA was extracted with TRIZOL (Shanghai Invitrogen Biotechnology, Shanghai, China). Then RTs were performed using TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (Beijing Transgenbiotech, Beijing, China) to prepare cDNA. The qRT-PCR experiments were performed using THUNDERBIRDTM SYBR qPCR Mix kit (TOYOBO, New York, NY, USA) according to the manufacturer's instructions. Specific primers used were: (5' ≥ 3')
Zonula occludens-1 (ZO-1): forward primer GCCGCTAAGAGCACAGCAA
Reverse primer TCCCCACTCTGAAAATGAGGA
Occludin: forward primer TTGAAAGTCCACCTCCTTACAGA
Reverse primer CCGGATAAAAAGAGTACGCTGG
Claudin-1: forward primer GGGGACAACATCGTGACCG
Reverse primer AGGAGTCGAAGACTTTGCACT
Claudin-2: forward primer CAACTGGTGGGCTACATCCTA
Reverse primer CCCTTGGAAAAGCCAACCG
Histology of intestinal tissue
Intestinal histology was evaluated by hematoxylin and eosin (H&E) staining. Samples of small intestine and colon tissue were harvest from TBI and sham mice. The samples were fixed in buffered formalin, then cut into 5-mm-thick sections (150 mm between each section, eight sections per fragment per sample), and stained with H&E.
Sample preparation for BA profiling
The fecal samples (10 mg) were added to 980 μL of extraction solvent (methanol/water [v/v] = 2:1, 0.005% HCOOH) and 20 μL of internal standards solution, followed by rapid freeze–thaw cycles in liquid nitrogen three times, followed by homogenization using TissueLyser at 50 Hz for 90 sec. The extracted supernatant was obtained by centrifugation at 12000 rpm, 4°C for 10 min, and injected into the ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) system. The moisture content was measured separately for each sample. For plasma samples, a volume of 50 μL of plasma was mixed with 490 μL of pre-cooled methanol and 60 μL of internal standards solution, and the mixture was vortexed and centrifuged at 12,000 rpm, 4°C for 10 min. The supernatant was then evaporated to dryness and redissolved in 50 μL of solvent (methanol/water [v/v] = 2:1, 0.005% HCOOH). Finally, the supernatants were injected into the UPLC-MS/MS system. The linearity of each BA calibration curve was evaluated by correlation coefficient, which was generated by least-squares linear regression analysis using the multiple reaction monitoring (MRM) peak area of each BA over the MRM peak area of the internal standards. Notably, because some of the BA internal standards were not commercially available, the internal standard with similar structures to these BAs was used. The profiling of BAs in this study was performed utilizing a service from LC-BIO (Hangzhou, China).
LC-MS
The UPLC-MS/MS system was made up of an Agilent 1290 UPLC coupled to an Agilent 6470 triple quadrupole mass spectrometer equipped with an Agilent Jet Stream electrospray ionization source (Agilent Technologies, Inc. Santa Clara, CA, USA). Samples (1 μL) were separated on ZORBAX Eclipse Plus C18 column (2.1 × 100 mm, 1.8 μm, Agilent, USA) at 45°C. Mobile phases consisted of A (water with 0.005% HCOOH, v/v) and B (acetonitrile with 0.005% HCOOH, v/v). The elution gradient was set stepwise as follows: 26% B to 32% B for 7 min, 32% B to 70% B for 5 min, and 70% B to 95% B for 1 min. The flow rate was 0.6 mL/min. MS detection of BAs was conducted in a negative mode. Fragmentor and product ions for every BA were optimized through direct infusion of available BA standards to improve detective sensitivity. Multiple reaction monitoring has been used for quantification of screening fragment ions. Peak determination and peak area integration was performed with MassHunter Workstation software (Agilent, Version B.08.00) whereas auto-integration was manually inspected and corrected if necessary. The obtained peak areas of targets were corrected by appropriate internal standards, and calculated response ratios were used throughout the analysis.
Statistical analysis
Microbial sequencing analysis was performed using R software package (Version 2.1). Operational taxonomic units (OUT) were clustered by Qiime (1.9.1) and Vsearch (1.9.6). Alpha diversity and beta diversity analysis were performed by Qiime and R software package. One-way analysis of variance (ANOVA) with Fisher's least significant difference multiple comparison tests was used to compare changes of microbiota composition in different groups. Data of inflammatory cytokines and tight junction proteins were expressed as mean ± standard error of the mean (SEM). Student's two tailed t test was used to compare differences in two groups, and for comparisons of more than two groups, one-way ANOVA followed by Tukey's post-hoc test for multiple comparison was used. Spearman's correlation analysis was used to investigate the association between bacterial general and BAs. Statistical significance was determined at a p value of <0.05.
Results
TBI induces intestinal inflammation and gut barrier impairment
Previous studies have suggested that focal TBI caused intestinal inflammation in animal models by using a controlled cortical impact (CCI) model. In our study, to investigate whether diffuse brain injury induced intestinal inflammation, small intestine and colon tissues were harvested at 1 and 3 days after TBI. Inflammatory factors including pro-inflammatory cytokines IL-1β, TNF-α, IL-6, and anti- inflammatory cytokines IL-10 were measured in the intestinal tissue by ELISA. In the small intestine, there were no significant changes in the levels of TNF-α at 1 and 3 days after TBI (Fig. 1A). Concentrations of IL-1β and IL-6 were significantly increased at 1 day after TBI, with no significant changes at 3 days after TBI (Fig. 1B, C). Levels of IL-10 were significantly increased at 1 and 3 days after TBI (Fig. 1D). In the colon, there were no significant changes in TNF-α (Fig. 1E) and IL-1β (Fig. 1F) at 1 and 3 days after TBI. Levels of IL-6 were significantly decreased at 1 day after TBI (Fig. 1G), while levels of IL-10 were significantly increased at 1 and 3 days after TBI (Fig. 1H).
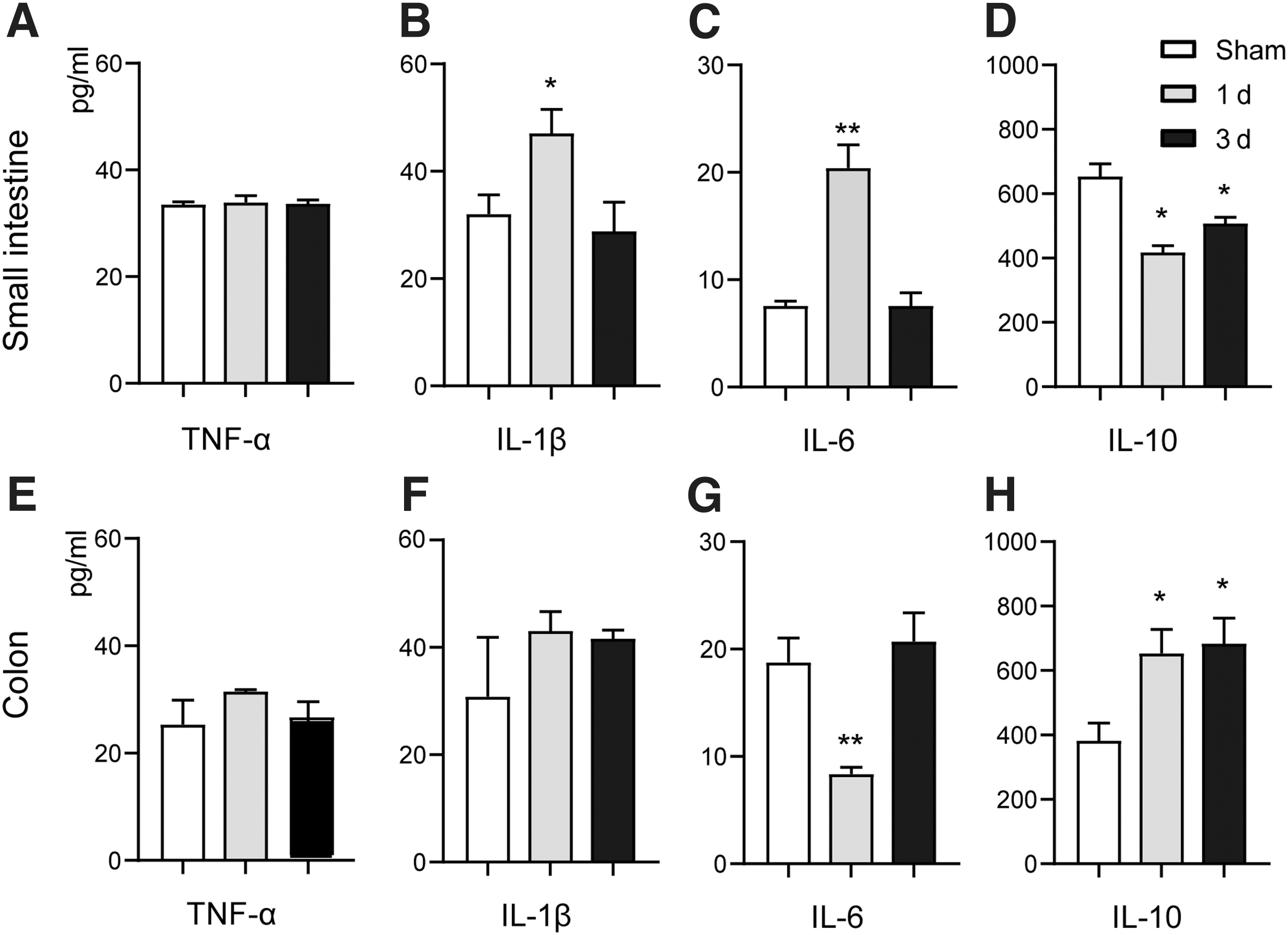
Traumatic brain injury (TBI) induces intestinal inflammation. In small intestine,
We further examined the alterations of intestinal barrier function by measuring the relative expression levels of mRNA of tight junction proteins including ZO-1, occludin, claudin-1, and claudin-2. No significant changes were observed in the mRNA levels of ZO-1 after TBI in either small intestine or colon tissues (Fig. 2A, B). We observed lower level of occludin mRNA in the small intestine at 1 day after TBI; however, this did not reach statistical significance. In the colon, the mRNA expression level of occludin was significantly decreased at 3 days after TBI. The expression level of claudin-1 mRNA was significantly lower in the small intestine at 1 and 3 days after TBI, and was also significantly lower in the colon at 1 day after TBI. The claudin-2 mRNA level was significantly decreased in the small intestine at 3 days after TBI and in the colon at 1 day after TBI. These results indicated that diffused TBI can impair the synthesis of tight junction proteins in intestinal tissue. H&E staining of the small intestine at 3 days after TBI showed that structures of intestine villa were impaired after TBI (Fig. 2C, D).
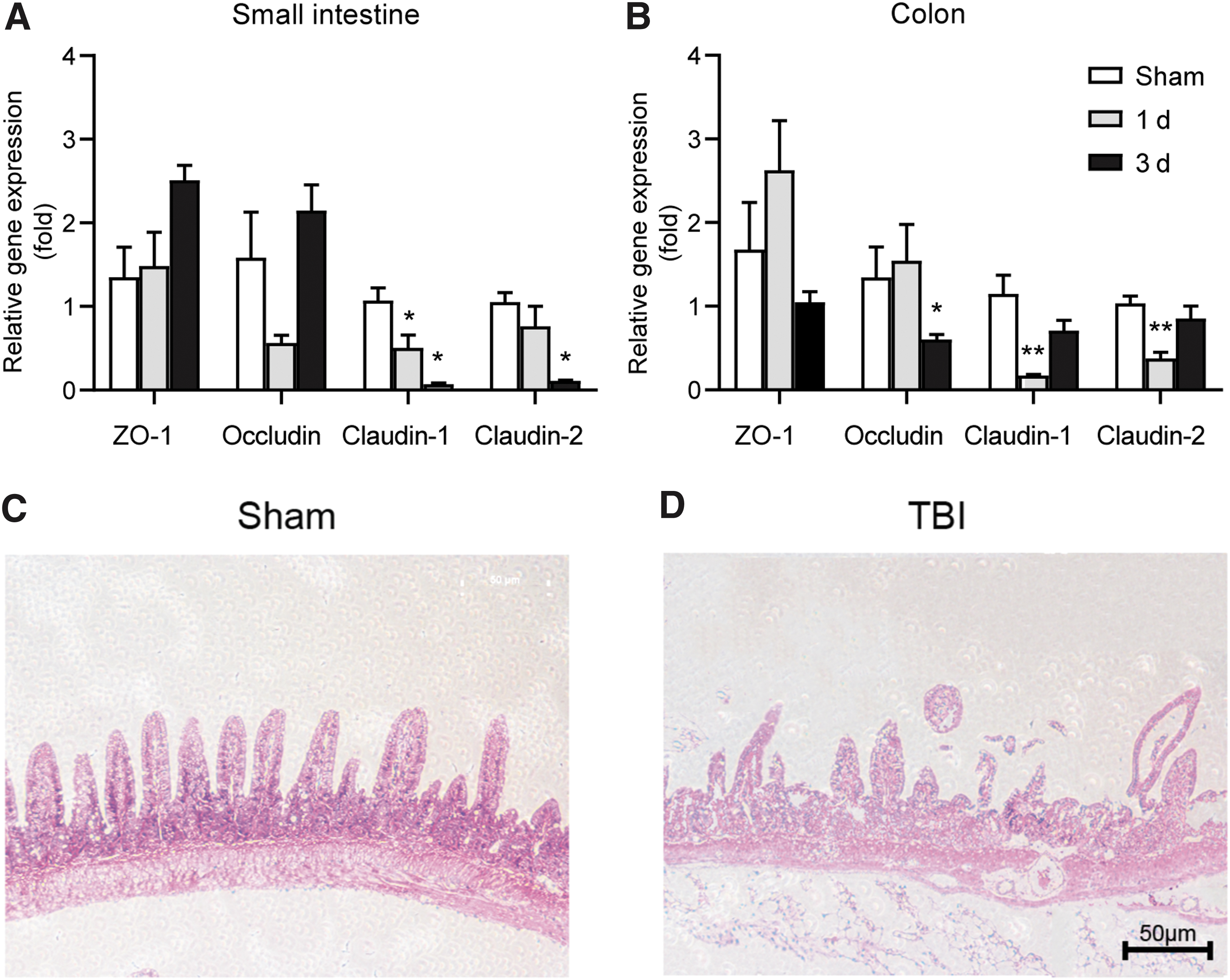
Traumatic brain injury (TBI) induces gut barrier impairment.
TBI decreases gut microbiota diversity in a time-dependent manner
In this study, gut microbiome diversity was measured by alpha diversity and beta diversity. Alpha diversity was estimated by Chao1, Shannon, and Simpson indexes. Gut bacterial community richness indicated by Chao1 index was significant lower after TBI (p < 0.001) (Fig. 3A). No significant differences in Chao1 index were observed at different time points after TBI, although there was a trend toward decreasing from 1 h to 3 days, and then a trend of increasing from 3 to 7 days after brain injury (Fig. 1A). Bacterial community diversity estimated by both Shannon (Fig. 3B) and Simpson (Fig. 3C) indexes both was significant lower in the TBI group than in the Sham group. In addition, Shannon and Simpson indexes showed a significant downward trend from 1 to 24 h post-injury, and then showed an upward trend from 3 to 7 days post-injury. These results suggested that the alpha diversity of gut microbiota may change dynamically in a time-dependent manner after TBI. It decreased gradually after TBI and reached its lowest level at ∼24 h to 3 days after TBI, and then recovered over time from 3 to 7 days after TBI.
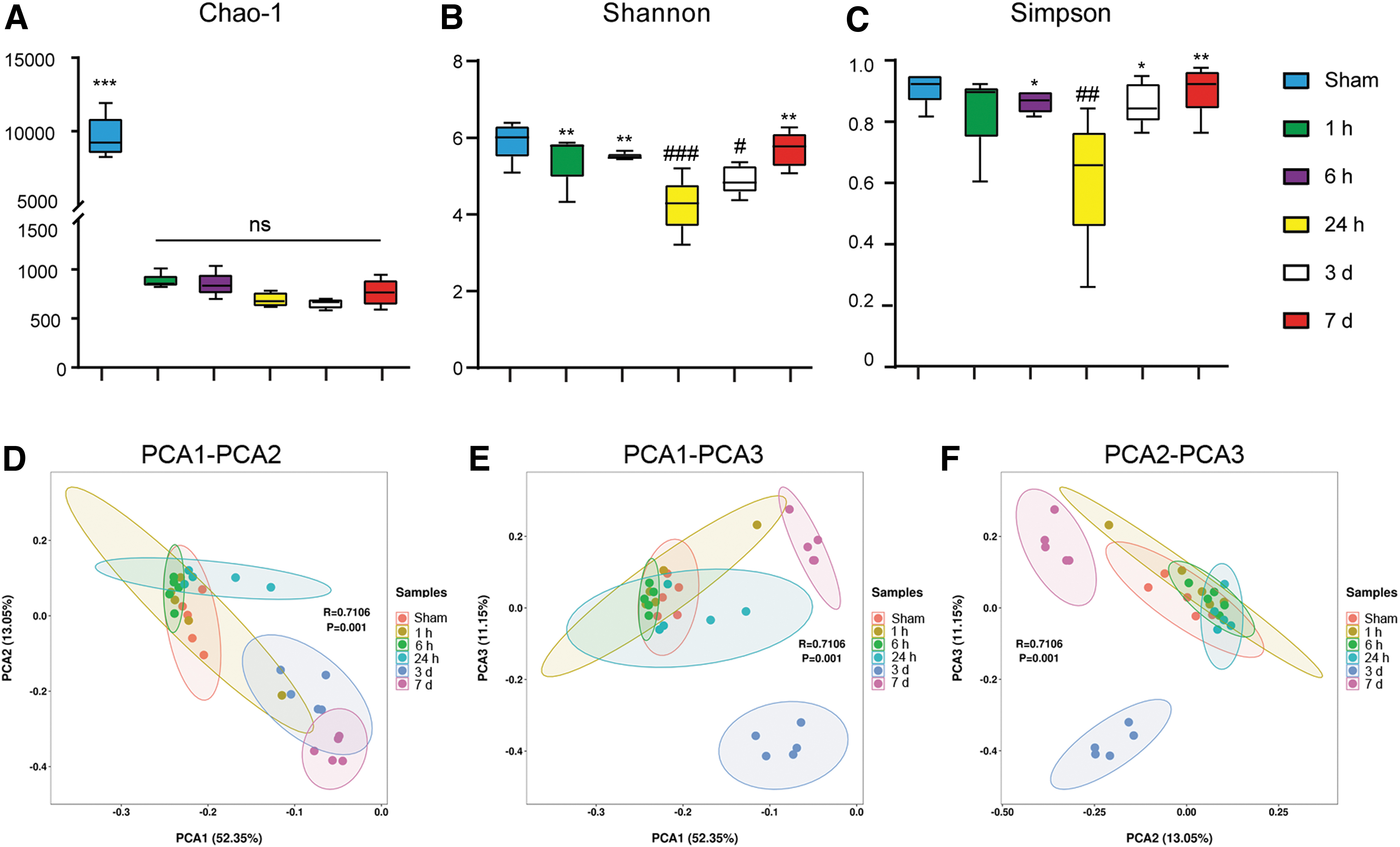
Alterations of gut microbiota diversity after traumatic brain injury (TBI).
Beta diversity analysis was performed to investigate the potential differences in bacterial community structure between the groups. Beta diversity was depicted by principal component analysis (PCA) plot. The microbial community structure did not change significantly at 1 or 6 h after injury. At 24 h post-TBI, a shift in microbial composition was observed in PC1, which accounted for 52.35% of the inter-sample variation. At 3 days post-TBI, shifts in microbial composition were observed in all three principal components (PC1, PC2, PC3), which together explained 76.55% of the variance. At 7 days after injury, shifts in microbial composition mainly occurred along PC1 and PC2, which together accounted for 65.4% of the inter-sample variation (Fig. 3D–F). These results indicated that gut microbial composition also changed over time after TBI, and that the most significant change may occur at 3 days post TBI.
Identification of discriminant taxa at phylum and genus levels after TBI
To investigate the alterations of gut microbiota after TBI, the discriminant taxa were identified at the phylum and genus levels. At the phylum level, no discriminant taxa were identified at 1 h after TBI. At 6 h after TBI, Bacteroidetes (p = 0.024) and Proteobacteria (p = 0.028) were significantly more abundant in the TBI group, while Firmicutes (p = 0.017) were less abundant in the TBI group (Fig. 4A). At 24 h after TBI, no specific taxa were found to be significantly different between the TBI and Sham groups (Fig. 4B). At 3 days after TBI, Firmicutes (p = 0.021) were less abundant in the TBI group (Fig. 4C). At 7 days after TBI, the abundance of Verrucomicrobia (p = 0.032) was significantly higher in the TBI group (Fig. 4D).

Discriminant bacterial taxa at phylum and genus levels after traumatic brain injury (TBI).
At the genus level, Oscillibacter (p = 0.042) were more abundant in the TBI group at 1 h post-TBI, while Ruminococcaceae_UCG-004 (p = 0.016) and Romboutsia (p = 0.044) were less abundant in the TBI group at the same time points (Fig. 4E). At 6 h post-TBI, the abundance of 13 genera was significantly different (p < 0.05) between the TBI and Sham groups. Of these, nine genera were more abundant in the TBI group, including the Lachnospiraceae FCS020 group, Bacteroidetes, Oscillibacter, Ruminococcaceae_UCG-009, Lachnospiraceae_UCG-001, Anaerotruncus, Ruminiclostridium, Negativibacillus, Escherichia-Shigella, and four genera were less abundant in the TBI group including Ruminococcaceae_UCG-004, [Eubacterium]_nodatum_group, Family_XIII_UCG-001, and [Eubacterium]_ventriosum_group (Fig. 4F). At 24 h after TBI, 11 genera were significantly different (p < 0.05) between the TBI and Sham groups. Of these, Tyzzerella was more abundant in the TBI group, while [Eubacterium]_ventriosum_group, Family_XIII_UCG-001, Lachnospiraceae_FCS020_group, [Eubacterium]_nodatum_group, Ruminococcaceae_UCG-005, Marvinbryantia, GCA-900066225, Lactobacillus, Romboutsia, and GCA-900066575 were less abundant in the TBI group (Fig. 4G). At 3 days post-TBI, 13 genera were significantly different (p < 0.05) between the TBI and Sham groups. f_Muribaculaceae_Unclassified, Dubosiella, and Faecalibaculum were more abundant in the TBI group, while Bacteroides, Lactobacillus, Ruminiclostridium_5, Family_XIII_UCG_001, Ruminococcaceae_UCG-009, Ruminococcaceae_UCG-005, f_Peptococcaceae_Unclassified, f_ Ruminococcaceae _Unclassified, Romboutsia, and [Eubacterium]_coprostanoligenes_group were less abundant in the TBI group (Fig. 4H). At 7 days after TBI, a total of 22 genera were significantly different (p < 0.05) between the TBI and Sham groups. Sixteen genera such as Alistipes, f_Muribaculaceae_Unclassified, [Eubacterium]_nodatum_group, Acinetobacter, and Staphylococcus were more abundant in the TBI group. Six genera including Bacteroides, Parabacteroides, Ruminococcaceae_UCG-005, Family_XIII_UCG_001, Ruminococcaceae_UCG-009, and Romboutsia were less abundant in the TBI group (Fig. 4I).
Production of BAs was impaired after TBI
BAs are one of the major metabolites of gut microbiota and are important media in microbiome–host interactions. To investigate whether gut microbiota dysbiosis after TBI affects the synthesis of BAs, they were profiled both in plasma and feces in the Sham group and 24 h after TBI. The results showed that the levels of primary and secondary BAs in plasma and feces were decreased at 24 h after TBI. In feces, concentrations of the primary BAs were decreased in the TBI group; nevertheless, the decrease did not reach statistical significance. Concentrations of secondary BAs including taurodeoxycholic acid (TDCA), DCA, lithocholic acid (LCA), and iso-LCA were significantly lower in the TBI group (Fig. 5A). In plasma, notably, levels of primary BAs were decreased after TBI, among which CA, TCA, tauro-beta-muricholic acid (T-β-MCA), taurochenodeoxycholic acid (TCDCA), and CDCA were significantly lower in the TBI group. Meanwhile, levels of all secondary BAs were also decreased after TBI, with TDCA, tauroursodeoxycholic acid (TUDCA), DCA, LCA, and w-MCA being significant lower in TBI group. These results indicated that TBI can significantly impair the production of BAs (Fig. 5B).
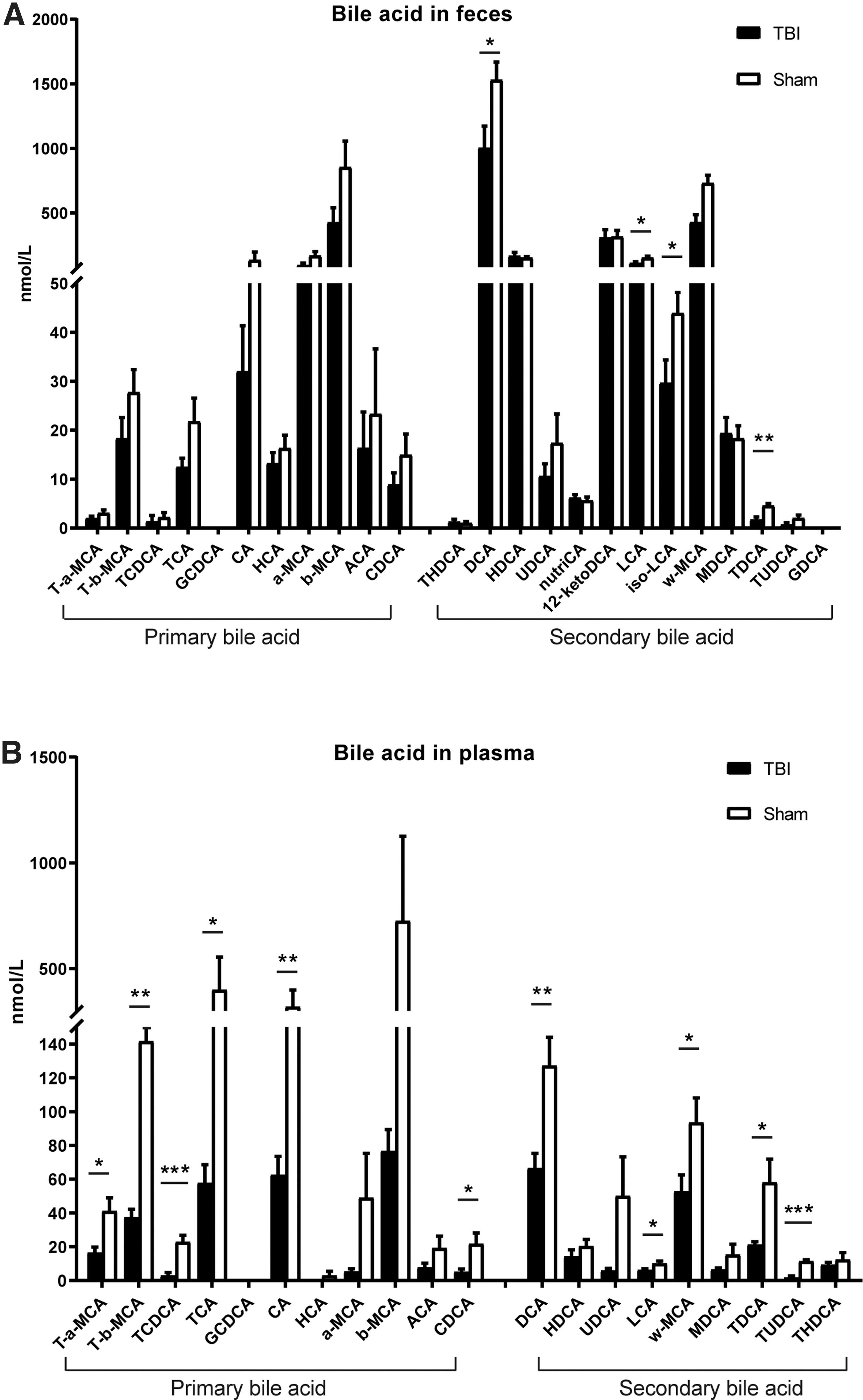
Alterations of bile acids in feces and plasma after traumatic brain injury (TBI).
Correlations between BA metabolism profile and gut microbiota
To further explore the relationships between BA metabolic profiles and gut microbiota, Spearman correlation analysis was conducted among BA levels in the fecal and plasma and gut microbiota at the genus levels, respectively. In feces, the decreased secondary bile acid TDCA was positively correlated with [Eubacterium]_nodatum _group and Lachnospiraceae_FCS020_group, and was negatively correlated with Tyzzerella, Coprococcus_3 and Ruminiclostridium_6. iso-LCA was positively correlated with Roseburia (Fig. 6A,B). In plasma, the decreased secondary BA LCA was negatively correlated with Staphylococcus (p < 0.01). T-a-MCA was positively correlated with f_Christensenellaceae_Unclassified (p < 0.01) and [Eubacterium]_xylanophilum_group (p < 0.05). TDCA was positively correlated with f_Christensenellaceae_Unclassified (p < 0.01) and [Eubacterium]_xylanophilum_group (p < 0.05), but negatively correlated with Candidatus_Arthromitus (p < 0.01). TUDCA was positively correlated with Lachnospiraceae_FCS020_group (p < 0.05), [Eubacterium]_xylanophilum_group (p < 0.01), Butyricicoccus (p < 0.01), Family_XIII_UCG-001 (p < 0.01), but negatively correlated with Candidatus_Stoquefichus (p < 0.01) and f_Muribaculaceae_Unclassified (p < 0.01). Similarly, TCDCA was also positively correlated with Lachnospiraceae_FCS020_group (p < 0.01), [Eubacterium]_xylanophilum_group (p < 0.05), Butyricicoccus (p < 0.05), Family_XIII_UCG-001 (p < 0.05), but negatively correlated with Candidatus_Stoquefichus (p < 0.01) and f_Muribaculaceae_Unclassified (p < 0.05) (Fig. 6C,D).
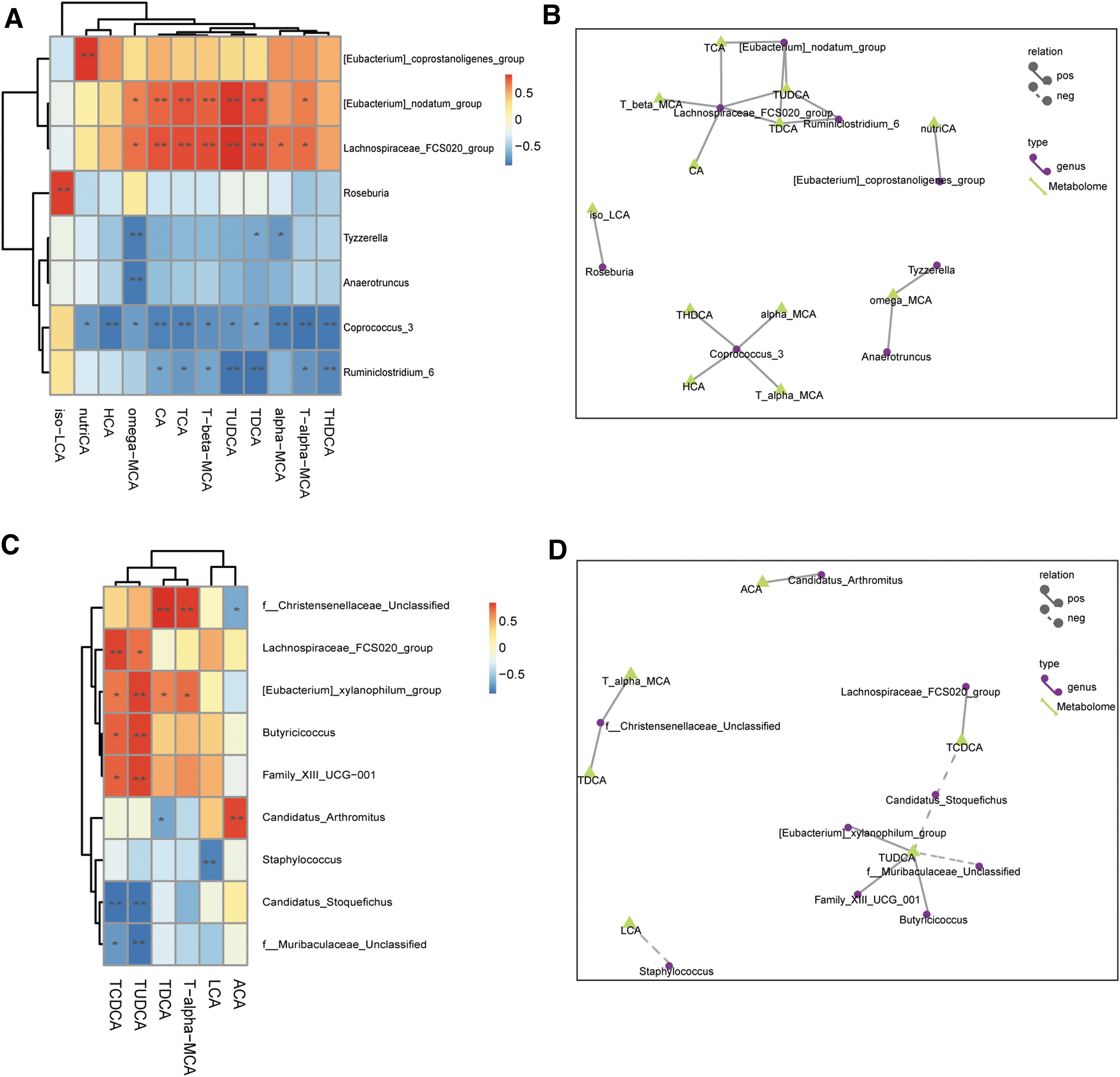
Correlations between bile acid metabolism profile and gut microbiota.
Discussion
Our study results showed that diffuse TBI led to significant time-dependent alterations of gut microbiota, including decreased gut microbiota diversity and changes of specific bacterial taxa. TBI also induced intestinal inflammation and impaired gut barrier function. In addition, we found that levels of BAs were decreased in feces and plasma after TBI. Correlations between BAs and specific gut bacterial genus after TBI were identified to reveal the interplay between them, which may lay a solid foundation for further researche on clarifying mechanisms of TBI- induced intestinal inflammation.
Emerging evidence has indicated a link between gut microbiota and brain function, well known as the “gut–brain axis.” 11,15,16 The gut–brain axis and their bidirectional interactions are being increasingly appreciated by researchers. Evidences showed that gut microbiota plays an important role in this axis and has been regarded as a potential biomarker and treatment target for a spectrum of brain diseases. 28 In recent years, several studies have reported that TBI can induce gut microbiota changes in mice. 20 –22 These studies are important in the field; however, some limitations should be noted. First, the method for establishment of TBI in these studies was control CCI model. It is known that both CCI and LFPI are widely used models for replicating TBI. However, some differences exist between the two models. Literature reported that CCI induced mainly focal brain injury, whereas LFPI induced mixed brain injury including focal cortical contusion and diffuse subcortical neuronal injury. 29 Damage from TBI can be classified as focal or diffuse injury; focal injuries include cerebral contusion and intracranial hemorrhage, whereas diffuse injuries comprise diffuse axonal injury (DAI) and cerebral swelling. 30 Therefore, we proposed that the alterations of gut microbiota after diffuse TBI remain largely unknown. For this purpose, an LFPI model was used for establishment of brain injury in this study. Similar to in previous study, 20 we observed a time-dependent change of gut microbiota diversity and bacterial community structure. The alpha diversity decreased after TBI and reached its lowest level at ∼24 h to 3 days after TBI, and then recovered over time from 3 to 7 days after TBI. It noteworthy that our results showed that gut microbiota altered as early as 1 h after TBI, which is earlier than in a previous study reporting alteration of gut microbiota at 2 h after TBI. This also indicated that TBI can alter of gut microbiota at a very early stage. Specific bacteria taxa that have significantly different abundance between the TBI and control groups were also identified at the phylum and genus level.
Second, these previous studies only provide preliminary results of gut microbiota changes after TBI, without probing the subsequent impact of gut microbiota dysbiosis on the outcome of TBI. Gastrointestinal dysfunction after TBI has long been observed and will greatly affect the outcome of TBI patients. It occurs in the acute phase after TBI and will persist for weeks. 31 Existing evidence showed that TBI-induced gastrointestinal dysfunction is characterized by intestinal inflammation and gut barrier impairment. 31 –33 As confirmed in our study, we found elevated inflammatory cytokines and decreased expression levels of tight junction proteins in the small intestine and colon tissue after TBI. Nevertheless, the underlying mechanism of TBI-induced gastrointestinal dysfunction is still not fully understood. The causes of intestinal inflammation and what is responsible for the persistent inflammation remain unknown. Until recently, studies on gut microbiota alterations after TBI provided new insights into the mechanisms of TBI-induced gastrointestinal dysfunction. It has been widely recognized that gut microbiota play a critical role in regulating intestinal function. 34 Gut microbiota dysbiosis contributes to intestinal inflammation in inflammatory bowel disease. 35 In addition, a recent study reports that antibiotic treatment prior to brain injury alleviated TBI-induced intestinal inflammation and gut barrier impairment. 36 Based on these findings, we proposed that alterations of gut microbiota after TBI may play a role in TBI-induced intestinal inflammation.
Accumulating evidence suggested that gut microbiota can interact with host physiology through their various metabolites such as BAs, short chain fatty acids, and tryptophan metabolites. 14,24,37,38 BAs are a major metabolite of gut microbiota, and evidence has shown that gut microbiota can regulate host physiology by modulating BA metabolism. 39,40 One important aspect is that BAs have been implicated in regulation of intestinal epithelial function. Altered BA profile was associated with intestinal inflammation in inflammatory bowel disease, and supplements of specific BAs such as DCA, LCA, and TUDCA alleviated gut inflammation and improved gut barrier function. 24,41,42 It is known that BAs can be divided into primary BAs and secondary BAs. The primary BAs are synthesized in the liver and then secreted into intestinal tract where they will be modified by gut bacteria to form secondary BAs. Therefore, we speculated that gut microbiota dysbiosis after TBI may contribute to intestinal dysfunction via the alteration of BA metabolic profiles. For verification, we further profiled the metabolic changes of BA in mice after TBI. The profile results showed an overall decrease of both primary and secondary BA concentrations in plasma and feces at 24 h after TBI, among which DCA and LCA were significantly lower after TBI in both plasma and feces, and TUDCA was significantly lower after TBI in plasma. These results well supported our previous conjecture and further indicate that TBI-induced gut microbiota dysbiosis contributes to intestinal inflammation by reducing the levels of secondary BAs. To our knowledge, this was also the first study describing the alterations of BA profile after TBI.
To identify the bacterial genera potentially implicated in the metabolism of BA profile, Spearman correlation analysis was conducted to reveal interactions between gut microbiota and BA metabolism. We found that LCA in plasma was negatively correlated with Staphylococcus, while TUDCA was positively correlated with Lachnospiraceae_FCS020_group, [Eubacterium]_xylanophilum_group, Butyricicoccus, Family_XIII_UCG-001, but negatively correlated with Candidatus_Stoquefichus and f_Muribaculaceae_Unclassified. Collectively, these results suggested that these specific bacterial genera were closely associated with BA metabolism. It has been reported that Lachnospiraceae and Ruminococcaceae were capable of performing 7α-dehydroxylation and were closely related to BA biotransformation. 43 Nevertheless, further studies employing bacterial transplantation techniques are needed to confirm the effects of these bacteria genera on the metabolism of BAs.
Conclusion
Our study results demonstrated that diffuse TBI can induce alterations of gut microbiota, and showed that bacterial diversity experienced a time-dependent change after TBI. Further, we found that TBI changed the profile of BAs: primary and secondary BAs were decreased in both feces and plasma. The decrease of certain secondary BAs may contribute to intestinal inflammation. Specific gut bacteria associated with BA metabolism were identified. This integrated analysis of gut microbiota and BA metabolic profile changes provided more insights into the mechanisms of TBI-induced gut inflammation, which is critical for the development of potential gut bacterial-based interventions. Further studies using fecal microbiota transplantation are warranted to clarify the mechanisms between specific gut bacteria and gastrointestinal dysfunction following TBI.
Footnotes
Acknowledgments
The authors thank the staff of the First Affiliated Hospital College of Medicine at Zhejiang University for their technical help.
Funding Information
This study was supported by the National Natural Science Foundation of China (No. 81971159, No. 82000489) and the Natural Science Foundation of Zhejiang Province (No. Y19H030059).
Author Disclosure Statement
No competing financial interests exist.