
Abstract
Abstract
Peripheral blood mononuclear cells (PBMCs) offer a significant promise for gene expression analyses as a substitute for tissues that are not easily accessible. The objective of this study was to validate the use of PBMCs for gene expression analysis as a marker of nutritional intervention as an alternative to skeletal muscle tissue (SMT) biopsies. We performed a transcriptome comparison of PBMCs versus SMT after an 8-week supplementation with n−3 polyunsaturated fatty acid (PUFA) in 16 obese and insulin-resistant subjects. Expression levels of 48,803 transcripts were assessed by the Human-6 v3 Expression BeadChips (Illumina, San Diego, CA). In SMT, 36,738 (75%) transcripts were detected, whereas 34,182 (70%) transcripts were detected in PBMCs. Further, 88% (32,341) of these transcripts were coexpressed in both tissues. Importantly, a strong correlation (r = 0.84, p < 0.0001) was observed between transcript expression levels of PBMCs and SMT after n−3 PUFA supplementation. In conclusion, PBMCs express the majority of transcripts expressed in SMT subsequent to n−3 PUFA supplementation and their expression levels are comparable. In the interest of practicalities and cost, these results support the use of PBMCs as a surrogate model for SMT gene expression in nutrigenomic studies. Further research on PBMC and SMT gene expression in response to other nutritional exposures is warranted.
Introduction
Recent studies have demonstrated the potential of using gene expression levels of PBMCs as a novel biomarker of nutritional interventions. Bouwens et al. (2009) showed that the intake of n−3 polyunsaturated fatty acids (PUFA), including eicosapentanoic acid (EPA) and docosahexaenoic acid (DHA), altered the gene expression profiles of PBMCs in the direction of a more anti-inflammatory and anti-atherogenic profile. Other studies have also used PBMC to predict weight loss regain in obese subjects (Crujeiras et al., 2008; Goyenechea et al., 2009), and to predict the inflammatory response to functional foods (Theuwissen et al., 2009). Taken together, PBMC profiling may indeed adequately reflect gene expression levels of metabolic changes due to short-term and long-term nutritional adaptations.
It is noteworthy that even though PBMCs are currently being used in nutrigenomic studies, no study has yet validated the use of PBMCs as a surrogate model for SMT after a nutritional intervention. Thus, the objective of this study was to validate the use of PBMCs for gene expression analysis in response to an n−3 PUFA supplementation as an alternative to SMT.
Materials and Methods
Subjects
Twenty-one males, as well as pre- and postmenopausal females participants aged 35–70 years were recruited in the Quebec City greater area. Participants had a body mass index (BMI) over 25 kg/m2, and had a fasting plasma insulin >90 pmol/L including a fasting plasma glucose <7.0 mmol/L and 2-h plasma glucose <11.1 mmol/L. Subjects were excluded from the study if they had: weight change ±10% within the 6 months prior to study onset, a major surgery in the 3 months prior to study onset, diabetes, familial or primary hyperlipidemia, hepatic or metabolic diseases, smoking, chronic hypertension (>160/100 mmHg), incompatibility with fish consumption (allergy, intolerance, or dislike) and medications known to affect lipid and glucose metabolism. To reduce metabolic variations, the tests were performed in the follicular phase for premenopausal women. Fasting blood samples were collected for plasma biochemistry and hematology. The Clinical Research Ethical Committee of Laval University Hospital Center and the Food Directorate of Health Canada approved the experimental protocol. Prior to the study entry, participants received a complete description of the protocol and their written informed consent was obtained.
Experimental design and diets
A clinical trial was carried out at the Institute of Nutraceuticals and Functional Foods (INAF) of Laval University. Individual dietary instructions were given by a trained dietician to achieve the National Cholesterol Education Program (NCEP) Step 1 diet guidelines (2001). Subjects were asked to follow these dietary recommendations and maintain their body weight stable throughout the protocol. After the 4-week run-in, each participant consumed the n−3 PUFA supplementation for 8 weeks. Each subject received a bottle containing all needed fish oil capsules for the 8 weeks, which provided the equivalent of a total of 1.8 g of EPA and DHA daily. Compliance was assessed from the return of capsules of n−3 PUFA. Subjects were asked to report any deviation during the protocol. In addition, food frequency questionnaires were assessed by a registered dietitian before the run-in period as well as before and after supplementation.
Fatty acids analysis
Fatty acid (FAs) analyses were performed by Lipid Analytical Laboratories (University of Guelph Research Park, Guelph, Canada). Briefly, lipids were extracted from red blood cells using a modified Folch extraction procedure (Folch et al., 1957). The n−3 FAs concentration in erythrocytes were measured following transmethylation of the extracted lipid based on previous work (Stark and Holub 2004) using a gas chromatograph (Varian 3800 gas-liquid chromatograph, Palo Alto, CA) and 60-m DB-23 capillary column (0.32 mm internal diameter) with nitrogen as the carrier gas. Erythrocyte FA profiles were expressed as wt. % for individual fatty acids based on the relative percentage areas of total FAs.
Muscle biopsy
The muscle biopsy was performed after the 8-week supplementation. Biopsies (approximately 50 mg) were obtained from the vastus lateralis muscle under local anesthesia using a needle with suction applied. Total RNA was extracted using the RNeasy Fibrous Tissue Mini Kit (Qiagen, Mississauga, ON, Canada) according to the manufacturer's instructions and stored at −80°C. After spectrophotometric quantification and verification of the total RNA quality via the Agilent 2100 Bioanalyser (Agilent Technologies, Palo Alto, CA), samples were sent for microarray analysis. Samples were excluded from additional analysis on microarray chips if they had poor RNA quality (RIN < 8).
PBMCs
Blood samples were collected into an 8-mL Cell Preparation Tube (CPT) (Becton Dickinson, Oakville, On, Canada) post- n−3 PUFA supplementation. PBMCs were separated by centrifugation (1500 × g, 20 min, at room temperature) and washed according to the manufacturer's instructions. Total RNA were extracted with RNeasy Plus Mini Kit (Qiagen, Mississauga, On, Canada) according to manufacturer's protocol. After spectrophotometric quantification and verification of the total RNA quality via the Agilent 2100 Bioanalyser (Agilent Technologies), samples were sent for microarray analysis. Samples were excluded from further analysis on microarray chips if they had poor RNA quality (RIN < 8).
Transcriptomic profiling
A total of 200 ng of total RNA was amplified and labeled using the Illumina TotalPrep RNA Amplification kit (Ambion, Austin, TX). cRNA quality was assessed by capillary electrophoresis on Agilent 2100 Bioanalyzer. Expression levels of 48,803 mRNA transcripts, to investigate 37,804 genes, were assessed by the Human-6 v3 Expression BeadChips (Illumina, San Diego, CA). Hybridization was carried out according to the manufacturer's instructions at the McGill University/Génome Québec Innovation Center (Montreal, Canada).
Statistical analysis
Data were expressed as mean ± SEM. Data for the n−3 FAs concentration in erythrocytes were analyzed using a paired t-test to determine significant changes between baseline and end point. Analyses were performed using SAS software version 9.1 (SAS Institute Inc, Cary, NC). A value of p ≤ 0.05 was used to determine significance.
Analysis of microarray data
First, filtering was done with the expression data of the lowest detection microarray score in each tissue type. A detection score on microarrays of a p-value threshold of 0.01 was used to define the number of transcripts detected, as recommended by manufacturer and implemented in FlexArray software (version 1.4.1) (Blazejczyk, 2007). The Lumi algorithm was used to normalize Illumina microarray data. Specifically, expression values were normalized by using Lumi via the Robust Multiarray Average (RAM) algorithm (Bolstad et al., 2003). This step was followed by quantile normalization and log2 transformation. For the analysis of the relation between the mean values of intensities of the expressed transcripts in SMT and PBMCs, the data were not normally distributed. Hence, a Spearman nonparametric rank correlation analysis with the PROC CORR procedure of SAS software version 9.1 (SAS Institute Inc.) was performed. Statistical significance was defined as p ≤ 0.05.
Second, the samples were then grouped according to tissues: muscle tissue and PBMCs. To assess which transcripts were differentially expressed between SMT and PBMCs, we performed a Significance Analysis of Microarrays (SAM) algorithm, an adaptation of a t-test for microarray data, on all probes. Further, this analysis was paired, that is, there was a correspondence between the dataset of probes imported for SMT and PBMCs for each subject. In general, the SAM application assigns a score to gene on basis of change in gene expression relative to standard deviation of repeated measurements. Then, SAM uses permutations of the repeated measurements to estimate the False Discovery Rate (Tusher et al., 2001). A cutoff of p ≤ 0.05 was used to select the regulated genes. In addition, a fold change cutoff was also computed by FlexArray software to assess the level and the direction of the gene regulation. This fold change is calculated as the absolute ratio of normalized intensities between the mean values of the samples. Thus, two cutoff values were used to minimize the chances of false positives. Fold changes at 1.2-, 1.5-, and 2-fold and p ≤ 0.05 were taken from each tissue to determine differentially expressed transcripts and transcript lists were generated. The SMT and PBMCs transcript lists were then merged via SAS, which generated a list of commonly differentiated transcripts. Percentage of commonly differentially expressed transcripts was calculated. Further, this transcript list was analyzed via SAS by correlation procedure as described above.
Biological pathway analyses
Pathway analyses allowed us to determine whether genes found to be differentially expressed belong to predefined networks more than expected by chance alone and help to add structure to the vast amount of data generated by microarrays. The Ingenuity Pathway Analysis System (Ingenuity® Systems, www.ingenuity.com) was used to visualize gene expression data in the context of biological pathways. First, three input files were uploaded in the Ingenuity Pathways Analysis system: (1) probe sets of intensities in SMT, (2) probe sets of intensities in PBMCs, and (3) probe sets of significantly different transcripts between tissues (n = 6,135), and dataset in Core Analysis was created. Further, the core dataset was analyzed using the general settings for Ingenuity Pathways Analysis system as “Ingenuity knowledge base (genes)” and “considered only molecules and/or relationships where species is humans.” Finally, the datasets were analyzed to identify the biological functions that were most significant to the genes in the datasets.
Results
Subjects characteristics
Sixteen subjects, seven men and nine women, aged 56.9 ± 2.4 years, and with BMI of 29.9 ± 0.9 kg/m2 (mean ± SEM), completed the study. Expectedly, subjects had suboptimal glycemic control: fasting plasma glucose of 5.98 ± 0.08 mmol/L, 2-h plasma glucose of 7.4 ± 0.4 mmol/L and fasting plasma insulin of 105.5 ± 9.2 ρmol/L (mean ± SEM). In addition, subjects had slightly elevated lipid levels: total cholesterol: 5.51 ± 0.30 mmol/L, low-density lipoprotein-cholesterol: 3.84 ± 0.25 mmol/L, high-density lipoprotein cholesterol: 1.02 ± 0.05 mmol/L and triglycerides: 1.51 ±0.11 mmol/L (mean ± SEM). There was no difference in baseline parameters in weight, glycemic parameters and plasma lipids between the men and women subgroups.
Fatty acids concentrations
In addition, as expected, the 4 week n−3 PUFA supplementation increased the level of total n−3 (from 7.31 to 10.84% of total FA) including significant increase in EPA (from 0.92 to 2.48% of total FAs) content in erythrocytes. Finally, the ratio of n−3 to n−6 PUFA increased significantly from 0.23 to 0.37 for the n−3 PUFA supplementation period. These data demonstrate compliance of subjects to the n−3 PUFA supplementation regimen.
Comparing genes expressed in PBMCs versus SMT
RNA extraction from PBMCs and muscle tissues was performed on 32 samples: 16 SMT and 16 PBMCs after the n−3 PUFA treatment. After verification of the total RNA quality, all samples were kept for further analysis on microarray chips. Results demonstrate that of the 48,803 transcripts present, 36,738 (75 %) were detected in skeletal muscle cells and 34,182 (70 %) transcripts were detected in PBMCs using the detection p-value threshold of 0.01. Of the detected transcripts, 32,341 (88 %) transcripts were detected in both skeletal muscle cells and PBMCs. Therefore, approximately 24,000 genes were coexpressed in skeletal muscle cells and PBMCs. Correlation analysis shows that the level of expression of transcripts in PBMCs were strongly related to expression of transcripts in SMTs (r = 0.84, p < 0.0001) (Fig. 1). Further, we explored the genes that were the most highly expressed in SMT (Table 1A) and PBMCs (Table 1B). The subanalysis by gender was also performed, and shows similar results in men and women (data not shown). Overall, these data indicate that PBMCs adequately reflect the expression of the majority of the genes in SMT.
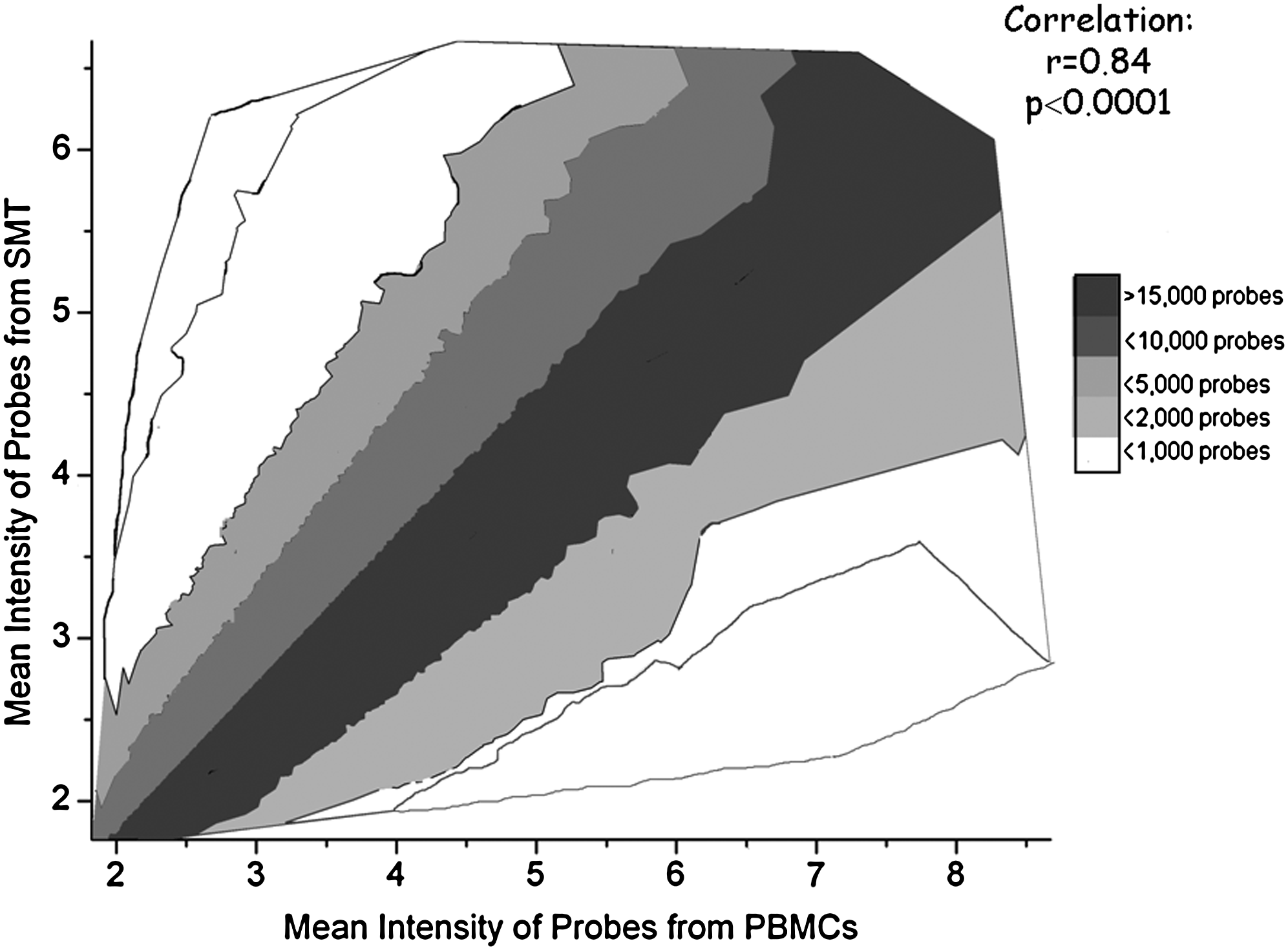
Correlation between intensity of transcripts expressed in SMT and PBMCs.
Comparing the effects of n−3 PUFA treatment on PBMCs and SMT
Further, we compared the differences in gene expression after an n−3 PUFA intervention in the SMT versus PBMCs. Results show that the whole-genome transcript expression in PBMCs compared to SMT were differentially expressed at 11, 25, and 42% for absolute fold change value of >5 or <0.2, >2 or <0.5, >1.2 or <0.83, respectively (Table 2). Of these approximately 46% were downregulated and 54% were upregulated in PBMCs compared to SMTs. However, when looking at transcripts that were coexpressed in both tissues, 0.5, 5, and 19% of transcripts were differentially expressed between PBMCs and SMT for absolute fold change value (two-sided) of >5 or <0.2, >2 or <0.5, >1.2 or <0.83, respectively (Table 2). Of the 6,130 differentially expressed transcripts, 3,987 were mapped to a mapped gene by the Ingenuity Pathways Analysis system (refer to Supplementary Table 1 for complete list of fold differences between tissues and gene identification). Of these genes, 376 were associated to physiological systems and development networks including: cell-mediated immune response, immune cell trafficking, hematological system development and function, respiratory system development and function, and hematopoiesis (Fig. 2). Overall, approximately 81% of transcripts were expressed in a similar matter in both SMT and PBMCs; even if there are some differences in expressed genes.
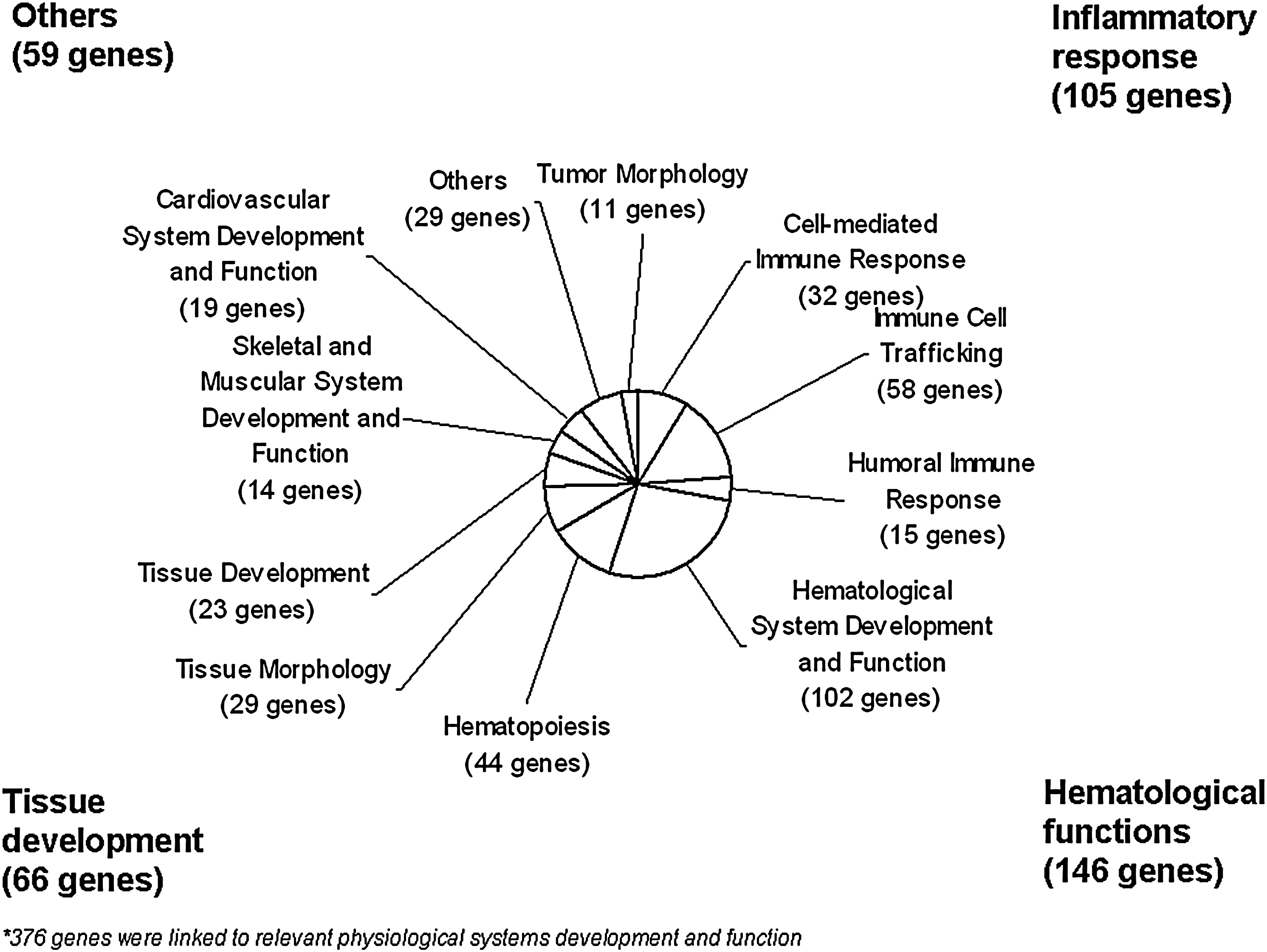
Differentially expressed genes between SMT and PBMCs related to physiological systems and development.
Discussion
PBMCs offer a significant practical advantage for gene expression analysis in nutrigenomic studies, because they are available in large quantities with minimally invasive techniques. Our findings collectively suggest that PBMCs can detect a large number of transcripts found in SMT, and that the intensity of gene expression is comparable after n−3 PUFA supplementation. Therefore, PBMCs deserve consideration as an appropriate surrogate marker for SMT gene profiling as observed in our study using n−3 PUFA supplementation.
Microarrays have the ability to simultaneously measure the expression of tens of thousands of transcripts enabling data-driven, holistic comparisons of groups or populations of cells, subtyping tissues, or predicting prognosis (Kitchen et al., 2010). It is well known that gene transcription is considered a process under strict control; only genes required by cells or tissues are expressed. Nevertheless, previous studies have reported that tissue-specific genes may be expressed in nontissue-specific manner (Liew et al., 2006). Results show that 75 and 70% of transcripts on the microarrays are expressed in skeletal muscle cells and PBMCs, respectively. Of the expressed transcripts, 88% were coexpressed in SMTs and PBMCs. Similarly, a recent comparison of genes expressed in PBMCs against a range of other tissues—liver, kidney, stomach, spleen, prostate, lung, heart, colon, and brain—revealed that over 80% of the genes expressed in other tissues overlapped with the PBMCs (Liew et al., 2006). Another study showed that 35 common genes downregulated both in adipose tissue and PBMCs from controls compared to obese subjects (Garcia-Amigot et al. 2005). Overall, PBMCs express the majority of transcripts for numerous tissues, including now skeletal muscle.
Second, our results also suggest that the expression levels of transcripts in PBMCs are highly correlated to that of SMT after n−3 PUFA supplementation. Previous studies with PBMCs gene expression have shown that the expression profiles of PBMCs do contain specific expression signatures in response to various physiological, pathological, and environmental changes (Liew et al., 2006). These studies hypothesized that the continuous interaction between PBMCs and the entire body gives rise to the possibility that subtle changes occurring in association with disease, may trigger specific changes in gene expression in PBMCs reflective of initiating stimulus (Liew et al., 2006). Recent studies showed that gene expression profiles of PBMCs were distinctive between individuals and changes in expression profiles of PBMCs were characteristic in a wide range of diseases, including: juvenile arthritis (Barnes et al., 2004), hypertension (Bull et al., 2004; Chon et al., 2004; Okuda et al., 2002), cancer (De Vos et al., 2002; DePrimo et al., 2003), chronic fatigue disease (Whistler et al., 2003), and neuronal injuries (Tang et al., 2001, 2003), lupus (Bennett et al., 2003; Rus et al., 2002), transplantation (Zhang et al., 2002), and under various environmental pressures, such as exercise (Zhang et al., 2002) and smoking (Ryder et al., 2004). It would have been ideal model to compare the change in gene expression levels from baseline to end point of intervention in two different tissues to determine if similar genes and pathways were affected by n−3 PUFA supplementation; however, due to ethical constraints this was not feasible in the current study. Future studies using baseline and endpoint tissues should be considered to reconfirm our findings. Taken together, these results suggest that gene expression levels in PBMCs are comparable to SMTs after n−3 PUFA supplementation.
Because genes that are chiefly expressed in microarray analysis of different tissues are related to their specific physiological function, we can expect some differentially expressed transcripts between PBMCs and SMT. Skeletal muscle highly expressed genes with specific biological activities related to muscle function. On the contrary, the highest expressed genes in PBMCs were related to immune response. Clearly, gene expression levels are related to specific functions—structural versus immune—of each tissue. In addition, 19% of coexpressed transcripts were significantly different between PBMCs and SMT. Therefore, the physiological networks, which were different between mononuclear cells and muscle tissue, were determined. These networks, including 376 genes, were incorporated in hematological functions (39% of genes), inflammatory responses (28% of genes), tissue development (17% of genes), and other physiological functions (16% of genes). Notably, these network differences between PBMCs and SMT clearly distinguish their specific functions. Therefore, if a particular role of a tissue is to be studied, for example, tissue growth, SMT may be a more appropriate biomarker for gene profiling. In the same way, PBMCs could be considered as a superior model for immune response. Overall, although the expression of certain genes varies between tissues, these results suggest that PBMC profiling can also reflect appropriately gene expression changes in muscle tissue, in particular, if examining the anti-inflammatory response to a nutritional intervention.
Conclusions
PBMCs express a large proportion of transcripts at comparable levels that are found in the SMT after n−3 PUFA supplementation. Overall, the present study supports the use of using PBMCs as a surrogate model for gene expression analysis of SMT. In the interest of practicalities and cost, these results suggest that PBMCs samples can be used to generate reliable data for transcriptomics in nutrigenomic studies. Further research on PBMC and SMT gene expression in response to other bioactive nutritional exposures is warranted.
Footnotes
Acknowledgments
We express our gratitude to the subjects for their excellent collaboration. The work of Julie Bisson, Éliane Picard-Deland, and Julie Marois in the coordination of the clinical trial is acknowledged.
This work was supported by a grant from the Advanced Foods and Materials Network (AFMNet). Iwona Rudkowska is supported by a Canadian Institutes of Health Research (CIHR) Bisby Postdoctoral Fellowship Award (200810BFE). André Marette holds a CIHR/Pfizer Research Chair on the pathogenesis of insulin resistance and cardiovascular disease. Marie-Claude Vohl holds a Canada Research Chair in Genomics applied to nutrition and health.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.