
Abstract
Blast-induced traumatic brain injury is typically regarded as a signature medical concern for military personnel who are exposed to explosive devices in active combat zones. However, soldiers as well as law enforcement personnel may be repeatedly exposed to low-level blasts during training sessions with heavy weaponries as part of combat readiness. Service personnel who sustain neurotrauma from repeated low-level blast (rLLB) exposure do not display overt pathological symptoms immediately but rather develop mild symptoms including cognitive impairments, attention deficits, mood changes, irritability, and sleep disturbances over time. Recently, we developed a rat model of rLLB by applying controlled low-level blast pressures (≤ 70 kPa) repeated five times successively to mimic the pressures experienced by service members. Using this model, we assessed anxiety-like symptoms, motor coordination, and short-term memory as a function of time. We also investigated the role of the NLRP3 inflammasome, a complex involved in chronic microglial activation and pro-inflammatory cytokine interleukin (IL)-1β release, in rLLB-induced neuroinflammation. NLRP3 and caspase-1 protein expression, microglial activation, and IL-1β release were examined as factors likely contributing to these neurobehavioral changes. Animals exposed to rLLB displayed acute and chronic short-term memory impairments and chronic anxiety-like symptoms accompanied by increased microglial activation, NLRP3 expression, and IL-1β release. Treatment with MCC950, an NLRP3 inflammasome complex inhibitor, suppressed microglial activation, reduced NLRP3 expression and IL-1β release, and improved short-term memory deficits after rLLB exposure. Collectively, this study demonstrates that rLLB induces chronic neurobehavioral and neuropathological changes by increasing NLRP3 inflammasome protein expression followed by cytokine IL-1β release.
Introduction
Blast-induced traumatic brain injuries (bTBIs) are caused by the impact of air pressure from the shock wave generated by an explosive device, which travels at supersonic speed outwards from the explosion site. 1,2 Blast exposures from explosive devices are the most common source of TBI sustained by soldiers during combat or training sessions. 3 –6 In active combat zones, soldiers more often experience high-intensity blast exposures, which occur sporadically and can cause pathological changes and cognitive impairments. 7 –11 In contrast, repeated low-level blast (rLLB) exposure is the most prevalent source of bTBI in service members training for combat readiness as well as law enforcement officers due to the use of heavy weaponry (i.e., 0.50-caliber sniper rifles, Carl Gustaf 8.4 cm recoilless rifles). 12,13 Standard military protocols limit blast exposures to ≤28 kPa peak overpressure to prevent damage to the tympanic membrane and subsequent hearing loss but do not have criteria related to neurotrauma. 14 However, incident blast overpressures (BOPs) were shown to reach 90 kPa in a study of U.S. Marines during a 2-week training protocol that included explosive breaching, demonstrating that occupational exposures can be within range to cause low-level TBI. 15 Unlike moderate and severe bTBI, rLLB does not induce overt symptoms in the acute stages but may result in neurocognitive impairments in service members or law enforcement personnel who have been exposed to rLLB over time. 15 –19
Despite numerous studies published on moderate single blast exposure and its influence on the brain, there has been limited progress in understanding the neurological effects of low-level blast, especially rLLB. 10 Of the limited studies on repeated blasts, most have been less relevant to service members in training due to significantly higher BOPs (> 70 kPa) and longer inter-blast interval times (i.e., hours to days) that do not directly replicate occupational exposures. 10,20,21 Recently, we developed a rat model of rLLB in which animals receive five 70 kPa exposures at 1- to 2-min intervals within a 10-min period to represent the repeated firing of Carl Gustaf 8.4 cm recoilless rifles or 0.50-caliber sniper rifles and use of breaching devices in combat or occupational training scenarios. 8,12,20,22 rLLB exposure produced chronic anxiety-like behavior, as well as motor and short-term memory impairments accompanied by increased microglial activation and proliferation and free radical generation. 8 In contrast, rLLB exposure to three blasts at 24-h intervals was shown to impair long-term but not short-term memory at 4 weeks post-injury. 23 Thus, the influence of rLLB on behavior and pathological changes appears to vary with the number of repetitions and time interval between blast exposures.
Earlier studies have indicated inflammasome complex involvement in initiating an immune response after a TBI event. 24 –29 In this study, we investigated the nucleotide-binding domain and leucine-rich repeat (NLR) family pyrin domain containing protein 3 (NLRP3) inflammasome as a component of an upstream mechanism that could contribute to chronic microglial activation and neurobehavioral changes, particularly short-term memory impairments. The NLRP3 inflammasome plays an important role in amplifying the immune response by facilitating the cleavage of caspase-1 to its active form and subsequent release of interleukin (IL)-1β from microglia. 30,31 Previous studies have demonstrated the influence of IL-1β on neurotransmitter release and turnover, endocrine functions and behavioral changes including spatial memory impairments, sleep disturbances, decreased exploratory activity, and anxiety through exogenous administration of IL-1β. 32 –38
In recent years, several inhibitors of NLRP inflammasomes have been developed, among which MCC950 is the most potent and specific inhibitor of NLRP3. 39 MCC950 (also known as CRID3 or CP-456,773) is a sulfonylurea compound with a specific binding site on the NACHT domain of NLRP3 inflammasome complex that prevents ATPase activity required for inflammasome complex activation, thereby inhibiting downstream release of IL-1β. 39,40 MCC950 has also been shown to bind to only the NLRP3 inflammasome and block all known forms of NLRP3 activation, including both canonical and non-canonical inflammasome pathways. 41 –44 Pre-clinical studies have demonstrated MCC950 to have good bioavailability and central nervous system (CNS) penetration, making it ideal for the treatment of inflammatory disorders 39,41,45,46 including Parkinson's disease, 47 Alzheimer's disease, 48,49 and stroke. 50
Several lines of evidence have shown that increased levels of proinflammatory cytokines, particularly IL-1β, can result in neurocognitive impairments in multiple acute and chronic neurological disorders including TBI. 51 –54 Acute microglial activation following TBI is accompanied by blood–brain barrier leakage as early as 30 min post-injury. 55 -57 It is noteworthy that MCC950 not only inhibits chronic neuroinflammation in various neurological conditions but also improves neurocognitive function in animal models of diabetes, 58 blunt TBI, 59 stroke, 60 and several other inflammatory conditions affecting the CNS. Here, we evaluated if MCC950 treatment would attenuate NLRP3-mediated chronic neuroinflammation and improve behavioral performance on an object recognition task following rLLB in our rat model (Fig. 1). To assess NLRP3-mediated inflammation, we examined changes in microglial morphology as well as expression of NLRP3 and downstream cleavage products of the inflammasome in the hippocampus and perirhinal cortex (PRh), two regions associated with object recognition memory. 61 –64
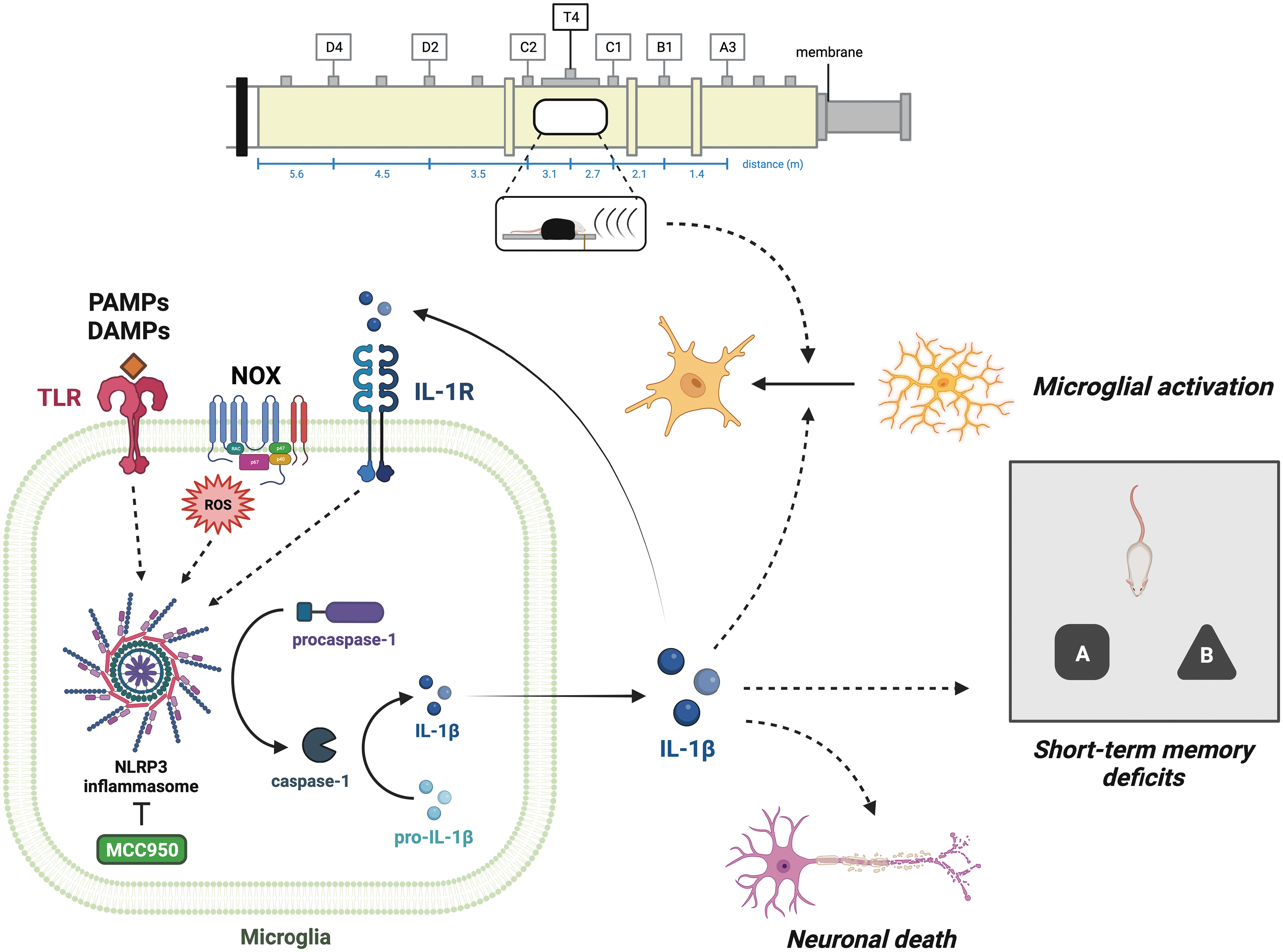
Schematic representation of repeated low-level blast (rLLB)-induced chronic neuroinflammation via NLRP3 inflammasome activation in microglia. Short-term memory deficits in rLLB may result from chronic microglial activation, elevated levels of proinflammatory cytokine interleukin (IL)-1β, and/or neuronal loss in the hippocampus and perirhinal cortex (PRh). NLRP3 inflammasome inhibition reduced microglial activation in hippocampus and PRh, suggesting that IL-1β release contributes to chronic neuroinflammation after rLLB exposure. Acute and chronic NLRP3 protein expression was increased in the hippocampus of rLLB rats. The number of NLRP3+ microglia increased by 30 days post-injury, suggesting that rLLB exposure induces NLRP3 expression and likely inflammasome activation in a larger population of microglia in response to various signals of damage including reactive oxygen species. Cleaved caspase-1 levels were increased at 1 day post-injury, demonstrating that NLRP3 activation occurs soon after injury. Acute and chronic IL-1β release was elevated in the hippocampus in response to rLLB exposure and decreased with treatment, suggesting that IL-1β plays a significant role in triggering microglial activation and neuronal loss that result in cognitive impairment. Acute and chronic neuronal loss in the hippocampus and PRh was reduced by treatment with MCC950, implicating the NLRP3 inflammasome as a major target for prevention of cell death and improvement of cognitive outcomes.
Methods
Animals
Adult male Sprague Dawley rats (10 weeks old, 300-350 g; Charles River Laboratories, Wilmington, MA) were used in this study. All animals were housed under standard room conditions (22°C temperature, 40% humidity, and 12-h dark-light cycle) with ad libitum access to food and water. Animals were randomly assigned to groups, and investigators were blind to experimental groups for the duration of the study. Rats were divided into three groups: control (sham+phosphate-buffered saline [PBS]), rLLB+PBS, and rLLB+MCC950. MCC950 (10 mg/kg; MedChemExpress, Monmouth Junction, NJ) in PBS or an equivalent volume of PBS was administered via intraperitoneal (i.p.) injection at 30 min post-injury and then every 48 h for the remainder of the study. 8 All procedures followed the guidelines established in the Guide for the Care and Use of Laboratory Animals and were approved by the Rutgers University Institutional Animal Care and Use Committee.
Rat model of repeated low-level blast exposure (rLLB)
Rats were exposed to five incident overpressures of 70 ± 5 kPa (duration: 4.0 ± 0.18 msec, impulse: 145 ± 5.5 kPa·msec) at 1- to 2-min intervals using a 6-meter 23 × 23 cm cross-section shock tube that has been well validated for field relevant blast exposures. 65 –68 Blast conditions included the use of ultra-high purity helium (99.99%) as the driver gas. The driver and driven sections of the shock tube were separated by a 1-mm thick Mylar® membrane which, upon bursting, developed pressure profiles with peak incident overpressures of 70 kPa. Incident pressure profiles were recorded at 1.0 MHz sampling frequency using ICP® pressure sensors (Model 132A24; PCB Piezotronics, Depew, NY). Animals were mounted in the middle of the shock tube (2.80 m from the breech, 3.05 m from the exit) in a prone position and were strapped securely to the aluminum plate using a cotton cloth wrapped around the body to prevent any excessive head motion. 65,69 To maintain continuous anesthesia for the entire period of blast exposure, animals received a continuous supply of 5% isoflurane into the exposure chamber via secured tubing with a hose for direct delivery into animal's nostrils. 8 Sham animals were kept in an anesthesia induction chamber next to the shock tube while blasts were fired to receive noise but not blast exposure. 70 Immediately after blast exposure, animals from both groups were monitored for any signs of apnea and/or respiratory distress. These symptoms were not observed, and no animals were removed from the study. Visible pathology including hemorrhage was not observed upon dissection.
Novel object recognition test
Short-term object recognition memory was assessed in rats (n = 8/group) following rLLB exposure using the novel object recognition (NOR) test at 2 and 31 days post-injury. 7 Briefly, the NOR test consists of three phases: acclimation (Day 1), familiarization (Day 2), and testing (Day 2). All phases were conducted in a 60 cm × 60 cm testing chamber. During acclimation, each rat was allowed to explore the testing chamber for 5 min. During familiarization, rats were placed in the chamber with two identical objects and allowed to explore for 5 min before returning to their home cages. After 1 h, one of the identical objects was replaced with a novel object, and each rat was allowed to explore the testing chamber for 3 min. The total time spent exploring objects was recorded using ANY-Maze software (version 7.1; Stoelting, Wood Dale, IL). A preference index for the novel object was calculated as (time spent with the novel object/time spent with both objects) × 100%.
Immunohistochemistry
Chronic microglial activation was assessed in CA1, CA3, and the dentate gyrus (DG) within the hippocampus and in PRh at 1 day and 30 days post-injury with immunohistochemistry. Rats (n = 4-5/group) were anesthetized with a cocktail of ketamine (100 mg/kg) and xylazine (10 mg/kg) at a 10:1 ratio via i.p. injection and subjected to transcardial perfusion with PBS (1X, pH 7.4) followed by 4% paraformaldehyde (PFA) in PBS. After perfusion, brains were extracted and post-fixed in 4% PFA at 4°C for 48 h prior to cryoprotection in 30% sucrose. Coronal brain tissue sections (20 μm thickness) were collected using a Leica VT1000S vibratome (Leica Biosystems, Deer Park, IL).70 All sections used for staining and analysis were between -3.12 to -3.60 mm from bregma.
Briefly, mounted sections were incubated with 10% donkey serum in PBS with 0.03% Triton X-100 (PBS-T) at 23°C for 1 h to block non-specific antigen binding. Slides were incubated overnight at 4°C in primary antibody solution containing either goat anti-Iba1 (1:250, PA5-18039; Invitrogen, Waltham, MA) or mouse anti-NeuN (1:400, ab104224; Abcam, Cambridge, UK) with 2% donkey serum in PBS-T. Sections were incubated for 1 h at 23°C with donkey anti-goat Alexa Fluor 488 (1:200, A11055; Invitrogen) with 2% donkey serum in Tris-buffered saline with Tween 20 (TBS-T) or with donkey anti-rabbit biotinylated antibody (1:200, NC0234201; Jackson ImmunoResearch, West Grove, PA) with 2% donkey serum in TBS-T for 1 h at 23°C followed by incubation with donkey-streptavidin Alexa Fluor 594 (1:200, S11227; Invitrogen) for 1 h at 23°C. The specificity of each antibody staining was validated in negative controls by excluding each primary antibody and visualizing for any nonspecific fluorescence.
Microscopy
Slides containing hippocampus and PRh were digitized at 20 × magnification using a Leica Aperio Versa 200 digital pathology scanner. Fluorescence intensities in each region were quantitated using Area Quant software (Leica Biosystems) and expressed as average fluorescence intensity*X unit area.70 Images for microglial morphology were acquired separately with a Fluoview 3000 laser scanning confocal microscope (Olympus, Center Valley, PA). Regions of interest were selected on a 1.25 × stitched map of the entire slide showing only the nuclear staining. Multi-channel Z stacks were collected by multi-area capturing under a 20 × objective with consistent light and detection parameters using 405, 488, and 561 diode lasers. Representative images for microglial morphology were captured as Z stacks using a 60 × oil immersion objective with 2 × zoom in the DG region.
Microglial morphology and counting
Morphological changes in microglia were quantified automatically by batch processing all images in ImageJ. Maximum Z projections were generated from multi-channel Z stacks for each image and then separated into images containing individual channels. Only images containing Iba1 staining (488 nm) were processed for morphology. All steps were carried out iteratively for each image using a macro. First, contrast was enhanced to improve Iba1 signal against background. Automated thresholding was performed using the built-in Triangle algorithm to create a binary image (Supplementary Fig. S1). 71 Objects were then skeletonized using the ImageJ Skeleton plugin, and any objects that were too small to be analyzed were removed using the Particle Remover plugin. Skeletons and branches were measured using the Analyze Skeleton plugin to record number of branches and mean branch length per cell in each image. 72 This procedure has been previously shown to identify reactive microglial morphology. 73 -75 Total process length was calculated per cell by multiplying the number of branches by the mean branch length. Overall mean process length for images of the same region in both hemispheres were averaged by sample. Sample averages were then used to analyze morphology by region for each group. All values for number of processes and process length per microglial cell are reported as mean ± standard error of the mean (SEM; Tables 1 and 2).
Mean Number of Processes Per Microglial Cell at 1 Day and 30 Days Post-Injury
PBS, phosphate-buffered saline; rLLB, repeated low-level blast; DG, dentate gyrus; PRh, perirhinal cortex.
Mean Process Length (μm) Per Microglial Cell at 1 Day and 30 Days Post-Injury
PBS, phosphate-buffered saline; rLLB, repeated low-level blast; DG, dentate gyrus; PRh, perirhinal cortex.
Total number of microglia was determined by manual counting of Iba1+ cells in ImageJ. Regions of interest (ROIs) were outlined using the polygon tool to define CA1, CA3, DG, and PRh in each image. Counts were normalized to each ROI area and reported as number of microglia per mm2 for each region. Normalized counts within each region in an image were averaged between both hemispheres for a given sample, and sample averages were used to analyze counts by region per group. All values for number of microglia are reported as mean ± SEM (Table 3).
Mean Number of Microglia by Area (per mm2) at 30 Days Post-Injury
PBS, phosphate-buffered saline; rLLB, repeated low-level blast; DG, dentate gyrus; PRh, perirhinal cortex.
Quantification of NLRP3+ microglia
NLRP3+ microglia were identified using an automated batch processing macro in cellSens Dimension (version 3.2; Olympus). Colocalization analysis was performed to generate a separate channel displaying colocalized pixels that were above threshold for both Iba1 and NLRP3. The channel containing only Iba1+/NLRP3+ pixels was then overlaid on the Iba1 channel to create an image that highlighted NLRP3+ microglia for consistent counting. Microglia containing colocalized pixels in the soma were manually counted in ImageJ. ROIs were outlined using the polygon tool to define CA1, CA3, DG, and PRh in each image. Counts were normalized to each ROI area and reported as NLRP3+ microglia per mm2 for each region. Normalized counts within each region in an image were averaged between both hemispheres for a given sample, and sample averages were used to analyze counts by region per group.
Neuron counting
Neurons were manually counted in ImageJ by identifying NeuN+ cells in CA1, CA3, DG, and PRh. ROIs were outlined using the polygon tool to define CA1, CA3, DG, and PRh in each image. Counts were normalized to each ROI area and reported as neurons per mm2 for each region. The granular cell layer of the dentate gyrus was omitted from counting due to difficulties in quantify densely packed neurons. All values for number of neuronal cells are reported as mean ± SEM (Table 3).
Western immunoblotting
NLRP3 and active caspase-1 protein expression in the hippocampus was quantified by Western immunoblotting (Supplementary Fig. S2). All groups of animals were divided evenly and sacrificed at 1 day or 30 days post-injury (n = 3/group). Following perfusion with PBS, the desired brain regions were excised and homogenized in radioimmunoprecipitation assay (RIPA) buffer (R0278; Sigma-Aldrich, St. Louis, MO) with Pierce protease inhibitor (A32963; Thermo Scientific, Waltham, MA) on ice using a sonicator. Lysates were centrifuged at 14,000 × g at 4°C, and the supernatant was reserved for further analysis. Protein concentration in the sample was estimated by bicinchoninic acid assay (Thermo Scientific).
A total of 20-30 μg of protein per lane were loaded into 4-20% SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) gradient gels (Bio-Rad Laboratories, Hercules, CA). Proteins separated according to molecular size were transferred onto polyvinylidene difluoride (PVDF) membranes using a Trans-Blot Turbo Transfer system (Bio-Rad). Membranes were cut in half at 60 kDa, blocked with 5% milk in TBS with 0.1% Tween-20, and incubated overnight at 4°C with rabbit anti-NLRP3 (1:500, NBP2-12446SS; Novus Biologicals) or rabbit anti-cleaved caspase-1 (p20; 1:500, PA5-99390; Invitrogen) followed by secondary antibodies. Bands were visualized using Western Pico Chemiluminescence Substrate (Thermo Scientific) on a ChemiDoc imaging system (Bio-Rad). PVDF membranes were stripped using Pierce Restore™ Western blot stripping buffer (21059; Thermo Scientific) and reprobed for β-actin (1:1000; PA1-46296, Invitrogen) as a housekeeping protein to normalize concentrations and identify any loading variations. For densitometric quantification of Western blots, images were digitized using a Bio-Rad GS800 calibrated densitometer and analyzed with Bio-Rad Quantity One software (version 5). 70 With a sample size of n = 3/group with power of 0.80, alpha = 0.05, mean standard deviation (SD) = 0.06 relative density units, two-tailed, our power analysis indicates that the minimum detectable change would be 0.19 relative density units.
Enzyme-linked immunosorbent assay
Pro-inflammatory cytokine IL-1β levels were estimated by enzyme-linked immunosorbent assay (ELISA). All groups of animals were divided evenly and sacrificed at 1 day or 30 days post-injury (n = 4/group). Following perfusion with PBS, hippocampal tissue was excised and homogenized in RIPA buffer (R0278; Sigma-Aldrich, St. Louis, MO) on ice using a sonicator and loaded onto ELISA plates (ab100768; Abcam). All steps of the ELISA protocol, including washes and incubation times, were conducted in accordance with manufacturer instructions. Plates were visualized and absorption was measured using SpectraMax i3 microplate reader (Molecular Devices, San Jose, CA) and analyzed using SoftMax Pro 6.5 software. With a sample size of n = 4/group with power of 0.80, alpha = 0.05, mean SD = 14 pg/mL, two-tailed, our power analysis indicates that the minimum detectable change would be 33 pg/mL.
Statistical analysis
Statistical analyses were performed using Prism 9 for macOS (version 9.4.1; GraphPad Software, San Diego, CA). Normal distribution of data was validated using the Shapiro-Wilk test. Homogeneity of variances was assessed using the Brown-Forsythe test. Behavioral data were analyzed using repeated measures one-way analysis of variance (ANOVA) with Tukey's multiple comparisons test. Immunohistochemistry, Western immunoblotting, and ELISA data were analyzed using one-way ANOVA with Tukey's multiple comparisons test or Welch's ANOVA with Dunnett's T3 multiple comparisons test for data sets with heteroscedasticity. Graphs and results are expressed as group means ± SEM.
Results
MCC950 mitigates rLLB-induced acute and chronic short-term memory deficits
To assess the effects of MCC950 administration on short-term object recognition memory after rLLB exposure, rats (n = 8/group) underwent a NOR task at 2 days and 31 days post-injury. Control uninjured rats had a stronger preference for the novel object than injured rats at both time-points. Preference index was significantly reduced in rLLB+PBS rats (x̄ = 48.3% ± 3.95) compared with controls (x̄ = 65.4% ± 5.56) at 2 days post-injury (p = 0.032) and in rLLB+PBS rats (x̄ = 38.7% ± 5.60) compared with controls (x̄ = 66.5% ± 5.57) at 31 days post-injury (p < 0.001). Lack of interest in a novel object as reflected by decreased exploration time is indicative of short-term memory deficits. rLLB+MCC950 rats showed preference for the novel object similar to that of uninjured controls at both time-points, suggesting that short-term memory deficits caused by rLLB exposure were mitigated by treatment (Fig. 2A). To investigate whether the decrease in novel object exploration in injured animals was due to impaired mobility, we examined total exploration time for both objects in the testing phase. We found that there was no difference in total exploration time between any groups at 2 days or 31 days post-injury, suggesting that the decrease in novel object exploration was solely due to recognition memory impairment (Fig. 2B).
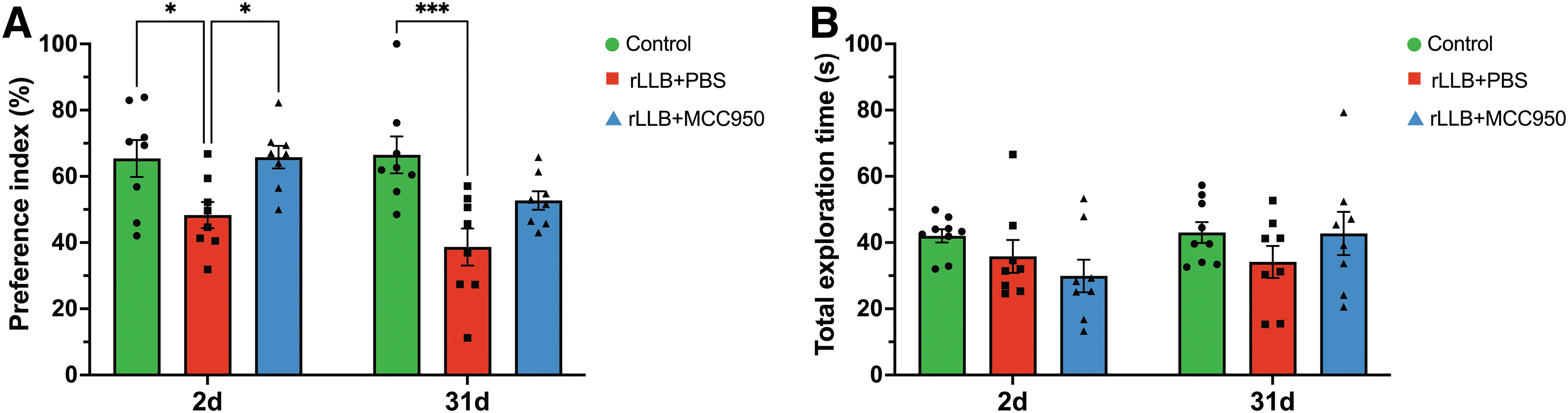
MCC950 improves repeated low-level blast (rLLB)-induced short-term object recognition memory deficits. Short-term recognition memory was assessed at 2 and 31 days post-injury by performance on a novel object recognition (NOR) task after a 4-h delay.
MCC950 decreases rLLB-induced chronic microglial activation
As surveillant cells of the CNS, microglia manifest different morphological characteristics which reflect their function relative to their environment. Activated microglia can be distinguished from resting microglia by their larger and oblong somas with shorter and thicker processes which enable phagocytosis of extracellular debris or apoptotic cells. 76 -78 To examine the effects of MCC950 on rLLB-induced microglial activation, we evaluated the total number of microglia and analyzed microglial morphology using immunohistochemistry based on the number of processes per cell (Table 1) and mean process length (Table 2) in the hippocampus and perirhinal cortex (PRh) at 1 day and 30 days post-injury (Fig. 3A, 3B; Supplementary Fig. S1). At 1 day, rLLB+PBS rats showed decreases in mean processes per cell (p < 0.001) and mean process length per cell (p < 0.001) in only the DG region compared with control, but these effects were not improved by treatment (Fig. 3C, 3D). At 30 days, rLLB+PBS rats showed a significant decrease in mean processes per cell in CA1 (p = 0.022), CA3 (p = 0.006), DG (p < 0.001), and PRh (p = 0.008) and mean process length per cell in CA1 (p = 0.044), CA3 (p = 0.003), DG (p < 0.001), and PRh (p = 0.024) compared with controls (Fig. 3E, 3F). MCC950 treatment increased mean processes per cell in CA1 (p = 0.037), CA3 (p = 0.022), DG (p < 0.001), and PRh (p = 0.002) and mean process length per cell in DG (p = 0.002) and PRh (p = 0.036) compared with rLLB+PBS rats at 30 days post-injury (Fig. 3E 3F). Further, rLLB+MCC950 rats were not different from controls in any region at 30 days post-injury. Together, these results indicate that microglial morphology is altered in a proinflammatory manner at both acute and chronic time-points following rLLB exposure and that treatment with MCC950 significantly prevented chronic microglial activation after rLLB exposure.
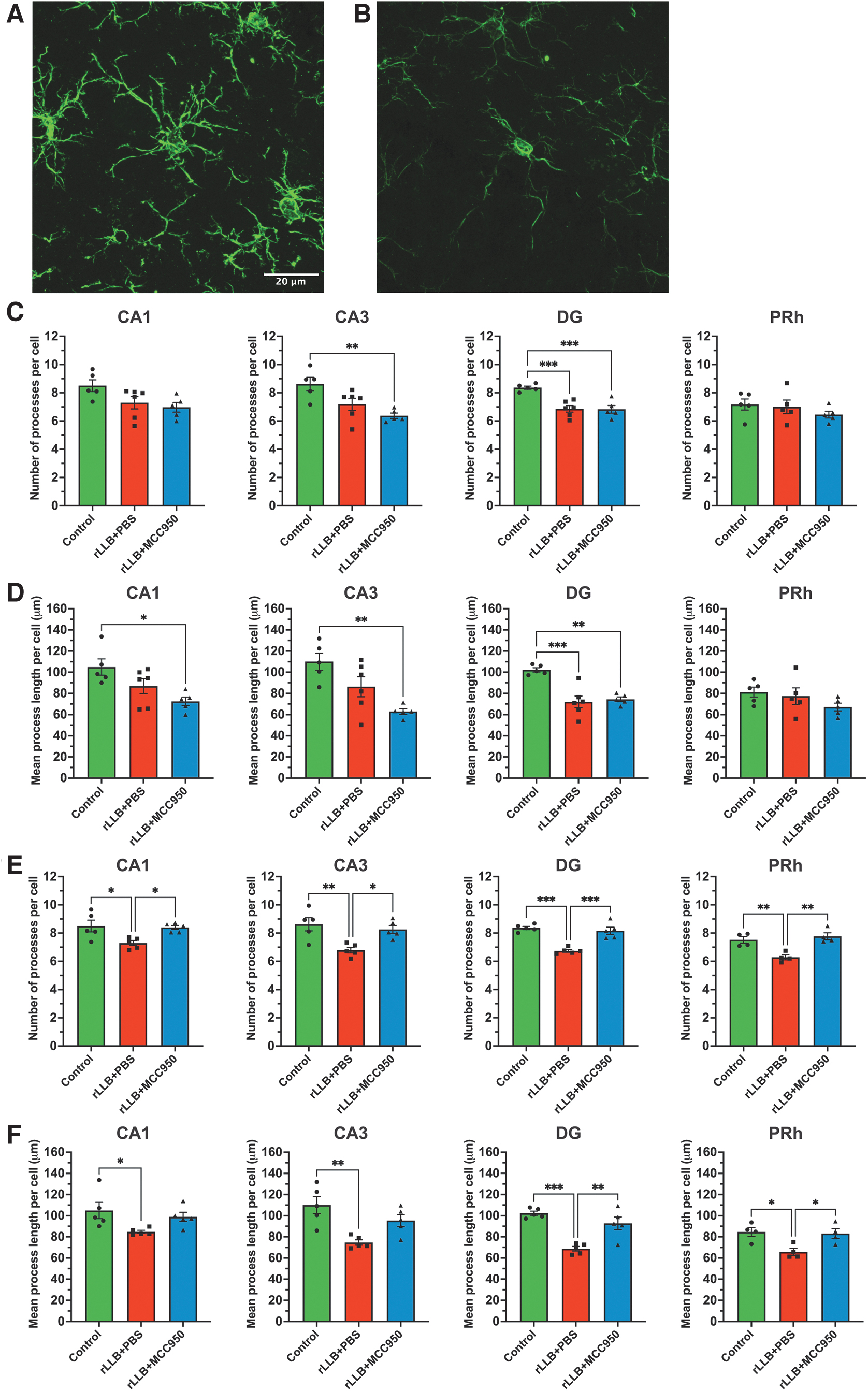
MCC950 suppresses repeated low-level blast (rLLB)-induced chronic microglial activation in the hippocampus and perirhinal cortex. Representative images of
MCC950 decreases total microglial counts after rLLB exposure
Based on the significant effects of MCC950 administration at our chronic time-point, we assessed the number of microglia present in the hippocampus and PRh at 30 days post-injury (Table 3). We observed a significant increase in the number of total microglia per mm2 in rLLB+PBS rats in CA1 (p = 0.028), CA3 (p = 0.007), and DG (p < 0.001) but not PRh compared with controls. However, MCC950 treatment had no effect on the total number of microglia in hippocampal subregions or PRh in injured rats (Fig. 4).
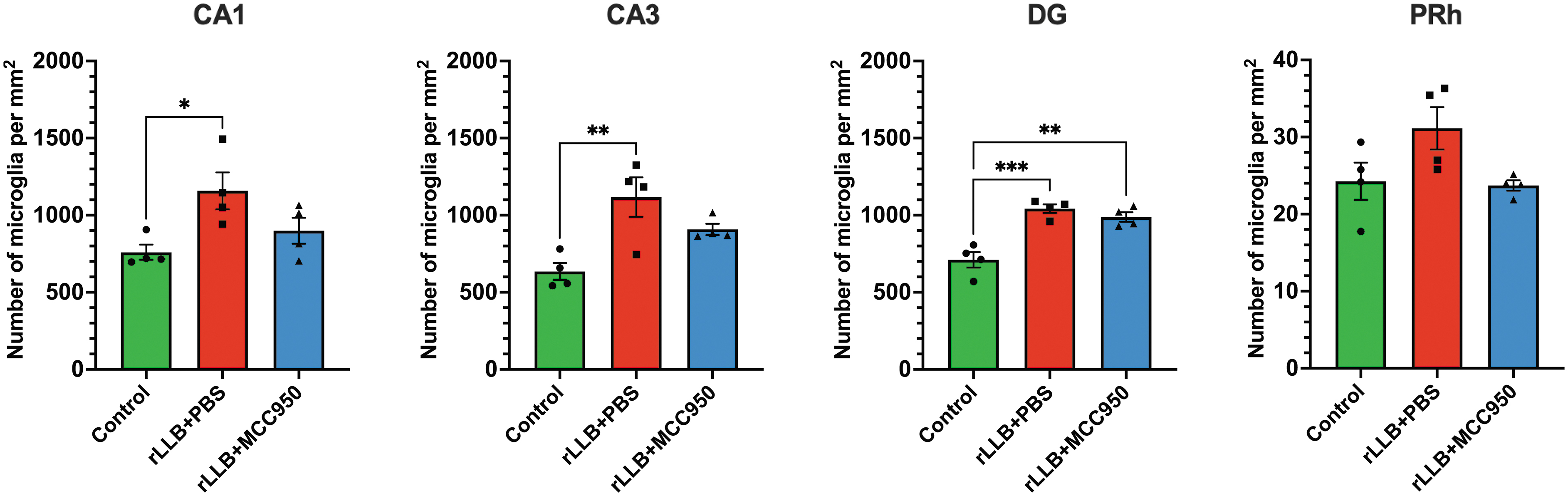
MCC950 suppresses repeated low-level blast (rLLB)-induced enhanced presence of microglia in the hippocampus and perirhinal cortex (PRh). Total number of microglia increased in rLLB rats in CA1, CA3, dentate gyrus (DG), and PRh at 30 days post-injury but did not decrease with treatment. Graphs depict mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001).
MCC950 decreases rLLB-induced acute and chronic NLRP3 protein expression
Due to changes in microglial activation in the hippocampus, we quantified NLRP3 protein expression in isolated hippocampal lysates at 1 day and 30 days post-injury using Western immunoblotting (Supplementary Fig. S2). rLLB+PBS rats showed a significant increase in NLRP3 protein expression as early as 1 day post-injury (x̄control = 0.0417 ± 0.0056, x̄rLLB+PBS = 0.112 ± 0.0079, p = 0.006) that persisted until 30 days post-injury (x̄rLLB+PBS = 0.096 ± 0.0042, p < 0.001) compared with controls. MCC950 treatment significantly decreased NLRP3 expression in rLLB rats at 1 day (x̄rLLB+MCC950 = 0.0586 ± 0.0141, p = 0.021) and 30 days (x̄rLLB+MCC950 = 0.0725 ± 0.00586, p = 0.047) post-injury compared with untreated animals, although NLRP3 expression in rLLB+MCC950 rats was different from control at 30 days (p = 0.014; Fig. 5). These results indicate that NLRP3 expression increases in response to rLLB exposure and remains elevated 1 month after injury, likely contributing to chronic neuroinflammation, and that MCC950 is effective at reducing NLRP3 expression with a stronger acute effect.
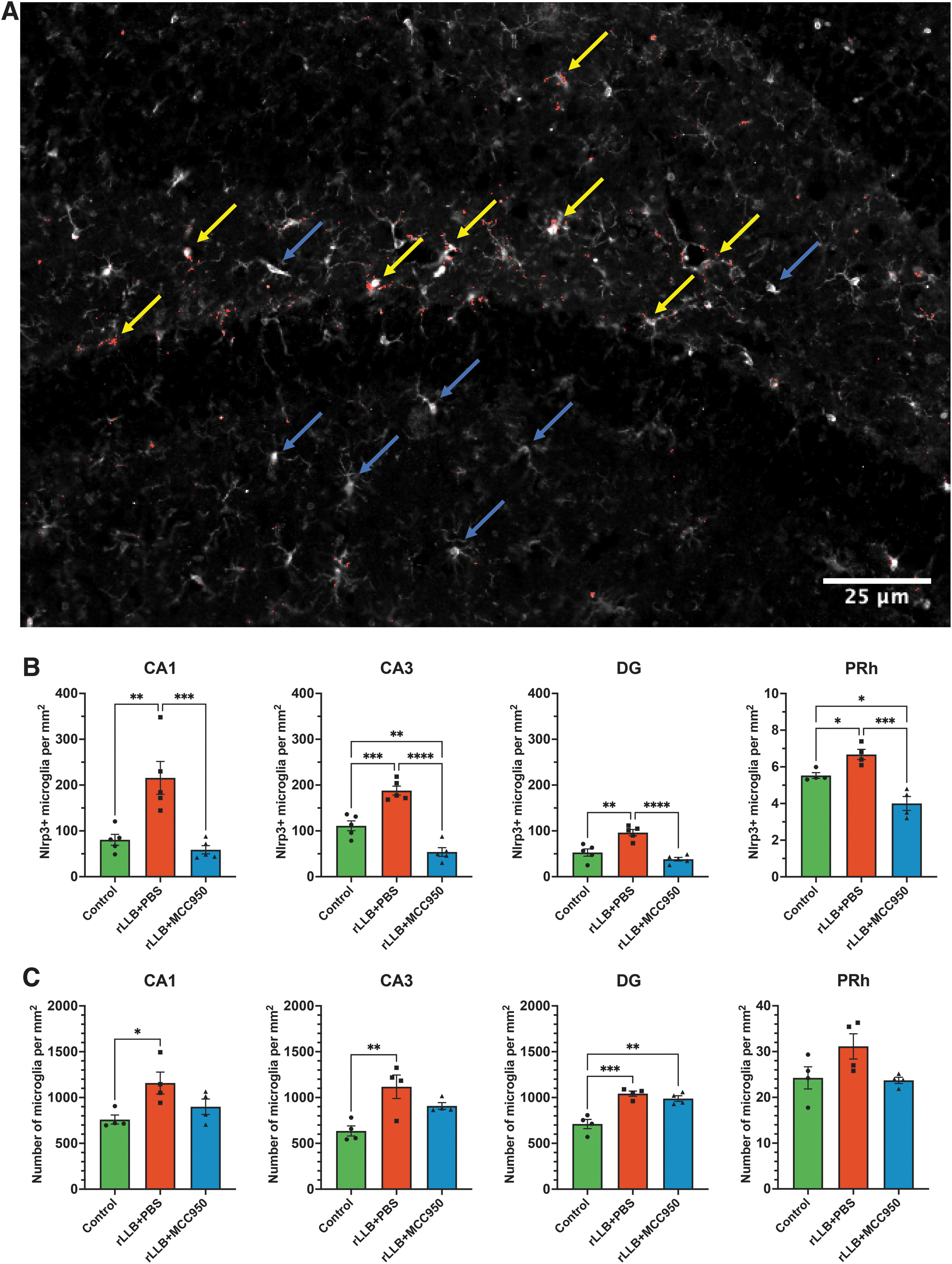
MCC950 decreases repeated low-level blast (rLLB)-induced acute and chronic NLRP3 protein expression in the hippocampus. NLRP3 expression was increased in rLLB rats but decreased with treatment at 1 day and 30 days post-injury. Graphs depict mean ± standard error of the mean (*p < 0.05, **p < 0.01, ***p < 0.001). Yellow arrows represents NLRP3 + microglia whereas blue arrows represents NLRP3-microglia.
MCC950 decreases rLLB-induced microglial NLRP3 expression
We performed immunohistochemistry to examine whether rLLB exposure induces chronic NLRP3 expression in a greater population of microglia. Sections were stained with Iba1 and NLRP3 to quantify NLRP3+ microglia in the hippocampus and PRh (Fig. 6A). At 30 days post-injury, we observed a significant increase in the number of NLRP3+ microglia in CA1 (p = 0.003), CA3 (p < 0.001), DG (p = 0.001), and PRh (p = 0.048) in rLLB+PBS rats compared with control. rLLB+MCC950 rats showed a significant decrease in the number of NLRP3+ microglia in CA1 (p < 0.001), CA3 (p < 0.001), DG (p < 0.001), and PRh (p < 0.001) compared with rLLB+PBS rats; however, the number of NRLP3+ microglia in MCC950-treated rats was also lower than control in CA3 (p = 0.004) and PRh (p = 0.011; Fig 6B). Thus, although NLRP3 was chronically expressed in a larger population of microglia in both the hippocampus and PRh, MCC950 was most effective at reducing microglial NLRP3 expression in DG and CA1 compared with other subregions.
MCC950 suppresses repeated low-level blast (rLLB)-induced chronic microglial NLRP3 expression and enhanced presence of microglia in the hippocampus and perirhinal cortex (PRh).
MCC950 decreases rLLB-induced acute active caspase-1 expression
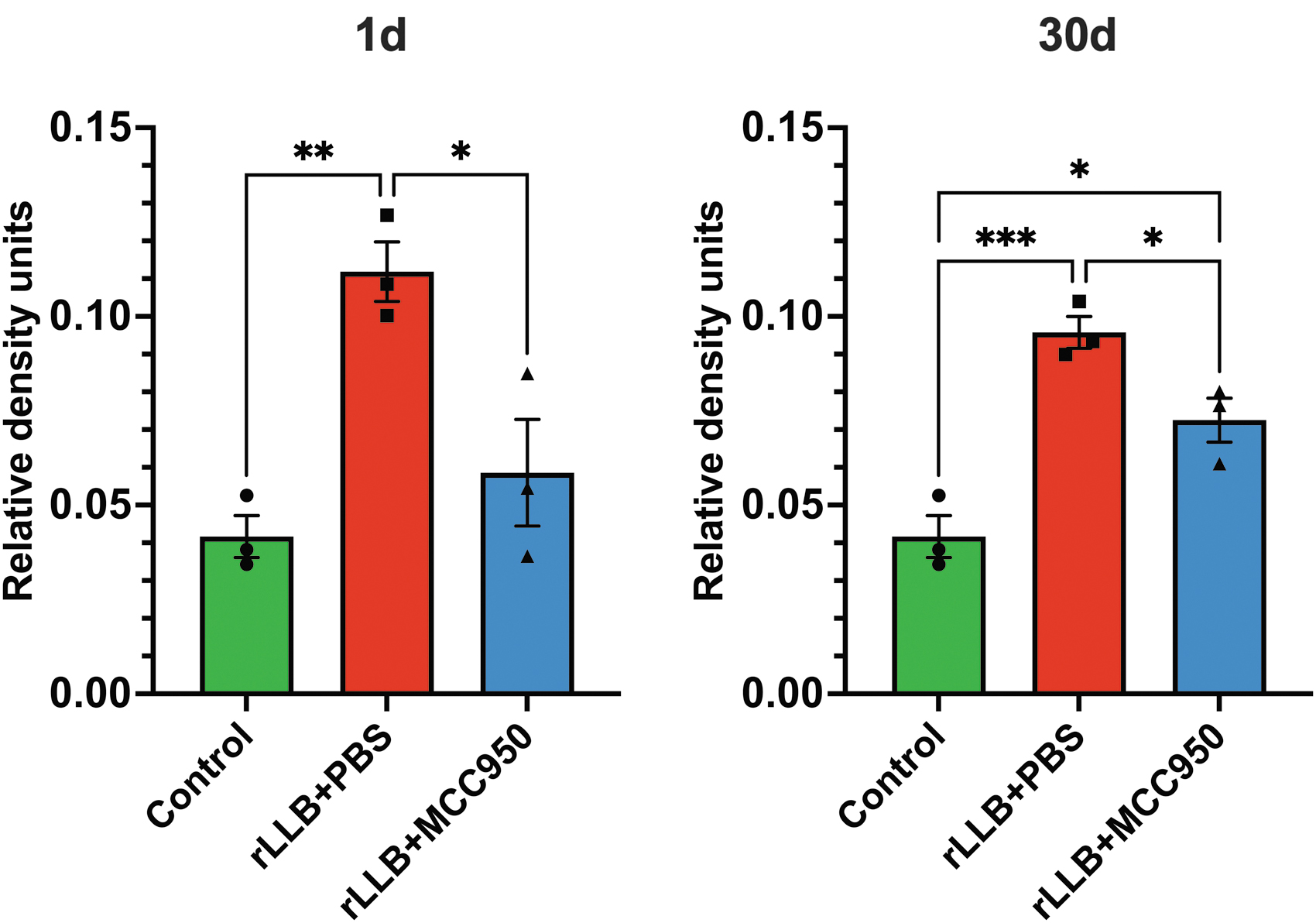
We measured expression of active caspase-1 (p20), a direct cleavage product of the activated NLRP3 inflammasome, in the rat hippocampus using Western immunoblotting (Supplementary Fig. S2). rLLB+PBS rats showed a significant increase in cleaved caspase-1 expression as early as 1 day post-injury (x̄control = 0.385 ± 0.0335, x̄rLLB+PBS = 0.778 ± 0.0179, p < 0.001), which remained greater than control at 30 days but did not reach significance (x̄control = 0.385 ± 0.0335, x̄rLLB+PBS = 0.492 ± 0.00837, p = 0.063). MCC950 treatment reduced cleaved caspase-1 expression at 1 day but did not reach significance compared with rLLB+PBS rats (x̄rLLB+MCC950 = 0.623 ± 0.0557, p = 0.069) and had no effect at 30 days post-injury (Fig. 7).
MCC950 decreases repeated low-level blast (rLLB)-induced acute active caspase-1 enzyme expression in the hippocampus. Cleaved caspase-1 (p20) expression increased in rLLB rats at 1 day post-injury but did not significantly reduce with treatment. Cleaved caspase-1 expression did not differ from control in rLLB+phosphate-buffered saline (PBS) or rLLB+MCC950 rats at 30 days post-injury. Graphs depict mean ± standard error of the mean (*p < 0.05, **p < 0.01, ***p < 0.001).
MCC950 decreases rLLB-induced chronic pro-inflammatory cytokine IL-1β release
We assessed pro-inflammatory cytokine IL-1β release in whole hippocampal tissue using ELISA. rLLB exposure induced an increase in IL-1β release as early as 1 day (x̄control = 221.4 ± 8.03, x̄rLLB+PBS = 277.8 ± 4.87, p < 0.001) that persisted even after 30 days (x̄control = 221.4 ± 8.03, x̄rLLB+PBS = 321.8 ± 17.35, p < 0.001) post-injury compared with controls (Fig. 8). MCC950 treatment significantly decreased IL-1β release at 1 day (x̄rLLB+MCC950 = 229.4 ± 8.00, p = 0.002) and 30 days post-injury (x̄rLLB+MCC950 = 258.4 ± 5.28, p = 0.009) compared with rLLB+PBS rats. Hippocampal IL-1β levels in MCC950-treated rats were not different from control at either time-point.
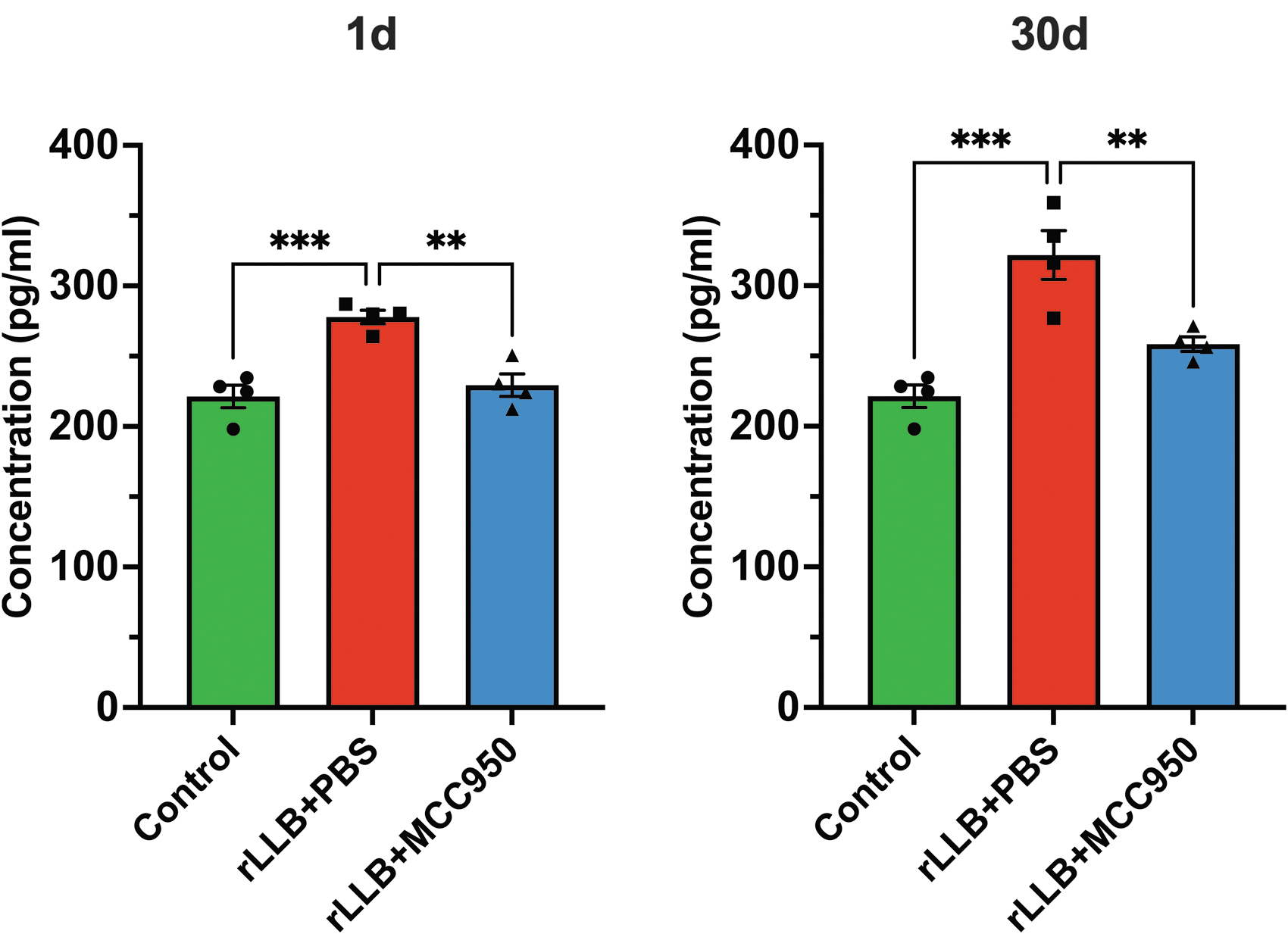
MCC950 suppresses repeated low-level blast (rLLB)-induced chronic pro-inflammatory cytokine interleukin (IL)-1β release in the hippocampus. rLLB exposure increased IL-1β release in the rat hippocampus at 1 day and 30 days post-injury. MCC950 significantly decreased IL-1β release in rLLB rats at 1 day and 30 days post-injury. Graphs depict mean ± standard error of the mean (*p < 0.05, **p < 0.01, ***p < 0.001).
MCC950 ameliorates rLLB-induced acute and chronic neuronal loss
To examine whether rLLB exposure results in neuronal loss, we quantified NeuN+ cells using immunohistochemistry (Fig. 9A). rLLB+PBS rats showed a significant decrease in NeuN+ cells in CA3 (p = 0.040) and DG (p = 0.003) at 1 day post-blast and in CA3 (p < 0.001) and DG (p < 0.001) at 30 days post-blast compared with controls (Table 4; Fig. 9B, 9C). MCC950 significantly reduced neuronal loss in DG (p = 0.005) at 1 day post-blast and in CA1 (p = 0.009), CA3 (p < 0.001), and DG (p = 0.006), at 30 days post-blast in rLLB rats; however, neuron counts were still lower than control at 30 days in CA3 (p = 0.004) and DG (p < 0.001; Table 4; Fig. 9B, 9C). There was no difference in the number of NeuN+ cells in PRh between sham and blast animals at either time-point and no effect of treatment. Together, these results indicate that neuronal loss was most significant in CA3 and DG and was somewhat reduced in the hippocampus by treatment with MCC950.
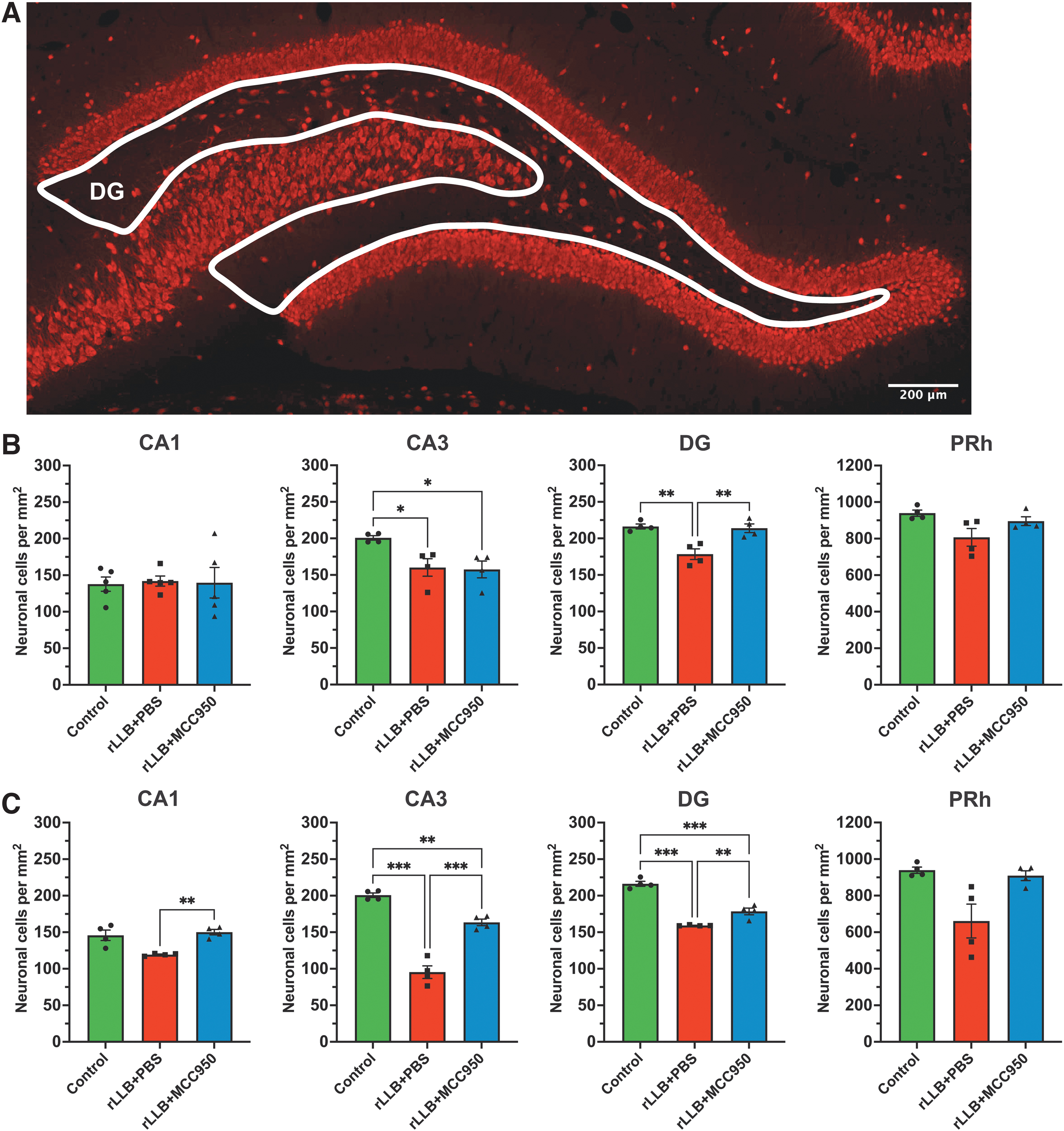
MCC950 ameliorates repeated low-level blast (rLLB)-induced acute and chronic neuronal loss in the hippocampus and perirhinal cortex.
Mean Number of Neuronal Cells by Area (per mm2) at 1 Day and 30 Days Post-Injury
PBS, phosphate-buffered saline; rLLB, repeated low-level blast; DG, dentate gyrus; PRh, perirhinal cortex.
Discussion
Recent studies have demonstrated the involvement of the NLRP3 inflammasome in chronic neuroinflammatory response following a TBI event. 24 -26,28,29 Previously, we reported that our model of rLLB with five consecutive blast exposures at 1-min intervals induced acute and chronic short-term memory deficits, anxiety-like behavior, and motor impairments. 8 Studies have shown that the hippocampus is highly susceptible to damage at a wide range of BOPs and that this vulnerability is increased with repetition. 79 Further, short-term recognition memory is impacted by exposure over a wider range of BOPs than spatial memory. 80 The current study explored the role of the NLRP3 inflammasome in rLLB and provided evidence that administration of a selective NLRP3 inhibitor, MCC950, can reduce rLLB-induced chronic neuroinflammation and restore short-term memory impairments in our rat model.
This study was a continuation of previously published work using the same rLLB model of five blast exposures with a peak overpressure of 70 kPa at 1-min intervals. 8 The use of five exposures was based on a field study of low-level blast overpressures in service members during multiday training sessions with 0.50-caliber rifles, which found that most subjects experienced 4-27 exposures per day with one to eight rounds fired within a 1-min period. 20 Ideally, we would be able to recreate this frequency using our shock tube model, but practical limitations, primarily with regards to replacing membranes in the blast tube between exposures within a short period of time, made it extremely difficult to do more than five successive exposures at 1-min intervals.
Several other studies investigating repetitive blast-induced in rodent models have used peak blast overpressures of 1-90 kPa at longer intervals, with the most common paradigm being 70 kPa blasts every 24 h. 10,21 Although a peak BOP of 70 kPa is on the higher end for subclinical exposures, we believe that the primary factor that differentiates rLLB-induced TBI from other blast-induced neurotrauma is the cumulative overpressure within a short time frame, and that the use of heavy weaponry is more accurately modeled by exposure to more low-level blasts at shorter intervals. The total cumulative peak pressure reported by Skotak and colleagues varied from 50-350 psi over the course of a 3-day training period, and our model was on the lower end of this spectrum (5 × 10 psi/70 kPa, or approximately 50 psi total). Additionally, we have confirmed in previous studies that one 70 kPa exposure is not sufficient to induce TBI pathology. 8 We concluded that 70 kPa was the maximum BOP that should be used to deliver a subclinical exposure while still representing the cumulative damage observed in occupational settings when limited to five blasts.
Using our rat rLLB model, we found that MCC950 treatment improved rLLB-induced short-term memory impairment at 2 days with a trend at 31 days post-injury. Neurocognitive outcomes have improved following treatment with several drugs targeting the NLRP3 inflammasome in other rodent models of TBI. MCC950 itself has been shown to improve neurological severity score at 1 day and 3 days post-TBI in rat and mouse injury models. 81 A study by Zheng and colleagues examined the effect of dexmedetomidine (Dex), an α2-selective adrenergic receptor agonist, on NLRP3 inflammasome expression after TBI and found that Dex administration improved spatial learning and memory. This effect was further enhanced when Dex was co-administered with the NLRP3 inhibitor BAY-11-7082. 2 Additionally, Lin and colleagues found a neuroprotective role of omega-3 fatty acid against NLRP3-mediated chronic neuropathological changes which improved memory following TBI. 82 Improvement in object recognition memory after MCC950 administration as reported in this study suggests that the NLRP3 inflammasome is an appropriate target for improvement of cognitive outcomes following rLLB as well as other forms of TBI. Although this effect did not reach significance at 31 days post-injury, other factors including oxidative stress and blood–brain barrier rupture can contribute to cognitive impairments following TBI, so treatments targeting the inflammasome would likely need to be co-administered with additional therapeutics to address related mechanisms at chronic time-points.
Activation of the NLRP3 inflammasome followed by caspase-1 cleavage and IL-1β release are key components in a neuroinflammatory mechanism that is more prominent in microglia compared with other cell types in the CNS. 59 We evaluated microglial activation based on process length and number of processes per cell and assessed the number of microglia expressing NLRP3 in the soma after injury and/or treatment. We found that rLLB-induced microglial activation was consistently reduced at chronic time-points following MCC950 administration in the hippocampus (CA1, CA3, and DG) and PRh. In contrast, acute microglial activation occurred predominantly within the DG and did not immediately respond to treatment. Additionally, we observed a significant increase in chronic NLRP3 expression following rLLB which was decreased by treatment with MCC950. Similarly, MCC950 has been shown to suppress microglial activation and decrease pro-inflammatory cytokine release, BBB permeability, and brain water content at 3 days post-injury in mice after cortical controlled impact (CCI). 59 Further, the same group evaluated the role of microglia-specific NLRP3 activation in chronic neuroinflammation and behavioral deficits by depleting microglial cells and did not observe MCC950-mediated neuroprotection, demonstrating that microglial NLRP3 plays a vital role in long-term neurological changes after TBI. 59
Our study also supports these findings, as we observed chronic microglial activation, increased NLRP3 expression, and colocalization of NLRP3+ microglia after rLLB exposure accompanied by short-term memory impairments. The number of NLRP3+ microglia was reduced by treatment with MCC950 at 1 day post-injury in CA1, CA3, DG, and PRh but was not significantly reduced at 30 days post-injury in any subregion. However, since we observed downward trends in NLRP3+ microglia at 30 days following treatment, we hypothesize that optimization of MCC950 dosage including more frequent administration (i.e., every 24 h rather than 48 h) may yield more promising results in future studies. Similarly, treatment with Dex has been shown to suppress microglial activation which was further enhanced upon co-administration with the NLRP3 inhibitor BAY-11-7082.2 We found that MCC950 administration had no impact on the number of microglia present in the hippocampus and PRh, suggesting that microglial proliferation or recruitment is triggered by something other than IL-1β release.
When examining the effects of treatment on proteins and cytokines involved in NLRP3-mediated neuroinflammation, we found that MCC950 decreased NLRP3 and cleaved caspase-1 expression and reduced IL-1β release in the hippocampus and PRh at 1 day and 30 days post-blast. Previously, Ismael and colleagues reported that administration of MCC950 mitigated TBI-induced NLRP3 and caspase-1 expression and IL-1β release at 1 day and 3 days post-injury and simultaneously decreased brain water content and neurological severity score in mice. 60 In rats, Zhao and colleagues found that MCC950 decreased apoptotic cell death, brain water content, NLRP3 expression, caspase-1 enzyme levels, and IL-1β release at 1 day and 3 days after TBI. 81 Dex administration decreased NLRP3 and caspase-1 expression, reduced IL-1β release, and increased neuronal viability in a rat model of CCI; these effects were also enhanced by co-administration of BAY-11-7082. Several other studies have also demonstrated NLRP3 inhibition-mediated neuroprotection following TBI. 83 –89 Although MCC950 reduced neuroinflammation following rLLB exposure by suppressing microglial activation and IL-1β release, this was not sufficient to provide full neuroprotection against factors contributing to short-term memory impairment.
Neurocognitive impairment after rLLB exposure could result from neuronal loss that can occur through several signaling pathways (i.e., apoptosis, necrosis, pyroptosis, etc.) and may be downstream of IL-1β mediated neuroinflammation. Single mild blast exposure at 130 kPa has been reported to result in reduced hippocampal neuron density. 90,91 In the present study, MCC950 suppressed IL-1β levels and reduced neuronal loss in CA3 and DG at 1 day post-injury and in CA1, CA3, and DG at 30 days post-injury, suggesting that NLRP3-mediated neuroinflammation and IL-1β release likely contributes in part to neuronal loss in the hippocampus. Recent studies have shown that the level of neuronal loss induced by blast exposure varies in different subregions of hippocampus. 92 Thus, the various mechanisms behind blast-induced neurodegeneration across hippocampal subregions remain unclear.
Based on our results, we postulate that microglia activation and IL-1β release may be the primary mechanism that underlies neuronal loss in CA3 region, which would be reflected by increased efficacy of treatment with MCC950 at preventing neurodegeneration. In contrast, neuronal loss in the DG could be due to both microglial activation and non-microglial mechanisms such as oxidative stress or apoptosis, in which case the treatment would not have been as effective at slowing or preventing overall neuronal loss due to injury. Since MCC950 treatment was beneficial across multiple subregions at 30 days, we hypothesize that IL-1β release is majorly contributing to chronic hippocampal neuronal loss overall following rLLB exposure, although this mechanism may impact specific neuronal populations to various degrees. Increased IL-1β release can lead to neuronal loss via glutamate excitotoxicity (overproduction by glutaminase-1), 93 caspase-3-mediated apoptosis, 94 -96 free radical generation by NADPH oxidases leading to lipid peroxidation, DNA oxidation, and protein nitration, 97 activation of NLRP1 in neurons to cause pyroptosis, 98 -100 and microglia-mediated neurotoxicity. 101 Besides neuronal loss, chronic microglial activation or elevated IL-1β levels alone have been shown to cause memory deficits. 7,34,102,103 Mechanisms of cell death that contribute to hippocampal neuronal loss as well as other possible downstream effects of increased IL-1β release need to be further investigated in the context of rLLB.
In this study, MCC950 was used primarily for proof of principle to demonstrate the role of the NLRP3 inflammasome in long-term sequelae of rLLB exposure. MCC950 is highly specific and does not interact with other known inflammasomes or TLR-driven pathways. MCC950 was previously tested in a phase II clinical trial for rheumatoid arthritis but was withdrawn due to elevated serum liver enzyme levels, which have prevented it from being implemented in the clinic; however, several other NLRP3 inhibitors are currently in the development pipeline. 42 Therefore, our findings also have translation relevance because we have established NLRP3 as a viable therapeutic target in the treatment of rLLB-induced TBI. Future studies could investigate whether TBI pathology can be mitigated by using other therapeutics which target the NLRP3-mediated neuroinflammatory pathway and whether prophylactic administration of NLRP3 inhibitors prior to rLLB exposure can improve functional outcomes in service members and law enforcement personnel who are at higher risk due to occupational exposures.
The physical mechanism by which a blast wave causes damage to the brain is still controversial. Like other forms of TBI, bTBI consists of primary injury caused by exposure to the blast wave and secondary injury which develops over time. 104 Microglial activation may be triggered by several different signals including the shock wave itself, free radical generation via NADPH oxidases (NOX), BBB damage, or DAMPs, and further activation may result from IL-1β release due to NLRP3 activation within microglia. 10,105 -107 In our previous study, we reported an increase in acute and chronic NOX1 expression after rLLB, which suggest that increased generation of reactive oxygen species in the extracellular likely contributes to microglial activation. 8 Here, we observed NLRP3 and IL-1β involvement in microglial activation which was suppressed by MCC950 treatment. Additionally, BBB dysfunction may also be involved in microglial activation 108,109 : in our previous studies on single moderate blast, we reported BBB dysfunction as early as 15 min to 24 h post-injury. 57 BBB dysfunction has also been reported after exposure to two moderate blasts at a 15-min interval 110 and three mild blasts at 30-min intervals. 111
In our study, we did not examine BBB dysfunction following rLLB, but we presume that our model could cause BBB compromise, which can lead to microglial activation through infiltration of DAMPs. 112 -114 Alternatively, acute microglial activation could be a direct result of physical manipulation of the cell membrane due to primary injury. One possible mechanism by which mechanical force from repeated low-level shock waves could induce microglial activation is via PIEZO1, a mechanotransduction ion channel expressed in neurons, astrocytes, and microglia which has been shown to activate the NLRP3 inflammasome in a Ca 2 ⁺/NF-κB-dependent manner in response to mechanical stress in other cell types. 115 –119 We speculate that repeated low-level shock waves could cause pressure changes within the brain that might be detected by PIEZO1 channels on microglia and cause acute activation leading to oxidative stress, BBB dysfunction, and cytokine release, which could in turn induce chronic microglial activation. Although PIEZO1 has not yet been evaluated in the context of TBI to our knowledge, studies have shown altered levels of PIEZO2 in the rat cerebellum following rLLB exposure. 10,120
Conclusions
Although there are a small number of studies referenced above using MCC950 as a treatment in rodent models of TBI, these studies only examined the therapeutic efficacy of MCC950 at acute time-points and did not evaluate its use in treatment of bTBI. Here, the effects of MCC950 were also assessed at chronic time-points (30 days post-blast) in our rat model of rLLB. We found that MCC950 treatment ameliorated short-term memory deficits and suppressed NLRP3 expression and microglial activation at both acute and chronic time-points. Hence, rLLB-induced chronic behavioral deficits in tasks associated with the hippocampus could be mediated through the NLRP3 inflammasome pathway.
This study did not investigate upstream mechanisms of NLRP3 priming and activation in rLLB. Several pathways have been demonstrated to trigger NLRP3 activation within microglia, including the binding of damage-associated molecular pathogens (DAMPs) to TLR4, which can induce transcription of NF-κB following TBI. 121 Further investigation into the mechanism by which microglia react to repeated shock wave exposures is necessary, as these pressures are lower in intensity than those employed to model single blast exposure. In both our previous and current studies on rLLB, we observed an increase in the total number of microglia in the hippocampus, but it remains unclear whether this increase represents proliferation or recruitment from other brain region. Additionally, this study did not evaluate sex differences in response to rLLB exposure, although a previous study on single blast exposure by members of our group did not observe sex effects following bTBI. 7 In summary, this study showed that repeated low-level blast exposure can induce chronic progressive neuropathological changes mediated in part by activation of the NLRP3 inflammasome in microglia.
Transparency, Rigor, and Reproducibility Summary
The study design and analytical plan were not formally pre-registered. For novel object recognition, sample size was eight animals per group based on power analysis to obtain >80% power. For immunohistochemistry, Western blotting, and ELISA, sample size was three to five animals per group based on previous studies. A total of 48 rats were randomly assigned to groups using a random number generator; 48 rats were subjected to experimental injury and two died after blast exposure due to apnea. A total of 46 rats received the allocated therapeutic, and one died while performing i.p. injection. Complete data was obtained for 45 animals. MCC950 administration occurred 30 min after injury and then once every 48 h for the remainder of the study. Dosing regimen was based on the optimal dosage of MCC950 for rodents available in the literature. MCC950 is commercially available and was obtained from the same lot number directly from the manufacturer. All reagents and antibodies used in this study are commercially available. Investigators were blind to groups for cell counting, and glial morphology was analyzed by automated processing to reduce investigator bias. Normal distribution of primary behavioral and molecular data was validated using the Shapiro-Wilk test, and homogeneity of variances was assessed using the Brown-Forsythe test. Data sets used and analyzed in the current study are available from the corresponding author upon reasonable request.
Footnotes
Acknowledgments
Thank you to Tulika Das, Aakash Gosain, and Thomas Dolalas for their assistance with manual counting of fluorescence images. Thank you to Anusha Reddy, Rama Devi, Mahendar Reddy, Seth Rosen, and family members for their support.
Authors' Contributions
Arun Reddy Ravula formulated the hypothesis, designed and performed biochemical and behavioral experiments, collected data, prepared figures, and equally contributed to manuscript preparation. Kathleen E. Murray performed confocal microscopy, skeleton analysis, and statistical analyses, prepared figures, and equally contributed to manuscript preparation. Kakulavarapu V. Rama Rao, Bryan J. Pfister, Bruce A. Citron, and Namas Chandra provided input during the course of the study. All authors read and approved the final manuscript.
Funding Information
This work was carried out with funding provided by grant no. 14059001, W81XWH-15-1-0303, titled “Primary Blast Injury Criteria for Animal/Human TBI Models using Field Validated Shock Tubes” (NC) and Grant 12744758 (RH180040) from Congressionally Directed Medical Research Programs (CDMRP; NC co-PI), the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development I01BX005015 (BAC), a VA Research Career Scientist award IK6BX006188 (BAC), and the Veterans Bio-Medical Research Institute.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.