
Abstract
Abstract
Global Catastrophic Biological Risks (GCBRs) refer to biological events—natural, deliberate, and accidental—of a global and lasting impact. This challenges the life scientists to raise their game on two hitherto neglected innovation frontiers: a veritable “futures” thinking to “think the unthinkable,” and “systems thinking” so as to see both the trees and the forest when it comes to GCBRs. This innovation analysis article outlines the promise of Omics systems science biotechnologies, for example, to deploy rapid fire diagnostics for health security crises at GCBR level, possibly involving neopathogens and/or incurring epidemics (e.g., severe acute respiratory syndrome [SARS] and Ebola) that collectively threaten the lives of global society and interdependent biological ecosystems. Moreover, Omics encourages thinking beyond immediacy and in long-term strategies for biopreparedness and response innovation when the timelines are aggressive and compressed in response to crises such as GCBRs, but also to non-global but surging, multiple threats occurring as successive, overlapping, or distinct events, rather than as distinct entities—a prospect enforcing a reboot in Bioresilience. We define Next-Generation Bioresilience as “a systems approach against natural, accidental and perpetrated GCBRs using Omics technologies, and a shift in mentality, whereby the systems approach is expanded to include multiple plausible futures and expose unchecked assumptions attendant to risks, beyond technological determinism.” In sum, it is time to think about the realistic potential of Omics biotechnologies beyond clinical practice and precision medicine so as to harness the opportunities and address the uncertainties associated not only with GCBRs but also with other emerging Omics applications in health and society.
Introduction
I
These conditions vastly perplex the healthcare surveillance and monitoring by expanding the scope of possible pathogens, practically to infinity. These pathogenic entities, the neopathogens, have diverse biological origins and niche/functions. Some of them become pathogenic in an opportunistic context, for example, when host immunity diminishes or fails; others, when they colonize hosts not previously exposed to their presence (Casadevall, 2017); and others still, on diverse evolutionary acquisition of virulence determinants.
At the same time, the diffusion of biotechnology not only to industry but also to individual users (Frinking et al., 2016; Koblentz, 2017), mostly legitimately but also within the realm of Black Biology (Casadevall and Pirofski, 2006), is bound to become the basis for human agency biorisk events (Millett and Snyder-Beattie, 2017b), which comprise both accidental and deliberate malicious biohazardous events.
All cases cited earlier represent potential dangers for humans, livestock, and crops, either exclusively or inclusively, depending on the agent and its dispersion characteristics. In some cases, escalation to Global Catastrophic Biological Risks (GCBRs) is plausible (Connell, 2017; Khalil and Shinwari, 2014; Millett and Snyder-Beattie, 2017b; Nuzzo, 2017; Schoch-Spana et al., 2017). The scope of threats may include metapathogens (agents custom-engineered for pathogenicity) or other, more mainstream microbiotes routinely used for legitimate applications or naturally existing in different habitats, but prone to exhibit pathogenicity (opportunistic or not) on release on susceptible populations.
Bioagents may cause GCBRs in terms of spatial and temporal magnitude of the impact, direct or indirect (Connell, 2017; Schoch-Spana et al., 2017; Yassif, 2017), and existential risks (Millett and Snyder-Beattie, 2017a) in terms of focused mortality: that is, the potential to exterminate a certain species or other taxon/taxa. Both these event categories are deemed the least probable, and the respective risk factor is deemed as being rather low (Millett and Snyder-Beattie, 2017a).
This prioritization seems reasonable, especially when the probability of lesser but still severe biorisks, termed subcatastrophic events (Lipsitch, 2017), is taken into account. Examples of the latter are the different kinds and scales of pandemics, which are made all the more virulent and morbid due to largely societal reasons. The population density, the centralization of services, and the need for commuting daily for billions of people in crammed transportation means, along with customs and social norms, promote the dissemination of disease (Cameron, 2017; Connell, 2017; Madad, 2014; Thelaus et al., 2017), possibly more than any natural virulence determinant.
The quick pace of emergence of these new threats may collectively generate many more instances of severe risk on a global scale, which would be exacerbated by the low probability but high impact of GCBRs. Thus, new approaches must be considered, developed, and implemented, taking into account the large scope, the unknown characteristics, and the simultaneous appearance of multiple new and diverse pathogens. Innovative approaches to GCBRs and such new generation of biothreats should also consider addressing intentional misinformation that is intended to increase the impact of events and forestall their containment, remedy, or management.
This expert review and innovation analysis introduces the concept of “Next-Generation Bioresilience” (BioR-NG) as a systems and operational philosophy and practice against GCBRs and next-generation biothreats using Omics systems science technologies. While debating the suitability of the available options and comparing them with actual options of “Standard Bioresilience” in light of the forthcoming threat characteristics, our analysis advocates the adoption of a futures thinking whereby the systems approach is expanded to include multiple plausible futures, and expose unchecked assumptions attendant to risks, beyond technological determinism.
Such a mode of thinking forms the basis of, and should become integral in, “Rebooting Bioresilience,” as the new challenges, some extremely sophisticated and others collective and/or diverse, will not be manageable by purely technical solutions, no matter how advanced. We, therefore, additionally propose that it is time to broaden the scope of Omics technology applications beyond clinical practice and precision medicine so as to address the uncertainties emergent from both GCBRs and other projected next-generation biothreats.
Rethinking Bioresilience: The New Playfield and Its Essentials
Novel biothreats, and especially GCBRs, challenge the life scientists to raise their game toward systems approaches, and to think “outside-the-box” as the key for dealing with the diversity anticipated to characterize intentional outbreaks of pathogens and neopathogens, possibly enhanced by deception measures. In this context, funding and research, the two pillars for developing vigilance and resilience against a threat, are understandably limited as more obvious and direct needs take priority (Lipsitch, 2017).
And in this fact lies the main problem: Extinction-level events due to bioagents are not probable, especially in mammalians, and any such event would most probably implicate new agents. There are usually no prior data, metrics, and markers to assess exposure of mammalians to novel pathogens, so as to determine the conditions that lead to the development of immune response as opposed to extinction events.
Moreover, the line of thought just cited takes into account mostly naturally occurring events, or accidents. If deliberate action, usually implied under the term “human agency” (Millett and Snyder-Beattie, 2017b), is taken into account, the frequency and impact of such novel bioagents will depend on operational and deterministic concerns rather than on natural and probabilistic parameters. Consequently, outbreaks may become frequent, diverse, dispersed, or surging. Moreover, they are likely to entail artificial pathogens, which are, by definition, novel and most probably customized for increased virulence and resistance.
The conventional approach to biosecurity threats and biological risks (Standard Bioresilience) is deterministic: Sensor systems seek directly, indirectly, or interactively a signature characteristic for an agent. The associated signature or bioagent may be degraded, however, and thus interpretation might be necessary, which introduces additional degrees of freedom, resulting, in turn, in increased uncertainty and unknowns.
This is the reason for the advent of “Advanced Bioresilience,” which introduced more robust approaches, for example, by using two or more signatures, either of the same type (multilocus sequencing) or of different ones, polyphasic identification (Arabatzis et al., 2011). Although spatial and temporal patterns (geography, season) may facilitate or complicate the risk and threat-calling process, the concept is straightforward and is limited by discerning the improbable agents, inaccessible bioagent-location, or cross-reaction degradation of signal.
This plausible brave new world may be filled by, if not congested with, engineered or fully artificial agents, for which current bio- or event signatures do not readily apply as a general rule, and their own respective signatures are unknown, and in some cases deliberately evasive. The ability to further modify in a targeted manner the engineered agents leads to whole ecosystems or taxa of new microbiotes, which shall overwhelm any notion of keeping track. Thus, the only applicable approach is agnostic formats of extremely high-throughput analyses and/or collective biosignatures made up from multiple similar signals; these lines define the concept of BioR-NG proposed herein.
BioR-NG offers the promise to resolve presence/identity queries in an agnostic fashion, and may possibly define the adaptation (“informing”) of current methods and protocols to detect the newly recorded agents in subsequent contacts. Thus, BioR-NG will supersede but may not immediately supplant Standard Bioresilience. The latter will remain superior in terms of cost-effectiveness, once it is informed on what exactly it is looking for by guidance from BioR-NG, which will likely be costlier.
The notion of protection: biosecurity
Biosecurity is a collective term that includes all concepts and measures against natural and anthropogenic—both accidental and perpetrated—events (Connell, 2017; Koblentz, 2017), and associated risk factors. The concept of (bio)risk underlines the possibility and probability of a hazard/threat entity to initiate an adverse event. The event, on the other hand, constitutes actuality or, conversely, a materialization of the (bio)risk (Lipsitch, 2017). Regarding anthropogenic biorisks/events, biosecurity is the most active component of bioresilience, is complemented by biosafety, and pertains to biodefense (or defensive biological warfare), antibioterrorism, and antibiocrime functions (Koblentz, 2017; Millett and Snyder-Beattie, 2017a).
One often neglected dimension of all three functions mentioned earlier (Fig. 1) is the microbial forensics (Koblentz, 2017), which is instrumental for prophylaxis, treatment, and further preventive action. The application intends to detect lineages and associations of microbiotes found in scenes of crimes or used for criminal purposes with environments and populations so as to establish causality between incidents (not necessarily of a biological nature) and perpetrators, locations, and objects.
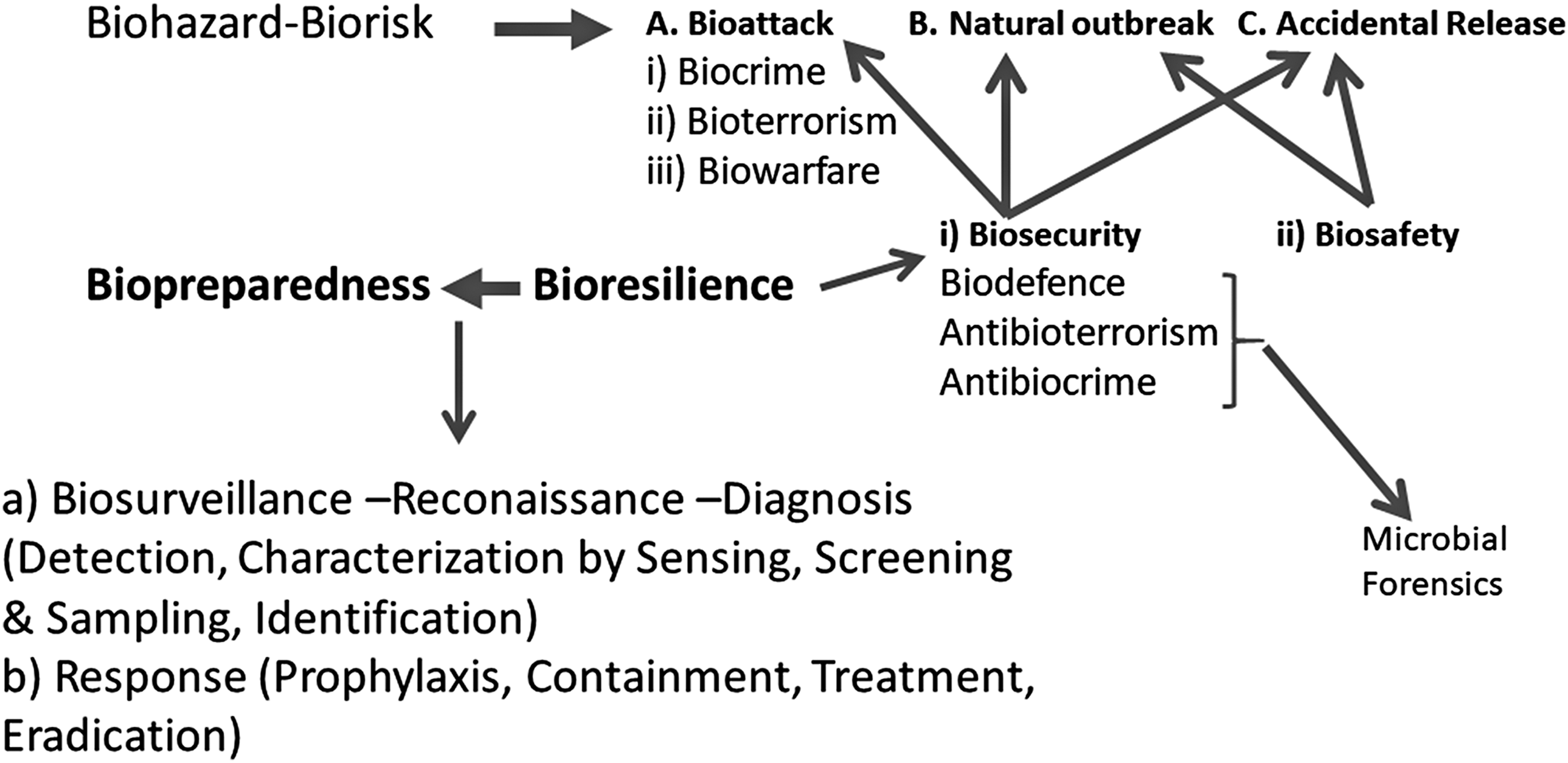
The conceptual network of Bioresilience and its constituents. Biopreparedness is the materialization of Bioresilience's conclusions, innovation, and optimization processes and contains Awareness (Biosurveillance, Reconnaissance, Diagnosis) and Response; whereas Bioresilience, as a cognitive, immaterial entity, can be broken down to Biosecurity and Biosafety. Each of the two latter applies to different biohazards.
Irrespective of the causal status, both human agency and natural occurring biorisks may be expanded, first to encompass similar entities in animal health, an eventuality anticipated under the “One Health” (Millett and Snyder-Beattie, 2017b) concept, since many human pathogens originate from zoonoses (Morens and Fauci, 2013). The addition of the agricultural aspect (Katara et al., 2012; Khalil and Shinwari, 2014; Madad, 2014; Nuzzo, 2017; Primmerman, 2000; Schoch-Spana et al., 2017; Thelaus et al., 2017), and of the broader environment including animate and inanimate objects as perceived by the “Environtome” concept (Hekim and Ozdemir, 2017) created, from the aspect of threat awareness, the all-inclusive and ever-expanding entity of “Total Biothreat.”
Consequently, to respond effectively to the total biothreat, the optimal prospect may be afforded by joint approaches (in terms of implicated services, sectors, and organisms) that will allow pooling of diverse, limited, and complementary resources, especially regarding biosurveillance and agent identification (Madad, 2014; Nuzzo, 2017; Primmerman, 2000; Thelaus et al., 2017). This notion of jointness and pooling of resources to any degree of integration through human, animal, plant, and environmental protection agencies is the first attribute of BioR-NG compared with the previous contexts, where all these agencies operated in a more or less isolated, if overlapping, manner, wasting resources in the name of expertise (Thelaus et al., 2017) but also compromising the chances to effectively identify and respond to the threats.
Objective: awareness
Situational awareness is the absolute prerequisite for responding to a threat. It can be implemented either by Surveillance, which entails a notion of temporal and/or spatial continuity, or by reconnaissance, a rather instantaneous, ad hoc entity. As an operational entity, (Bio)surveillance may be understood as the constant or repeated inspection of a space or object to establish drifts from a factual baseline; the latter is defined by the operator and consists of the “normal horizon of events” (Velsko and Bates, 2016). Events diverging from or disrupting the normal horizon must be acknowledged, validated, and processed (awareness) to determine whether to “sound the alarm” for activating reaction protocols, usually of a confirmatory nature but occasionally of an interceptive/interventionalist nature as well.
The Biopreparedness in a social entity (Cameron, 2017; Thelaus et al., 2017) consists of (1) the network of Biosurveillance, in terms of methods and resources; (2) the earmarked and available response elements, which, in an integrated format, constitute the concept of Iatromics (Hekim and Ozdemir, 2017); and (3) every and any material or immaterial resource directly implicated. The biopreparedness can be understood as the constant but changing result of the application of Bioresilience, which is a cognitive rather than operational entity. Evolution and transformation of bioresilience are necessary to keep biopreparedness vigilant, updated in real time, and operationally relevant (Fig. 1).
The implementation of surveillance, in general, focuses on, without being restricted to, providing early warning (Doggett et al., 2016), which allows responses (Wolicki et al., 2016), either in the form of protection or in the form of prophylaxis (proactive action) and then containment, decontamination/disinfection, and eradication (reactive action). The operating sequence applies to all three measures: biodefense, antibioterrorism, and antibiocrime (Fig. 1).
However, the timing in this scheme may be tricky: Detection might not be early enough to allow proactive measures, resulting in much less prevention (“detect to protect”) (Petrovick et al., 2007). If such is the case, its usefulness might be limited to the selection and initiation of targeted reactive measures to contain and manage the impact of an event (“detect to warn”) (Petrovick et al., 2007; Primmerman, 2000). Far from being “too little too late,” the latter offers a situational and threat awareness (Cameron, 2017; Nuzzo, 2017) that enables better distribution and delegation of the available resources, coordination of efforts, and optimized application of countermeasures.
In the detect-to-protect mode, the methods and sensor instrumentation (detectors) of choice (Petrovick et al., 2007; Primmerman, 2000) can detect agents preferably (Halász et al. 2002; Joshi et al., 2013; Kauchak, 2006; Ludovici et al., 2015; Primmerman, 2000), but not necessarily (Petrovick et al., 2007) at a distance (stand-off detection), so as to provide early warning for imminent biohazards. These sensors are, however, usually unable to implement any degree of identification (Halász et al. 2002; Joshi et al., 2013; Pak, 2008). Detectors can be deployed in fixed installations to provide constant surveillance (Kauchak, 2006) or mounted on different mobile platforms or physically loaded to vehicles to increase their surveillance footprint and range (Kambouris, 2018; Ludovici et al., 2015; Primmerman, 2000).
In surveillance, there are two difficulties: first, to establish a divergence from normality, normality must be defined; usually, it takes the form of a background image, or in a more primitive context, a value/figure, which must be determined, calculated, or compiled beforehand. This is not as easy as it sounds, since background parameters may change over time, and not always in a recurrent fashion. These changes must be both updated and projected/predicted by special algorithms.
Thus, to establish a divergence, which may well be due to a covert release of a biowarfare/bioterrorism agent, the suspect signal must be detected and discriminated against the current background, which is the second difficulty. In the context of biosurveillance, that in theory may be accomplished by collecting information from diverse data sources, including syndromic surveillance (Deshpande et al., 2016), this means discrimination from non-living entities, as is dust and sand, and of live (vegetative or dormant) entities incorporated in the normality and/or benign (as in commensal and mutualistic formats) (Sintim and Gursoy, 2016). The latter makes identification a must.
Moreover, the identification of an agent is essential to project number and severity of health casualties and for administering the correct treatment; it is also infringing on decisions for public health measures (quarantines, directions to the public) for the detect-to-warn formats (Wolicki et al., 2016).
Identification is achieved, in any depth and discrimination range, by a series of instruments, the identifiers (Petrovick et al., 2007; Primmerman, 2000). Identifiers of different stock use diverse technological and scientific principles to function, but all of them operate on live or ex vivo samples of the analyte. These samples are collected by still another breed of instruments, the collectors or samplers that may be embedded to the identifier or operate separately (Petrovick et al., 2007; Primmerman, 2000) (Fig. 2).
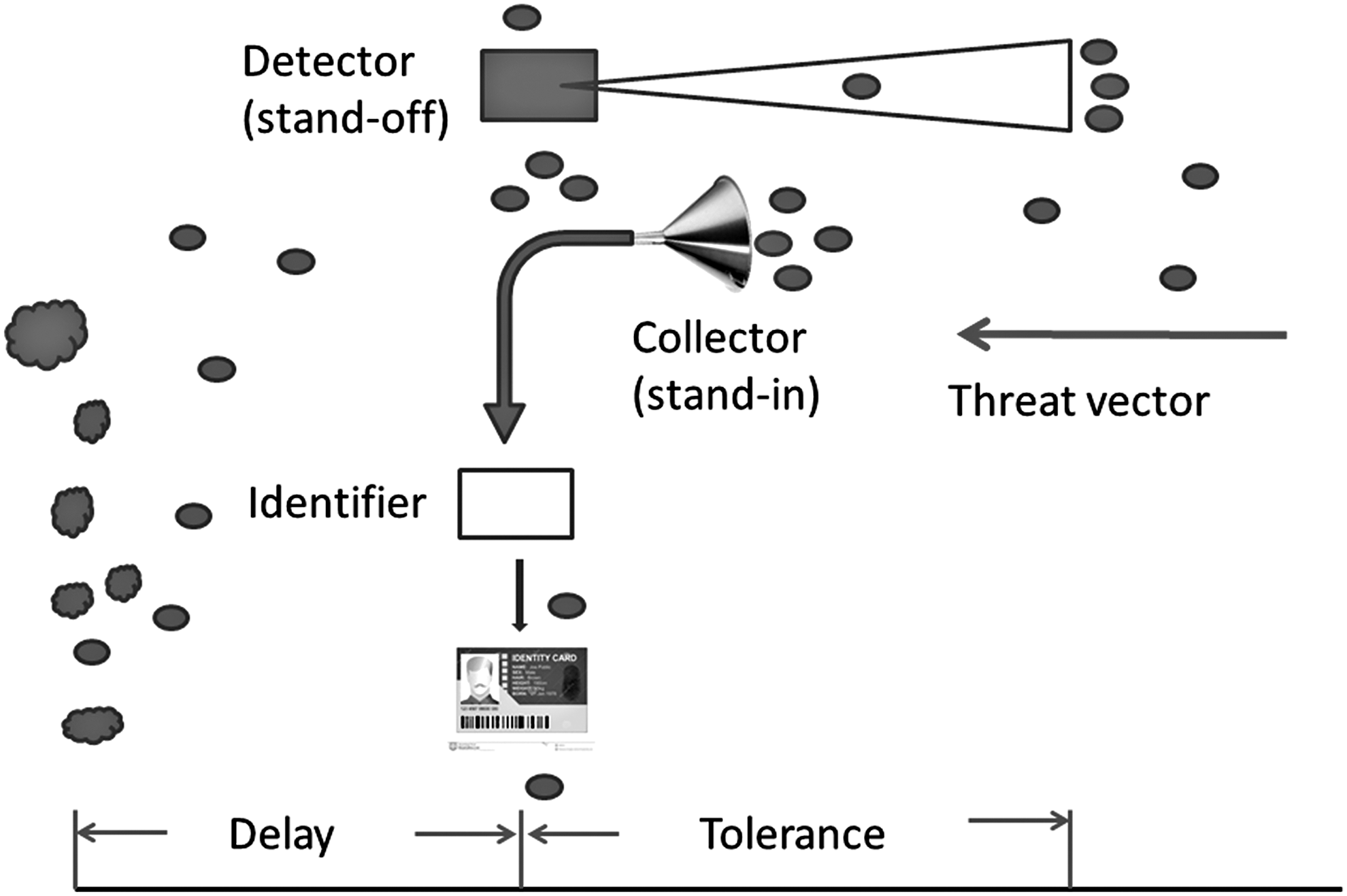
The operational and temporal phases of Biosurveillance. The Detector is a stand-off sensor, seeking signatures from a distance and validating them as potential threats or not. Tolerance is defined from the moment of signature detection to the moment of the physical emergence of the agent at the location of interest (where the Detector may or may not be positioned) and provides a rough estimate for the available time for deploying protective measures. The Delay starts at the moment of the physical emergence of the agent and ends with the onset of disease. It defines the time available for the determination of the threat, by the combination sampler-identifier, which operates in physical contact with the agent (stand-in) and for the deployment of MCMs and decontamination protocols. MCMs, Medical Countermeasures.
Contrary to biosurveillance, reconnaissance refers to a singular observation event, possibly but not necessarily of ad hoc nature. Reconnaissance aims at obtaining as much information as possible. The usual application of reconnaissance in medical and health promotion operations is the pair S-S (screening and sampling). Obviously, repeated and cohesively planned multiple reconnaissance events, as in screening, may lead to actual surveillance if integrated cohesively and put into context. It is also obvious that reconnaissance does not have to establish any background or horizon. It simply collects data.
Thus, an overt release of bioagent, by an explosion or in any other visible manner, should result in a reconnaissance effort that is aimed at detecting and identifying the agent involved. Initially, at least, the agent is concentrated near the release point, and is expected to undergo a vectored distribution. As a result, samples collected or images acquired promptly from the vicinity or of the instrument of the release are expected to be highly enriched in the analyte. In this way, identification methods of lower sensitivity and specificity may be used.
A further application of identification, beyond surveillance and reconnaissance, pertains to diagnosis. Diagnosis is a retroactive cognitive process that determines the cause of health casualties. In the context of infectious disease, it allows the proper medical countermeasures (MCMs) to be implemented through identification and analysis of the causative agent. In diagnosis, identification (direct or indirect) is a must; the background is restricted to the host biological signatures, augmented in some cases by natural microflora.
The background usually compromises the signal either by producing noise or by directly interrupting/dissipating or suppressing/cancelling it. To increase the signal-to-noise ratio to usable and confident levels, there are two distinct strategies, applicable exclusively or in combination: One is signal enhancement, achievable mainly by sample enrichment or by signal amplification. The other is background depletion, a task not always possible and never easy (Doggett et al., 2016).
Remaining relevant: the temporal dimension of countering biothreats
The specifications and performance of detection-sampling-identification equipment define the temporal correlation of the processes of biopreparedness in relation to the phases of an outbreak, which is crucial for the success of the containment effort. The former include detection, sampling, identification, protection and suppression, prophylaxis, diagnosis, and treatment, although different agencies produce different lists. The latter are, in short, emergence or dispersion, approach, infection, germination, and disease; each phase is possibly divided into subphases.
Delay is the time lapse from the actual appearance of the agent and the concomitant infection of the host or the contamination of its environment (habitat, resources) until the onset of agent-induced adverse effects (disease). Within the Delay, the spreading, the infecting, the germinating, and the incubating phases of the agent, the disease and related outcomes take place. Delay depends largely on the properties of the agent itself (germination time, multiplication rate, toxin production curves) and defines the available time to alert pertinent authorities and responders so as to initiate response procedures (“detect-to-warn”). The latter include issuing bulletins and directives for diagnosis, prophylaxis, and treatment, and activating MCMs that are both diagnostic and prophylactic-therapeutic (Ravi and Adalja, 2017).
Tolerance, on the other hand, is afforded by stand-off detectors and is measured by the time lapse between detection of the agent and its actual appearance at the locus of the sensor (contamination). It defines the ability to evade infection proper by proactive protection measures (“detect-to-protect”). Longer detection ranges for stand-off detectors increase the tolerance and, thus, the margin for early reaction.
On the contrary, stand-in/point detection exacerbates the temporal challenges of biorisk by minimizing tolerance. The management of the temporal dimension is, therefore, instrumental. Extended and persistent surveillance techniques, rapid bioassays, and intelligence reports are all partial solutions to the problem of temporal management of bioagent detection and response. A high degree of networking and wireless reporting either through smartphone applications or through embedded or dedicated networks, routes, and nodes can bolster such an effort. By making instantly available database assets and transducing raw data between sensors and processors (which might be extremely distant in spatial terms), an online surveillance capability may be readily achievable (Doggett et al., 2016).
A Matter of Performance
Methodological considerations and hardware specifications
Biosurveillance draws on standard biomedical advances to achieve situational awareness (Nuzzo, 2017), so as to enable suitable responses to specific biothreats. The “suitability” clause includes: the time, nature, location of application/use; the intensity/scale of possible countermeasures of different kinds (in perpetrated events); and the respective executives/enforcers.
The gradual narrowing of the scope of the probable identity for a detected agent, might this be emerging or de novo appearing, is the most valuable tool in staged identification for both diagnosis and surveillance applications. But for the reasons mentioned earlier, this process is likely to become unattainable. Thus, the present, high specificity and high cost assays, performed by specialized personnel using state-of-the-art equipment, will be both irrelevant and unaffordable.
For such use, candidate solutions are constantly improving in terms of availability/accessibility, user friendliness (Doggett et al., 2016), reliability, discriminatory power (Ludovici et al., 2015), throughput (Wang et al., 2005), dependency on infrastructure and supplies (Bartholomew et al., 2017; Ludovici et al., 2015; Mayboroda et al., 2016; Ramage et al., 2016; Rohrman and Richards-Kortum, 2012), and turn-around time (TAT) (Doggett et al., 2016). The latter is especially important in all cases of malicious events, whereas the specific gravity and importance of all the other parameters mentioned earlier depend on the actual context. Thus, among different Omics disciplines, culture-dependent assays, as in culturomics (Kambouris et al., 2017b), may seem inapplicable in principle; others, focusing on direct analysis of the core biosignatures, are conditionally considered (Ludovici et al., 2015).
In addition, for consideration for BioR-NG schemes in the new, more severe, challenging, and broadly framed environtome (Hekim and Ozdemir, 2017) of the emerging and artificial pathogens, the selected approaches ought to fulfill two key performance parameters (KPPs).
The first is the capability for agnostic identification, to cover any possible evolved, engineered, or artificial microbiote/pathogen, which most probably will be customized not only for increased virulence but also for undetectability (Millett and Snyder-Beattie, 2017a). As a result, a detection method should be capable of characterization as well. In contrast to the common wisdom (Deshpande et al., 2016), for truly novel agents, functional characterization may be achieved before full identification is reached, or even without it. The second is the capability for simultaneous multi-agent resolution; an eventuality is rarely considered except in the context of an agent released in the urban or rural environment or of spiking supplies (food, feed, water). In these cases, the resolution usually concerns one agent and the sample or environmental background signal.
Still, in every perpetrated/offensive application, it is common wisdom to use at least two, if not multiple, assets to capitalize on any possible synergy and to thwart surveillance and countermeasures; thus, multiple diseases or at least multi-agent release should be expected and taken into consideration (Ema et al., 2016; Jakupciak and Colwel, 2009; Mayboroda et al., 2016; Millett and Snyder-Beattie, 2017a; Pak, 2008; Song et al., 2005). Be as it might, the candidate approaches must tackle issues in two main areas should they aspire to satisfy the pair of the KPPs mentioned earlier.
Signature
Of course, genomics, in a broad sense, initially appears as an ideal candidate approach. This is due to the availability of a wide range of different technologies and methods offering different output, performance, and logistics options and, thus, permitting a choice of solutions that are tailor-made for specific needs. Genomics focuses on a very robust biosignature, the DNA, a chemically resilient molecule that allows both differential identification of agents and detection of some virulence factors within them (i.e., resistance genes, biotoxin production). Thus, genomics is recognized to bear an extensive array of advantages in the effort to detect biological entities, characterize, classify, and record them for future contacts and encounters.
Nucleic-acid-dependent approaches, irrespective of encompassing amplification steps or scope of targeting, have one important drawback: They are unable to detect biotoxins in cell-free contexts. Toxins are transmissible by various ways but remain non-contagious and non-reproducible and, thus, do not pose grave threats to the tune of epidemics/pandemics; they pose much less existential risks or GCBR. Only the use of extreme quantities of toxins in a concerted manner by extremely wide distributions/delivery networks or perpetrators might increase the toxin threat to the levels mentioned earlier. Still, biotoxins are counted within the biorisks because they are produced with biowarfare technology and facilities rather than synthetic chemistry, as it happens with chemical weapons (Anderson, 2012).
Biotoxins remain feared agents because (1) of their very high specific activity and potency that lead to low drastic doses, (2) because of their extreme variability, and (3) because they may be used to enhance the lethality of other, more or less destructive means. The latter include live septic bioagents (Anderson, 2012; Jakupciak and Colwell, 2009) and conventional, kinetic energy piercing or slashing objects. The latter constitute a lethal option with a long tradition, starting at the latest from the poisoned arrows of Ulysses (as referred to the Odyssey of Homer, vs. i.260–i.262) all the way to the Bulgarian Umbrella (Kendall, 2008).
As a result, structure-dependent assays or constitution analyzers seem more appropriate, for biosurveillance, as they cover the toxins along with the live agents. The former category contains different applications and formats of immunodiagnostics (Bartholomew et al., 2017; Petrovick et al., 2007; Sapsford et al., 2008), and, lately, aptamers (Pak, 2008). Both the structure-cognitive approaches depend on a three-dimensional (3D) structure coupled with electric (micro)charges to achieve binding and, thus, produce positive reaction and result. The latter category uses the newest breakthrough in mass spectrometry (MS) to analyze large moieties (currently up to cell size) at atomic and molecular level by fracturing molecules and finely discriminating mass-to-charge ratios (Jakupciak and Colwell, 2009; Ludovici et al., 2015; Madad, 2014; Sapsford et al., 2008).
Purity
In different applications, incidents, and events, pure samples might be available. Pureness is relative, but it affects mainly the specificity of any given method and occasionally its sensitivity as well. A sample enriched so as to keep signals from any contaminants below the detection threshold of the method may be considered “pure” for practical purposes. If pure samples are obtainable, agnostic methods are feasible, and, thus, unknown agents can be processed and characterized. In such cases, Next-Generation Sequencing (NGS) shows the content of even the most diverse genetic/genomic assembly.
If samples are not pure, then calling methods can be used, which examine the analyte for matching signature(s) with a list of high priority taxa, as the ones included in Select Agents Lists (Fierer and Kirkland, 2002). Clinical diagnosis developed in this manner, as did most biosurveillance methods up to now in the realms of Standard and Advanced Bioresilience. It is clear though that novel artificial pathogens cannot be tackled in this way, as their respective signatures are nor known and recorded so as to be called.
Current and Projected Approaches in Bioresilience
Current military diagnostic approaches, fast and hardened to comply with strict time sensitiveness and windows of opportunity on the one hand and different, adverse field conditions on the other, brought about Advanced Bioresilience. The latter is characterized by multi-potential, special-purpose equipment for surveillance and reconnaissance field operations. As a result, such approaches had attracted considerable interest and funding; but as each of them can cope with very limited ranges of pathogens, usually up to a score (Bartholomew et al., 2017; Ema et al., 2016; Ozanich et al., 2017), they remain basically of little usefulness in BioR-NG.
Metagenomics
In principle, metagenomic analysis of different environmental samples has been devised to tackle mixed/impure samples and implement identification of unknown entities. So, it satisfies the needs for multi-agent resolution and for agnostic application, which are focal in BioR-NG, but not for both simultaneously. For samples possibly containing totally novel, artificial pathogens, the approach is unattainable as a one-off test, in detect-to-warn formats, for samples possibly containing a detected threat that must be identified or at least characterized.
There are no solid principles to subtract in silico known genomes from a metagenomic mix in these conditions. This is an absolute prerequisite to deduce the sequences of the novel agents and then assemble them into elucidating the engineered/artificial genome(s); the latter may contain sequences stemming from none, any, or many of the background microbiotes.
Thus, the fully agnostic approach in metagenomics solutions is practically limited to either cases where regular screening had been performed or cases where targeted reconnaissance might be performed through sampling efforts executed on request. The former context enables the agnostic approach by providing a substractable background so as to discriminate new signatures and contacts. The latter fulfills the same purpose by allowing for the specific collection of highly enriched samples.
Mass spectrometry
The issue of agnostic applications is exacerbated in the case of MS; state-of-the-art MS approaches such as matrix-assisted laser desorption ionization–time-of-flight (MALDI-TOF) and electrospray ionization (ESI) can always take an objective read from an environmental or multi-species sample (Wieser et al., 2012). This read is expected to fully catalogue the molecular context of the sample at the required resolution.
As the sums of molecules are associated through respective, occasionally proprietary libraries to existing, analyzed, and recorded agents, they do not help to characterize an unknown agent, as phylogenetic distance and molecular ratios and context are not proportional. Similarities eventually do show proportionality, but this applies to different taxa and in different degrees (Emami et al., 2016; Ferreira et al., 2011). Thus, there is neither universal depth of comparison to identify totally unknown/unidentified isolates nor any algorithm to customize it in a case-by-case basis. In artificial agents, neither assembly of the molecular content nor classification of any assembled contents can be expected to conform to standard rules and patterns that may apply in naturally occurring and evolving microorganisms.
Immunoassays
Antibodies are notorious for their tendency to cross-react with different epitopes (Schroeder and Cavacini, 2010; Vojdani, 2015; Vojdani and Cooper, 2004), and more so as more possible target microbiota emerge. This handicap limits their potential for point-of-care assays that are designed for sensitive detection and highly specific identification of known analytes in unknown and even challenging or compromised samples (Bartholomew et al., 2017). The prospect worsens if multiple analytes must be resolved. But even in standard, single-analyte applications, the use of specific, conventional antibody-based methods cannot tackle novel agents emerging for the first time, nor artificial agents; thus, the prospect of immunoassays for BioR-NG applications is dismal.
Still, if pure samples of novel, artificial agents are available, a detailed profile (as proposed later) can be compiled by antibody microarrays containing thousands of paratopic loci. In such a case, the collective signal may compensate for the perceived lack of specificity of the individual specific ones, providing a better alternative to differential detection of known pathogens than current formats with mutually exclusive and highly specific loci. By fusing the binary signals of the inquired loci, epitopic imaging may be achievable and even coarse 3D mapping of the epitopes of an interrogated microbiote.
Thus, for novel, unknown, and unrecorded pathogens, typing becomes a realistic possibility. This is not so for identification or characterization: Evolutionary proximity does not establish similar antigenicity, not even a steady association between the two. The relationship between phylogenetic distance and immunoprofile is opportunistic and not causal—thus, no proportionality can be expected. Quantitative comparisons in such profiles constitute very weak association bases. Consequently, fused epitopic imaging, properly developed and utilized, is not prone to identify unknown agents in agnostic mode: “agnostic” in this case implying “analytes not previously processed and typed by the given panel of antibodies.”
Immunophenomics
High-throughput differential immunotyping for BioR-NG
Immunoassays have been the method of choice in every kind of Bioresilience applications, because they are adequately specific and can detect target epitopes in the high clutter (low signal-to-noise ratio) of mixed samples (immunodetection) (Bartholomew et al., 2017; Morel et al., 2012). They can positively identify known pathogens and can be used in clinical samples to diagnose syndromes (Ramage et al., 2016). Fast-track immunology can cope with emerging pathogens as long as the epitopes of the new analytes remain steady and stable over time.
Evidently, this is a medium-term option as new monoclonal antibodies are developed in 1–2 months (Ouisse et al., 2017; Press Release, 2017), which means even more prolonged assay development. Moreover, immunoassays bear the vital advantage to detect biotoxins and other conformation-dependent, acellular agents such as viruses and prions and possibly viroids (McCutcheon et al., 2014; Petrovick et al., 2007). This is coupled to their abilities not only to discriminate such analytes in complex samples but also to combine so as to resolve multiple agents of any kind (Bartholomew et al., 2017; Yen et al., 2015).
Fast-evolving threat agents, though, might be able or designed to change their epitopic signature through any applicable mechanism of antigenic variation, including, but not restricted to, antigenic shift and antigenic drift (Janeway et al., 2001; Palmer et al., 2016), to foil immunodetection efforts by false negative results (Finlay and Mcfadden, 2006). As a result, the potential of immunoassays for BioR-NG is compromised, at least in standard formats. Still, the current, highly specific, and targeted immunodiagnostics might shift toward multiple, non-deterministic, low-specificity bonding interactions, bearing reduced informativity.
In such an event, antibodies are going to reemerge as prominent biosensors for the processing of unknown samples and the characterization of unknown pathogens (Bartholomew et al., 2017; Petrovick et al., 2007). Irrespective of the platform technology, the operative idea for this comeback is to have thousands of antibodies arrayed in homogenous monovalent loci. This idea is based on the understanding that antibody paratopes interact and bind on compatible epitopes of the antigens; the compatibility is a function of 3D and electrostatic charge conformation, within a degree of respective tolerance. This is the reason for cross-reactions among antibodies and highly different antigens (Schroeder and Cavacini, 2010; Vojdani, 2015).
Thus, cross-reactivity is expected for most of the antibodies, without compromising their usefulness and reliability in highly specific contexts, as in many clinical and laboratory conditions.
Moreover, the number of possible gene codes for paratopes within a human cell genome is huge but finite at any given time (Schroeder and Cavacini, 2010; Tonegawa, 1983) and constitutes the cellular idiotypome. The Somatic Hypermutation mechanism increases this number by introducing new alleles to the gene modules that contribute to the paratope formation and thus rejuvenate, or recast the cellular idiotypome. In its turn, the individual idiotypome (all possible paratopes coded within one individual organism) is enriched as a result. At population level, this arrangement results in an enormous number of possible idiotypes (Eichmann and Berek, 1973) (collective idiotypome), which is the basis for the different efficiencies of the immune systems of individuals of a population toward a given antigen.
Any novel biological particle should, thus, present a binding profile by attaching to a finite number of different antibodies within an extended panel of available/known ones; differential binding is realized by binding by different epitopes or/and at variable affinities. None of these bindings is expected to be highly specific, and even if a few are, it matters little. The fused result of the binary signal from many such bindings might be highly specific and plot a very robust and detailed picture.
It is obvious that this kind of array is inclusive and binary. “Inclusive” can be understood as producing results as a function of the combination of positive loci. On the contrary, “exclusive” means that any positive locus presupposes negativity of other loci, and “differential” implies that all loci are expected to be positive but with qualitatively different signals. On the other hand, “binary” implies that each locus can only be positive or negative, as opposed to “differential,” where, as already mentioned, loci can be positive with different qualitative values.
If standard, high-density antibody microarrays—or other high-throughput parallel formats of attached antibodies—can be massively produced in a consistent manner, pure samples of even few cells or particles could be processed. Each sample/agent would produce a differential pattern of binary signals, highly characteristic. This immunosignature is agnostic in nature and carries no proportionality to phylogeny, as small genotype differentiations may lead to very extensive conformational changes of epitopes. The immunosignature does not identify in a comparative fashion an unknown agent, much less a novel one; similarities in profiles are much more likely to be opportunistic rather than causal (Doggett et al., 2016; Vojdani, 2015) but they type it most robustly, for subsequent encounters.
Of course, this immunosignature is expected to change through the different phases of the lifespan of an agent. The most basic examples are the immunoprofiles of vegetative versus dormant status, and the ones of different vegetative conditions and forms of the same agent, such as planktonic as opposed to sessile forms. This creates the need for a dynamic, multiple tag of such epitopic profiles, which are provisionally termed immunophenomic. Its purpose will be to record and chart any change in the epitopic profile of an agent/organism through all its possible developmental phases, stages and forms, and substantiate associations. But to tackle different agents in similar forms, as is the context of a suspicious white powder (Morel et al., 2012; Ramage et al., 2016) or the remnants and residues on a dispersion system, the concept can be accurate and handy, producing only one or a limited number of binding profiles.
The usefulness will be even greater if the assay becomes available in a point-of-care format, possibly but not mandatorily as a chip/strip, lateral flow immunoassay (Bartholomew et al., 2017; Koczula and Gallotta, 2016; Ludovici et al., 2015; Ramage et al., 2016). Although lateral flow assays are not as sensitive as other tests, immunoassays (enzyme-linked immunosorbent assay [ELISA]) or others (real-time polymerase chain reaction [RT-PCR]) (Deshpande et al., 2016; Doggett et al., 2016), they remain handy in point-of-care and out-of-area applications.
This collective, multi-signal pattern signature is, by definition, unsuitable for corrupted samples and impure or intentionally mixed, multi-agent samples. Robust software tools may allow multiple assignments of called paratopes at a later day, perhaps under a future Bioresilience guise, and thus lead to resolution of samples containing known, or at least recorded and typed multiple agents.
The main challenge for the immunophenomic approach is that any developed, tested, optimized, and fielded assay must be producible massively and consistently for a considerable depth of time. On top of this, it must be produced by many facilities around the world, to comply with the Global Health Security Agenda (GHSA) concept. The latter proposes to standardize equipment, infrastructures, and procedures globally, so as to detect and promptly contain outbreaks and other mass infection events before disseminating to pandemic, or even epidemic status and develop into GCBR (Koblentz, 2017; Millett and Snyder-Beattie, 2017b; Wolicki et al., 2016).
Arguably, doubts regarding identical performance of assays produced in different facilities and times/batches may arise. Moreover, concerns about the stability margin of immortalized cell lines, and about the self-life and storage requirements of the assay platforms and expendables justifiably emerge. The real challenge is the long-term availability of identical preparations of many thousand antibodies in terms of quality and performance. No matter the exact technology, antibodies are produced in batches and there is considerable variability among batches of the same antibody, making the long-term consistency of the assay a tricky issue. The very high number of loci and the need to use such assays in point-of-care context (Deshpande et al., 2016) exacerbate the inherent limited self-life of the antibodies, due to low stability (Doggett et al., 2016).
It must be underlined that point-of-care use includes, but is not restricted to, sensitive applications under adverse conditions, such as sampling an exploded shell or a sprayer can, or screening the whole population of a city exposed to accidental or deliberate biorisks (Connell, 2017; Schoch-Spana et al., 2017; Yassif, 2017). As a result, the odds are against maintaining good condition, and, most importantly, steady and reproducible performance of the antibody population, both within a locus and among different loci, thus impairing consistency in the collective signature.
On another level, the phylogenetically unbiased results of an immunophenomic assay may readily find applications in two more areas within the BioR-NG context: first, in determining the affinity of different paratopes to an antigen so as to formulate a customized cocktail of antibodies for immediate treatment of infected subjects; second, in identifying the candidate epitopes on the new pathogen for multitasking/retasking existing antibodies. The universal influenza vaccines constitute an application of this very principle of relaxed specificity (Lipsitch, 2017).
The immunoassay described here is scalable: It is easily upgradable by printing additional loci. Given the abundancy of the sample (over two orders of magnitude more than the saturating dose for one locus), new loci do not interfere with old ones. This is a distinct advantage over nucleic acid microarrays, especially the ones requiring amplification steps (Kambouris, 2016). To keep the array from indefinitely growing, and keep it in proportion, subtractive customization may be employed, which consists of selective deletion of old, uninformative, or irrelevant/obsolete loci as needed.
The optimized loci makeup for an antibody microarray entails a spatial/geoepidemiological dimension: It is entirely possible to need regionally customized arrays for different geographic locations (Deshpande et al., 2016; Gkantouna et al., 2015). Customization by age groups, by different, collocated populations of distinct ethnic origins, or even by expected syndromes may be sought for, as well. But given, as already mentioned, the inherent lack of interference among loci, highly automated and flexibly programmed robotic printing is bound to readily solve such issues (Kambouris, 2009, 2016; Song et al., 2005; Wang et al., 2005).
The immunophenomic profiling can be used as a stand-alone method in screening applications for some time, as it is robust. Still, as pointed out already, it is not specific due to lack of causality between in the sensory molecules and their ligands. Eventually, just as fast but considerably more reliable, dependable, and informative assays shall be needed, to contain an event. The development of the latter might be concomitant with the progress of the event and specific for the involved agent(s) (“development on request”). This concept exemplifies the integration of BioR-NG methods, concepts, and processes with Standard and Advanced Bioresilience technologies and methods to an affordable and effective ensemble.
Genomics in Bioresilience
Although the term “genomics” appeared only at the turn of the century, the “molecular” approach in identification of pathogens had already been established as a choice once molecular genetics came of age. It afforded detection and identification of reproducible biological agents with high sensitivity and specificity and in a timely manner, much faster than culture-based methods, and, occasionally, more objectively, as it was not affected by phenotypic shifts and antigenic variation (Velegraki et al., 1999a, 1999b).
Detecting and analyzing a DNA, or, more precisely, a nucleic acid sequence—hence Nucleic Acid Amplification Tests (NAATs)—is not of any value but for a number of known/existing sequences, pinned to a number of organisms so as to infer specificity (Doggett et al., 2016). So, initially, to implement molecular identification and corresponding molecular diagnosis of infections, detected and analyzed microbiote sequences from pure cultures were used as targets for oligonucleotide polymerase chain reaction (PCR) primers and probes were designed accordingly. Subsequent tests with clinically or phylogenetically relevant or similar organisms determined specificity and discriminative power (Kambouris et al., 1999). But in an era of unprecedented wealth of sequences available in public databases, the previous drill is perhaps simplified and both oligonucleotide design and assay specificity can be predicted, or rather determined in silico (Kambouris, 2009; Wang et al., 2005). This fact has long ago permitted optimization of the NAAT methods to allow more detailed output and almost simultaneous reading of results (real-time).
Simultaneously, the informational context of the NAAT assays was increased. The first axis has been the relative—and, with the use of proper standards, absolute—quantification of the target sample, most dramatically achieved by RT-PCR (Doggett et al., 2016; Kambouris, 2010; Sapsford et al., 2008).
The second axis was high throughput, achieved by multiplexing, which is conveniently defined as the ability to ask more than one question about a sample at the same time, or, in other words, the simultaneous interrogation of multiple markers per sample (Deshpande et al., 2016). Initially it was achieved in PCR formats (Lin et al., 1996) and, most importantly, with microarrays (Pastinen et al., 2000), to be promptly followed by their combination (Song et al., 2005; Wang et al., 2005), which brought forth the term “Genomics.” However, a robust way to combine these two axes was not devised, as the microarrays proved too cumbersome and were never very popular, despite offering a very appealing feature: genetic discretion (Kambouris, 2016).
The capability of molecular approaches—or rather NAAT—to detect dormant and uncultivable/fastidious pathogens (viruses being the best example of the former) made molecular diagnostics the most attractive Culture-Independent Diagnostic Tests (CIDTs) (Doggett et al., 2016) for military/security applications. The propensity of the viruses within the select agents lists (Federal Select Agents Program, 2017) furthered this attractiveness. Thus, the new technological breakthroughs in molecular biology found their way into Military Specification (MILSPEC) instruments (Christensen et al., 2006; Ozanich et al., 2017) belonging to the identifier category and designed for detecting and identifying a number of bioagents. This tendency has been the birth-moment of Advanced Bioresilience. Initial technological progress was implemented by an increase in throughput and the adoption of parallel formats, under the guise of multiplexing.
This progress went to high-order multiplexing, or “hi-plex,” which may reach four-digit numbers of loci/markers (Doggett et al., 2016; Wang et al., 2005). Hi-plexing was considered essential for unknown and multiple agent(s) resolution (Dod, 2006), and its prospects improved further with the advent of panmicrobial arrays (Doggett et al., 2016; Liu et al., 2009; Palacios et al., 2007). However, the prospect of massive genomic engineering, both to obtain access to effective pathogens (by inserting virulence factors to apathogenic strains) and to customize existing and available ones (Millett and Snyder-Beattie, 2017b) undermined its projected usefulness. And thus, the quest for BioR-NG has been initiated.
A third axis of evolution led to actual improvements in sensitivity (down to single-digit genomes/cells) and TAT (decreased to 30 min or less) along with quantification. The catalyst has been the invention of RT-PCR (Doggett et al., 2016); but in this case, low plexing (low double-digit of markers) is as far as technology can get. And this comes at an investment cost of nearly $50,000 for state-of the-art benchtop stand-alone amplifiers, with double that for automated formats that integrate sample preparation and nucleic acid extraction processes (Deshpande et al., 2016; Doggett et al., 2016).
Subsequently, portability and ruggedization ensued, ideal for expeditionary use of RT-PCR, which became available in portable, battery-operated instrumentation (Ozanich et al., 2017). Fully automated formats followed suit, such as the GeneXpert System, which integrated extraction, amplification detection/reading in the reaction mix prepackaged in a single cartridge (Doggett et al., 2016). In this way, NAAT tend to become point-of-care solutions, complementing or competing with immunologic, antibody-based assays (Deshpande et al., 2016; Doggett et al., 2016; Petrovick et al., 2007).
On the other hand, the availability of NAAT for massive, population-scale use is somewhat questionable. Low-plexing processing of samples introduces a bottleneck in the surveillance/screening processes for cases of risk factors that entail extreme time sensitivity (Ramage et al., 2016). High cost, which currently is more or less manageable in diagnostic assays for testing—possibly insured—individuals, becomes an issue in massive applications. Consequently, laboratory settings, with the capability to perform characterizations with higher resolution and throughput (e.g., by using multiplex PCR, Nucleic Acid microarrays), become a must, in terms of both operational needs and optimized management of available resources. Consequently, they should be included within the planning of any national or international biosurveillance concept (Deshpande et al., 2016; Velsko and Bates, 2016).
In a slightly different context, isothermal chemistries are much more prone to miniaturization and independence of energy and utilities. In effect, portable or even instrumentation-free assays, both being suitable as point-of-care tests for in situ screening (Doggett et al., 2016; Euler et al., 2013; Mayboroda et al., 2016; Rohrman and Richards-Kortum, 2012), are likely to prove rather affordable to acquire and use and easy to develop.
The actual problem with NAAT is that nucleic acid analysis depends on comparison with one or more known sequence(s) and thus on a scored phylogenetic distance from a known entity, which keeps them outside of the BioR-NG realm. And this limitation applies to technologies focused on nucleic acids (NA) sequence detection and analysis by any conceivable manner, such as, but not limited to, ELISA, ESI, electrophoresis, hybridization, and fluorescent in-situ hybridization (FISH) (Ema et al., 2016).
The next step—or leap forward: metagenomics
The ultimate step in this context is the metagenomic discipline. Metagenomics, mainly implemented through NGS, offer the best mid-term prospect: They implement detection of novel, hitherto unchartered agents, in both environmental and clinical samples. The latter application is admittedly less of a challenge, as background depletion for optimization of microbiote sequence reads is easier (Nakamura et al., 2008). Once a genomic entity is compiled by software from the wet reads, it can be compared with genetically known entries in databases. The procedure is, thus, likely to perform detection of new genomic signatures that may substantiate migrational, evolutionary, or release events, or to establish links, relationships, and interactions with other, known taxa, thus implementing typing and characterization (Doggett et al., 2016).
Further, environmental samples can be processed with metagenomics (and culturomics) beforehand, before any notion of biohazardous event, to create a background picture of microflora (Bardet et al., 2017; Doggett et al., 2016; Lagier et al., 2012) and all respective genomes. In this way, new signatures can be detected against this background and assessed.
The wealth of sequences currently available increases the utility of metagenomics. An almost universal accessibility of databases of submitted raw sequences may be readily expected under cooperation agreements, with GHSA (Forzley, 2017; Koblentz, 2017; Millett and Snyder-Beattie, 2017b; Wolicki et al., 2016) being the best example. The issue first arose through the One Health-One Humanity concept, which identified global-scale vulnerabilities to emerging biohazards due to individual states' dismal healthcare capabilities, which created “weak links” (Barbeschi, 2017) in terms of global dispersion, be that at the level of GCBRs or not.
On the contrary, metagenomic analysis is not fast: The time lapse from sampling to results is considerable and dependent on computer power, availability, and quality of databases. The core issue, availability and affordability of the necessary hardware, allows no optimism: Metagenomics-capable platforms are expensive and available in benchtop formats only (Doggett et al., 2016), mainly intended for specialized sequencing hubs and not for interested end users. As things now stand, portable automated workstations are not expected down the line at any foreseeable future, with a possible exception in the evolutionary line of the long-range sequencing approach (Heather and Chain, 2016).
Moreover, metagenomics, as such, remains singularly unsuited when artificial agents are concerned (Doggett et al., 2016). The algorithms of current software tools align and assemble output sequences following patterns and logic possibly not applicable in synthetic genomes, especially in ones custom-built for illicit purposes. This diversion may either forestall assembly of unknown genomes or lead to erroneous assemblies, resulting in missing the intrusive agent or, at the very least, mischaracterizing it. In both these cases, surveillance fails and the next step(s), (sampling and) identification analysis, may not even be triggered, especially in the former case.
NGS: the Lydian Stone for anthropogenic GCBRs
NGS, on the other hand, if applied to abundant and essentially pure samples in a whole genome sequencing (WGS) context, shows impressive potential (Voelkerding et al., 2009). Initially, it may be used to classify every possible specimen harvested in sampling events, much like the famous Lydian Stone was doing with ores resembling or hiding gold. Outperforming the latter by quite a margin, NGS also deciphers encoded virulence factors of different origins by resorting to trans-omic resources, such as transcriptomic, proteomic, toxinogenomic, and metabolimic libraries.
The NGS breakthrough was anticipated in public imagination since the mid-1990s, with Sci-Fi movies showing direct sequencing of pure biosamples producing whole genomes in a few hours. Its merit lies in deciphering the whole genome of an organism in a medically or industrially acceptable budget and TAT. This is achieved mainly by new breeds of reagents, ground-breaking instrumentation, and much more powerful and dedicated software tools (Voelkerding et al., 2009). The expected limit of evolution, in terms of flexibility and user friendliness, might be benchtop equipment of modest requirements in terms of utilities, logistics, expertise, and preparation so as to allow inclusion in mobile laboratories (Doggett et al., 2016).
The use of WGS in direct surveillance and screening is restricted mainly by cost. The price for accessing through a smartphone application the assembled sequence and processed information is just below the $1000 mark for a human genome at the moment (Regalado, 2016). This means a price for a microbiote, with perhaps 1/1000 of the size of the human genome (Land et al., 2015), at the low hundreds of dollars per sample. But even so, it remains unaffordable for massive use, especially for eukariotes. Moreover, WGS runs are not rapid by any notion.
This becomes an issue, as the temporal dimension is of focal importance in both routine and expedited surveillance and/or reconnaissance (Bartholomew et al., 2017; Doggett et al., 2016; Joshi et al., 2013; Petrovick et al., 2007; Primmerman, 2000; Sapsford et al., 2008). In some cases, it adversely affects background determination runs as well. The needed pace of health security operations, especially in events of human agency with an impact potential at the class of existential risk, dictates for wet processes TATs of 4 h or less for operational relevance.
WGS, though, comes at its best against pure samples of genomically engineered microbiotes, which have already emerged and will continue to do so in accelerated rates, mainly within the framework of rapidly advancing applications, sectors, and disciplines of biotechnology. The synthetic genomics sector already exhibits synthesized viral genomes from chemical expendables (Jackson et al., 2001; Koblentz, 2017; Smith et al., 2003), a fact directly hinting at the capability to increase the virulence and/or resistance of infectious agents. Moreover, such capabilities suggest the ability to resurrect eradicated pathogens such as smallpox or polio by an increasing number of facilities around the world (Cello et al., 2002; Koblentz, 2017).
In terms of systems science, synthetic genomics tacitly introduce the concept of the metagenome. The creation of a metagenome presupposes the use of minimal undergenomes, incorporating the most basic and necessary housekeeping gene circuitry to maintain generic cell platforms (Gibson et al., 2010; Hutchison et al., 2016; Umenhoffer et al., 2010). Onto this substrate the user can have overgenomes “pasted” (either transfer or transplant)—so as to acquire custom-built microbiotes with desired characteristics.
The overgenomes may be of various natures (Fig. 3). The unitary overgenome, the most basic category, refers to a whole genome being transplanted, or, alternatively, to a part of a specific genome. A more dedicated and challenging option will be the combined overgenome, the assembly of an overgenome from a number of subgenomes. The latter are—more or less large—parts of different genomes. And the third category, the compiled overgenome, might be the product of massive pasting toward a custom assortment of genes and regulatory sequences either synthesized de novo or excised and harvested from scores of different source genomes. In this third approach, the selected parts and pieces should undergo one or more homogenization, standardization, integration, and qualification phases, which can precede or follow the pasting.
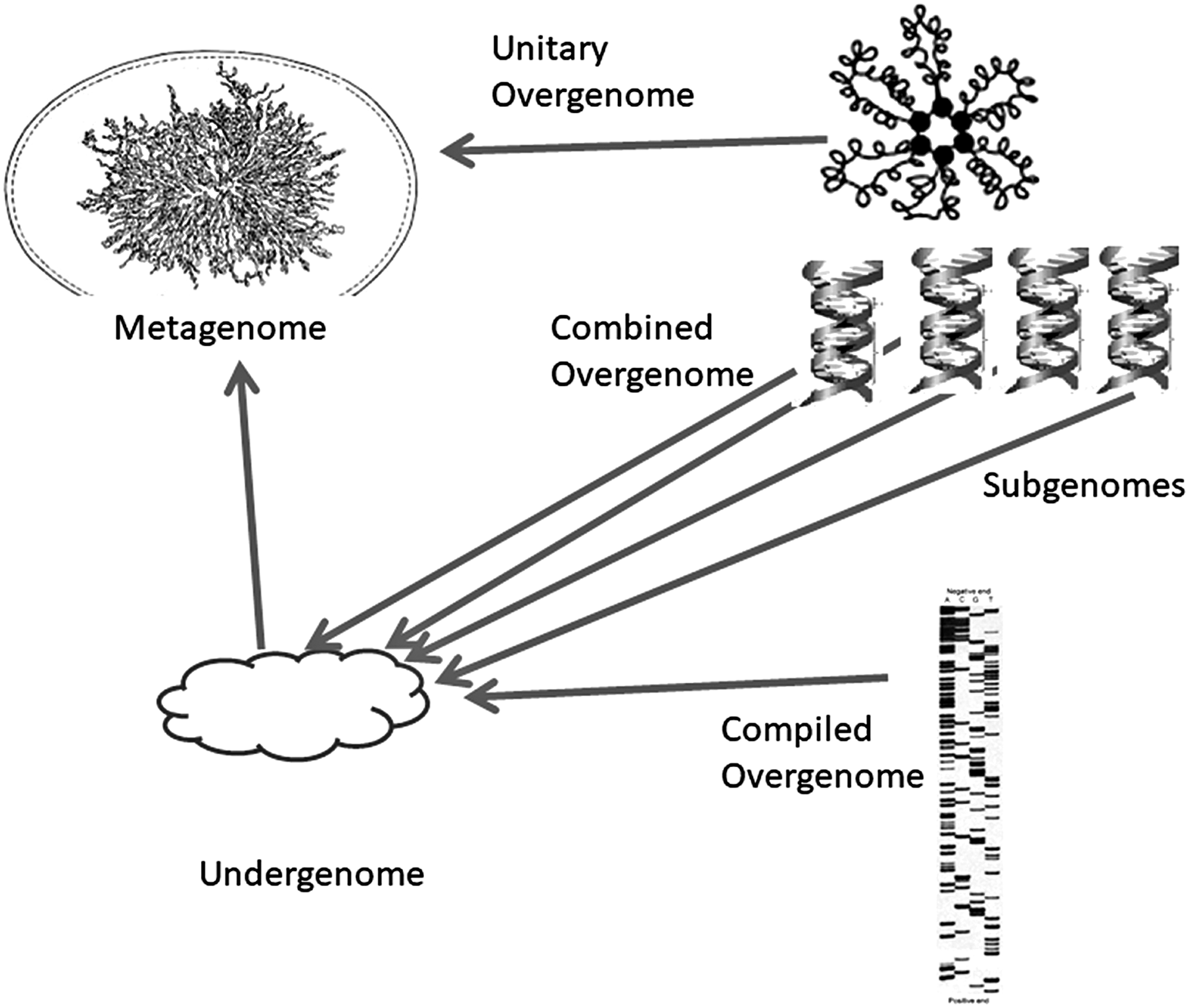
Genomic engineering: basic types of artificial genomes (Metagenomes) according to the origin of genomic elements. The minimal Undergenome maintains a cellular, undifferentiated platform and allows pasting of overgenomes, which will endow the new microbiote with the desired haracteristics. The overgenome may originate from (a part of) a single genome of an allogen (Unitary Overgenome), which is the simplest form. It also may be composed of bits and pieces of different genomes (Combined Overgenome) to obtain a heterogenous panel of characters. A third case is the Compiled Overgenome, where synthetic sequences are used for a thoroughly customized, integrated, and proprietary offspring.
The concept of metagenomes, especially when using unitary overgenomes, allows prompt introduction of fully artificial, totally novel, and, thus, literally unknown microbial populations with selectable degrees of lethality. In addition, they may be able to thwart diagnosis based on NAAT and/or clinical algorithms (symptoms and signs).
Given that novelty itself, especially in immunologic terms, might be a factor enabling mass extinction (Barbeschi, 2017), the prospect of massively emerging metagenomes in pathogenic agents practically epitomizes the idea of Anthropogenic GCBR hypothesis: “The biggest existential risk to humanity is due to technology advanced enough to drastically manipulate either human or/and environmental biology.” Of course, this is a rather expected conclusion if the Optimal Virulence Theory, which maintains that in nature emerging pathogens display a balance in transmissibility and virulence, really holds water ( Millett and Snyder-Beattie, 2017a, 2017b).
In such a scenario, WGS is the only way to characterize a novel pathogen. Generic, unbiased priming can ensure that multiple sequences are amplified simultaneously whereas the depth of sequencing provides a filter against in vitro or, more importantly, in vivo contaminants. Keeping in mind that a metagenomic picture is not the object here, but the characterization of a novel agent IS, sampling, as said earlier, becomes of paramount importance to furnish as abundant and pure samples as possible. In some cases, flow cytometry might be a sample processing option to ensure analysis of single cells, thus circumventing the issue of contaminants in the cellular level, before interrogating the sample with WGS.
The WGS approach mentioned earlier in the NGS context has a number of drawbacks. Focal among them is its need for advanced, dedicated, and heavy software to enact metagenomic sequences classification, since sequences cannon be discarded by being determined as uninformative. The latter is a common approach for software tools designed to be compatible with standard computers' performance (Doggett et al., 2016). The only valid reason to discard sequence data in our case is the low copy number, and this only in the simple sample preparation case; if the preparation of the sample includes the flow-cytometry step, to ensure single-cell baseline, no sequence can be discarded.
Moreover, the wet steps cannot be performed in field labs and of course not in mobile or portable devices. Major, heavy, and expensive equipment, handled and overseen by experts and adequately supported in terms of logistics, is necessary (Doggett et al., 2016). Sequencing depth, achievable by unbiased amplification if single cells are used, or by abundance of sample if not, is paramount for correct assembly of sequences. It must be stressed that genomic engineering might follow different rules and patterns for the synthesis of a genome than nature does.
Once the full genome is assembled, fusing and mining capabilities of assorted software tools and networked resources become vital. Detecting and comparing undergenome elements in databases might produce forensic evidence on the perpetrator(s) or on the acquisition pipelines used. Doing the same with the overgenome, the source organisms of (the modules of) it might be identified and the metabolomic, toxinogenomic, and resistomic profiles deduced. Genes and regulatory sequences can be compared with database entries for homologues to predict function, activity, and potency of the engineered agent so as to indicate possible protective, preventive, and suppressive actions as well as treatment options.
The key element for the sustainability of the concept might be the Long-Range Sequencing offshoot. It is often referred to as Third-Generation Sequencing, with the typical NGS formats being the second generation and the Sanger-based solutions being the first. As the name implies, it produces extremely long runs with excellent accuracy. This accomplishment reduces accordingly the assembly errors and the processing power needed. In addition, the sequencing depth necessary for assembly is drastically reduced, which further affects the software and processing power needs. But even more important, it allows reliable runs with minimal sample sizes, without amplification steps, mostly thanks to the excellent sensitivity provided by new technology and chemistries, but also due to the decreased depth required.
This fact not only allows minute amounts to be processed but also resolves the problem of amplification errors and biases, especially with repetitive sequences. In this way, flow-cytometry treated and segregated samples can be reliably processed. Such performance does change the game, even if the projections of achieving very high-speed reads with unprecedented affordability prove exaggerated. But the most useful feature is that the respective platforms are developed for civilian users of a very wide and differentiated base, much to the like of the simple, manual, first-generation sequencing devices.
This fact, which reverses the tendency of the NGS platforms to be addressed to dedicated sequencing centers, implies easier operation, enhanced user friendliness, more compact and adaptable design, and lower requirements for support, both in terms of interfaces, resources, and supplies, and in terms of information technology support—that is software, processing power, and computer hardware. With such specifications, mobile applications, starting with dispersed hubs and facilities, proceeding with expeditionary field labs and perhaps achieving integration on mobile platforms for near real-time analysis in stand-in formats, can be hoped for and actually expected (Heather and Chain, 2016).
Lastly, there is the “informant effect”: WGS deciphers a genome/metagenome, and the data produced may allow the development of specific NAATs for the new entity in as short a time as a fortnight. Thus, the genomic signature of an agent gets recorded by WGS and becomes detectable and possibly identifiable by clinical diagnostics and environmental surveillance NAAT amenities in the event of any further or subsequent encounter (Doggett et al., 2016).
Culturomics: Too Late but Not Too Little
Culturomics obviously seem out of place in cases where time sensitivity is supreme and CIDTs are developed with extreme toil and expenditure. Labor intensive, with a large logistical footprint in human and material resources and long or very long incubation times and TAT (Bardet et al., 2017; Lagier et al., 2012), this approach seems really incompatible with the specifications of biosurveillance for biosecurity purposes (Bartholomew et al., 2017; Doggett et al., 2016; Joshi et al., 2013; Petrovick et al., 2007; Primmerman, 2000; Sapsford et al., 2008).
But this is not as it seems. First, when processing environmental, impure, multi-agent samples that are suspect for the presence of artificial microbiotes, culturomics is the only option allowing multiple agent resolution, so that WGS may be applied to segregated isolates—a function that is vital for BioR-NG. Second, in a trans-OMIC context (Hekim and Ozdemir, 2017), culturomics may form a data generation and processing complex along with proteomics, transcriptomics, and metabolomics.
This complex, if fed with adequate quantities of the isolated agent, an exemplary task of Analytical Culturomics (Kambouris et al., 2017b), can establish similarities with other taxa; predict affinities, mannerisms, and dissemination dynamics; and elucidate virulence factors, production of metabolites and toxins. But, most importantly, due to what has been mentioned earlier, it may determine the septic or toxic mode of action, the drastic moieties along with their possible targets, and any unfavorable or, even better, adverse conditions in terms of physical and chemical challenges (Kambouris et al., 2017a).
The detailed parametric studies of Descriptive Culturomics (Kambouris et al., 2017b) can associate or dissociate a novel agent and characterize it accurately in vitro or, in some cases, project in vivo results, if high-fidelity cultures are used. This detailed characterization represents the ultimate diagnostic output, which may confirm or contradict genomic predictions and proteomic- or transcriptomic- and metabolomic-based assumptions.
Personalized Medicine: Any Room?
It is self-evident that the massive character of GCBRs allows no room for the fine-tuned and resource-intensive personalized medicine. There is though one exception: the individually targeted bioweapons that make use of the concepts of personalized medicine but in the opposite sense (Hessel et al., 2012), dubbed “personalized disease.” The toolkit of the black mirror image of personalized medicine may include inducible instead of “drugable” molecular targets in cancer transcriptomics (Nam, 2017) and individual characteristics in host-pathogen interactions (Culibrk et al., 2016). The compilation of the latter generates the differential vulnerability profile of an individual to panels of existent and engineered pathogens, toxins, and allergens.
Bioresilience NG, Next-Generation Bioresilience; ESI, electrospray ionization; GCBR, global catastrophic biological risk; IA, immunoassay; MALDI-TOF, matrix-assisted laser desorption ionization–time-of-flight; MCM, medical countermeasure; MS, mass spectrometry; NAATs, nucleic acid amplification tests; TAT, turn-around time; WES/WGS, whole exome/genome sequencing.
Assassination by bioagents has always been a valid, if not preferred, option (Kendall, 2008; Schoch-Spana et al., 2017) in the underworld. The target selection and acquisition were nominally performed by the operative or the vector of the bioagent. Precisely targeted, personalized bioweapons, where the bioagent itself performs the selection of one among many potential individual targets, are more subtle than usual “cloak-and-dagger” practices and represent a quantum leap in capability. They present perpetrators with a spatially long reach, a concealable means, and selectable initiation of activity in temporal terms. As an end result, such means may provide the opportunity to magnify even a limited destructive event to a massive disruptive one (Connell, 2017; Schoch-Spana et al., 2017), due to the current structure of economy, state, and society.
The extermination of a handful of key individuals entails the potential to magnify the disruption to massive proportions, perhaps even to Global Crisis events, with subsequent immense loss of life, social destabilization, or even collapse. In such a context, personalized medicine, as an opposite force to personalized disease, is expected to play a vital role, especially in prevention, and it is a safe bet that it has already started, tacitly and discretely, doing so for quite some time.
Conclusions and Outlook
GCBRs are attracting interest anew, but on a limited scale, whereas relevant funding is even more limited (Cameron, 2017; Schoch-Spana et al., 2017). One of the principal reservoirs of GCBR, the Biowarfare research programs are not prohibited by any convention. Still, confirmatory actions of even modest mandate such as the Biological Weapons Convention being underfunded (Millett, 2017), the main source of risk remains unchallenged. Research on tackling GCBR is understandably limited as they are considered highly unlikely and probably manageable within existing frameworks (Millett and Snyder-Beattie, 2017b).
The history of naturally occurring existential risks, extinction events, and pandemics backs up the former assumption and the events of the SARS and Ebola epidemics during the past 7 years validate the latter (Cameron, 2017). Moreover, GCBR research is expensive, speculative, and not prospectively lucrative: Such events may never happen (Cameron, 2017; Lipsitch, 2017; Schoch-Spana et al., 2017). After all, the stakeholders vehemently wish it so and enact intelligence, regulatory, political, and even social actions so as to avert materialization of similar risks of human agency into events.
On the other hand, social and environmental changes are likely to facilitate emergence and spreading of Biohazardous agents. Still, the focus of the threat is human agency (Cameron, 2017; Millett and Snyder-Beattie, 2017b). Biotechnology is easy to master and affordable (Cameron, 2017; Frinking et al., 2016), making the containment schemes used in Nuclear Non-Proliferation initiatives utterly useless.
Thus, perpetrated, intentional events must be expected in an unprecedented scale, at least in terms of frequency if not of severity as well, without taking into account accidents (perpetrated or even spontaneous) involving biotechnology facilities. Intentional events of any scale—Biocrime, Bioterrorism, Biowarfare—are likely to involve engineered or fully artificial microbiotes (Millett and Snyder-Beattie, 2017a, 2017b), and the extremely high number of possible perpetrators foretells an even higher combination of possible customization approaches and baseline agents. Thus, the outcome may be drastically enhanced agents, with qualities and performance completely outside the scope projected by legitimate bioscience: Optimal Virulence Theory (Millett and Snyder-Beattie, 2017a) and other natural constants may not apply.
The focal point is the timing of determining the GCBE status of an unfolding event in terms of magnitude and impact (Lipsitch, 2017): Any delay may result in global dispersion before containment measures and response protocols are initiated (Cameron, 2017). In this context, BioR-NG comes to play, introducing well-planned, multi-layer biosurveillance, enacted by a combination of Omics and conventional bioscience methods integrated with different technologies (Hekim and Ozdemir, 2017). Once biosurveillance can be made joint in scope, expanded in volume, and relevant in time parameters, it may buy the time needed for effective response. And response measures will be developed, procured, stockpiled, distributed, and fielded more rapidly and affordably if a robust bioresilience doctrine translates into a biopreparedness policy.
Such a scheme would suggest, encourage, or enforce multitasking (Cameron, 2017). This means that preferable development and fielding of countermeasures (materials, methods, and skills) are applicable not only to multiple—if not to the full spectrum of—exceptional biothreats but also to conventional, more mundane needs and challenges. In this way, research and development and production costs may be dissipated, a prospect vital for the viability of such endeavors (Lipsitch, 2017; Millett and Snyder-Beattie, 2017a).
Definite biocompatibility results, including therapeutic and decontamination options, will become available on completion of culturomics analyses, a task likely to take from a week to a few months, depending on the agent (Kambouris et al., 2017b). On the other hand, customized antibodies for developing new immunoassays need much longer to produce (Ouisse et al., 2017; Press Release, 2017). Still, once this task is accomplished, network and virtual amenities (Doggett et al., 2016) might allow immunophenomic profiling to implement prompt differential detection, for detect-to-warn and detect-to-treat applications, preferably in point-of-care formats (Petrovick et al., 2007).
The amenities needed for BioR-NG are expensive as they are capital-, technology-, and expertise intensive. These qualities make them unpopular with auditing and planning authorities that always seek low total costs, or at least to lower them. The competitive advantage they bear is their cost-effectiveness; a very expensive approach can pay off in multiple by preventing irreplaceable losses and damage (Millett and Snyder-Beattie, 2017a).
But this is a superfluous argument: Suffice to underline that without using the multi-omic integrated approach, while striving to improve its affordability and applicability, probably with long-range sequencing (Heather and Chain, 2016), BioR-NG simply cannot take effect and any surge on neopathogens might trigger a collective Global Catastrophic Biological Event. The dissemination of instrumentation inherent in the Long-Range Sequencing approach will allow dispersion of WGS, thus allowing better global coverage and/or improved expeditionary intervention, as visualized under the One Health—One Humanity Concept (Barbeschi, 2017), to timely contain events irrespective of their spatial coordinates.
BioR-NG intends to tackle multiple bioagents simultaneously or engineered bioagents throughout the spectrum of microbiology. Multiple engineered bioagents can be resolved only with prior segregation. The direct resolution of such challenges will be the objective of subsequent releases of Bioresilience concepts, along with better cross-talking among processes and more flexible and affordable instrumentation.
Footnotes
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.