
Abstract
Abstract
Breast cancer is one of the leading causes of morbidity and mortality that is in need of novel diagnostics and therapeutics. Meta-analysis of microarray data offers promise to combine studies and provide more robust results. We report here a molecular classification of pathological subtypes (estrogen receptor [ER], progesterone receptor [PR], and Human Epidermal Growth Factor Receptor 2 [HER2]) of breast cancers with microRNA (miRNA)-dependent signatures. A ranking-based meta-analysis approach was applied to eight independent microarray data sets and meta-miRNA lists were obtained that are specific to each breast cancer subtype. The comparison of the lists with miRCancer and the PhenomiR 2.0 databases pointed out nine prominent miRNAs: let-7b-5p, let-7c-5p, let-7e-5p, miR-130a-3p, miR-30a-5p, miR-92a-1-5p, miR-211-5p, miR-500a-3p, and miR-516b-3p. Further analysis conducted with the TCGA data showed that these miRNAs can differentiate tumors from normal samples as well as discriminate the molecular subtypes of breast cancer. According to the PAM50 classification, three of these miRNAs (let-7b-5p, let-7c-5p, and miR-30a-5p) downregulated significantly, whereas miR-130a-3p, miR-92a-1-5p, miR-211-5p, and miR-500a-3p upregulated in tumors from the luminal A to the basal-like subtypes. When the prominent meta-miRNAs and their targets were analyzed, they appeared to be taking part in important signaling pathways in cancer such as the PI3K-Akt signaling and the p53 signaling pathways. Furthermore, the regulatory genes, which are key players for ER, PR, and ErBb signaling pathways, were found to be under control of several meta-miRNAs. These meta-miRNAs and the genes they are regulating offer new promise for future translational research and potential targets for precision medicine diagnostics.
Introduction
B
The classification of breast cancers is based on tumor location (histological), receptor expression levels (pathological), differentiation degree (grade), and gene signatures (molecular subtypes). Breast cancers have heterogeneous characteristics such as altered expression levels of the estrogen receptor (ER), progesterone receptor (PR), and Human Epidermal Growth Factor Receptor 2 (HER2) receptors. Determination of the expression levels and presence of the estrogen, progesterone, and HER2/neu receptors, which characterize pathology of breast cancers, are indispensable to adopt a treatment method since therapeutic approaches are generally targeting these receptors (Osborne, 1998; Ross et al., 2008). Recent expression-based studies have provided evidence that different pathological subtypes show varied expression profiles (Rakha et al., 2010).
For the past few years, microRNAs (miRNAs) have emerged as significant members of gene expression regulators. Mature miRNAs are single-stranded and small noncoding RNAs, which are ∼20 nucleotides long. They regulate gene expression negatively at the post-transcriptional level by binding to sequences (mostly it is in 3′UTR region) with partial complementarity on its target messenger RNAs (mRNAs). It is known that miRNAs play a crucial role in several diseases such as breast cancer (Mulrane et al., 2014; Serpico et al., 2014). It has also been shown that miRNA expression profiles are associated with pathological features such as ER and PR expression (miR-30) and tumor grade (miR-213, miR-203, and let-7 family) (Mattie et al., 2006; Oztemur et al., 2015).
miR-195 and miR-154 expressions were found to be inversely correlated with the presence of ER (Lowery et al., 2009). In addition to these, miR-206 targets ERalpha and represses ERalpha mRNA and protein expression in breast cancer cell lines (Adams et al., 2007).
Meta-analysis is an important statistical approach that helps to qualitatively and quantitatively combine the results of various studies, including the microarray data (Normand, 1999). In this study, our aim was to find out miRNA signatures that can classify pathological subtypes of breast cancer, which can be used as a molecular signature to decide treatment strategies. Thus, we identified meta-miRNAs that can classify specific breast cancer pathological subtypes, such as ER+, ER−, PR+, PR−, HER2+, and HER2−. To achieve this, we applied a ranking-based meta-analysis approach to different microarray data sets, which was developed by our group previously (Oztemur et al., 2015).
Materials and Methods
Selection and reannotating of miRNA data sets
MiRNA expression data sets were identified using the search terms “miRNA,” “microarray,” and “breast cancer” together by using Gene Expression Omnibus (GEO). Studies with human primary breast cancer tissue samples, which had the pathology characteristics (ER, PR, and HER2 status), were extracted from the database. Eight miRNA data sets (Table 1) were selected. The data sets were generated by using cell lines. The formalin-fixed paraffin-embedded (FFPE) tissue samples were excluded.
ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; miRNA, microRNA; mRNA, messenger RNA; PR, progesterone receptor.
Pathological characteristics were restructured for standardization. Probe-naming was based on different miRBase releases in independent studies. In the meta-analysis, data sets were merged by using the miRNA names and so the annotations were produced according to the last miRNA version; version 21. For reannotation of the miRNAs in the data sets, miRBase Tracker (Van Peer et al., 2014), which is a web tool that allows researchers to keep their miRNA annotation up to date, was used. This step was important to prevent data loss. While using miRNA names, miRNA nomenclature guidelines were considered as well (Desvignes et al., 2015).
Meta-analysis
We used an analysis of variance (ANOVA)-dependent ranking-based meta-analysis program, which had been developed in our previous study (Oztemur et al., 2015) to identify pathological subtype status-specific meta-miRNAs. In brief, in the meta-analysis program, ANOVA tests were performed to find the p-values and the miRNA lists were generated accordingly for each study, and miRNAs were ranked according to their p-values. Finally, for each miRNA the mean of its rank in all studies was calculated and noted as its real rank in the meta-list.
Prediction of miRNA-target interactions
The mRNA targets of the pathological subtype status-specific meta-miRNAs and ESR, PGR, and ERBB2 targeting miRNAs were identified bioinformatically by using miRWalk (Dweep et al., 2011) (http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/index.html). miRWalk is a comprehensive database that contains predicted and/or experimentally validated targets. In this study, only the predicted targets of the miRNAs were used.
Selection of mRNA data sets and differential expression analysis
mRNA expression data sets were investigated using the search terms “mRNA,” “microarray,” and “breast cancer” together by using GEO. A study with human breast cancer tissue samples, which had all the pathological characteristic information (ER, PR, and HER2 status), was determined. The data sets that were generated by using cell lines and FFPE tissue samples were excluded. The data were downloaded as raw data files and were analyzed by using BRB-Array Tools (v4.5.1), which is an integrated package for statistical analysis of expression data (Simon et al., 2007).
Robust multichip average (RMA) normalization, which is one of the most widely adopted methods for analyzing Affymetrix data, was used for normalization (Irizarry et al., 2003). Class comparison test, which provides a powerful method for finding differentially expressed (DE) transcripts when controlling ratio of false positives, was performed to find out DE mRNAs between samples for each pathological subgroup [fold change (FC): 2, p ≤ 0.05] (Simon et al., 2007).
Common lists (named as meta-miRNA-related mRNAs) between the meta-miRNA targets and DE mRNAs were identified by using Venny (Oliveros, 2007) (http://bioinfogp.cnb.csic.es/tools/venny/).
Meta-miRNAs in diseases, biological processes, and pathways
Different open access bioinformatics tools: mirCancer (http://mircancer.ecu.edu/), PhenomiR 2.0 (http://mips.helmholtz-muenchen.de/phenomir/), DIANA-mirPath v.3 (http://snf-515788.vm.okeanos.grnet.gr/), and WebGestalt (http://www.webgestalt.org/webgestalt_2013/) were used for characterization of the meta-miRNAs and meta-miRNA-related/target genes (Ruepp et al., 2010; Vlachos et al., 2015; Xie et al., 2013). The Cancer Genome Atlas (TGCA) data analysis was performed by using Xena Browser (Goldman et al., 2018).
Results
The miRNA and mRNA data sets
The eight miRNA microarray studies and an mRNA study were selected from GEO (Barrett et al., 2013). The total number of breast cancer samples was 927 for miRNA studies and 131 for the mRNA study. The details of the platforms, the pathological information, and the sample numbers are given in Table 1.
ER, PR, and HER2-status-specific meta-miRNAs
As provided in Supplementary Table S1, 697 miRNAs for the classification of ER+/ER−, 677 miRNAs for PR+/PR−, and 469 miRNAs for HER2+/HER2− subtypes were listed according to the mean of their ranks and they were ranked according to their real rank scores. Since the real rank in the meta-list was used instead of the significance value, we focused on the first 20 meta-miRNAs listed in Table 2.
DE mRNAs between different pathological subtypes
Class comparison analysis performed with GSE17907 data using BRB-Array Tools in 47 breast tumor samples revealed that 77 genes for ER, 47 genes for PR, and 59 genes for HER2-status were DE (Supplementary Table S2).
Pathway enrichment analysis of the DE mRNAs was performed with WebGestalt and the results were found to be significantly regulative in breast cancer pathway (Supplementary Fig. S1) for all three subtypes (ER, PR, and HER2).
DE mRNAs have common elements with meta-miRNA targets
For each pathological subtype, status-specific meta-miRNA lists predicted that target genes of each miRNA were found by miRWalk. Meta-miRNA targets and DE genes obtained from an independent study, GSE17907, were intersected and 30 of the 77 genes for ER-status, 22 of 47 genes for PR-status, and 30 of 59 genes for HER2-status were found to be common; these common genes were named as “meta miRNA-related mRNAs” (Fig. 1; Supplementary Table S3). Since the miRNAs affect their targets both at mRNA and translational levels, it was expected to be found out DE genes common with the miRNA-targets under same conditions (i.e., ER comparison), which also validated our meta-analysis results.
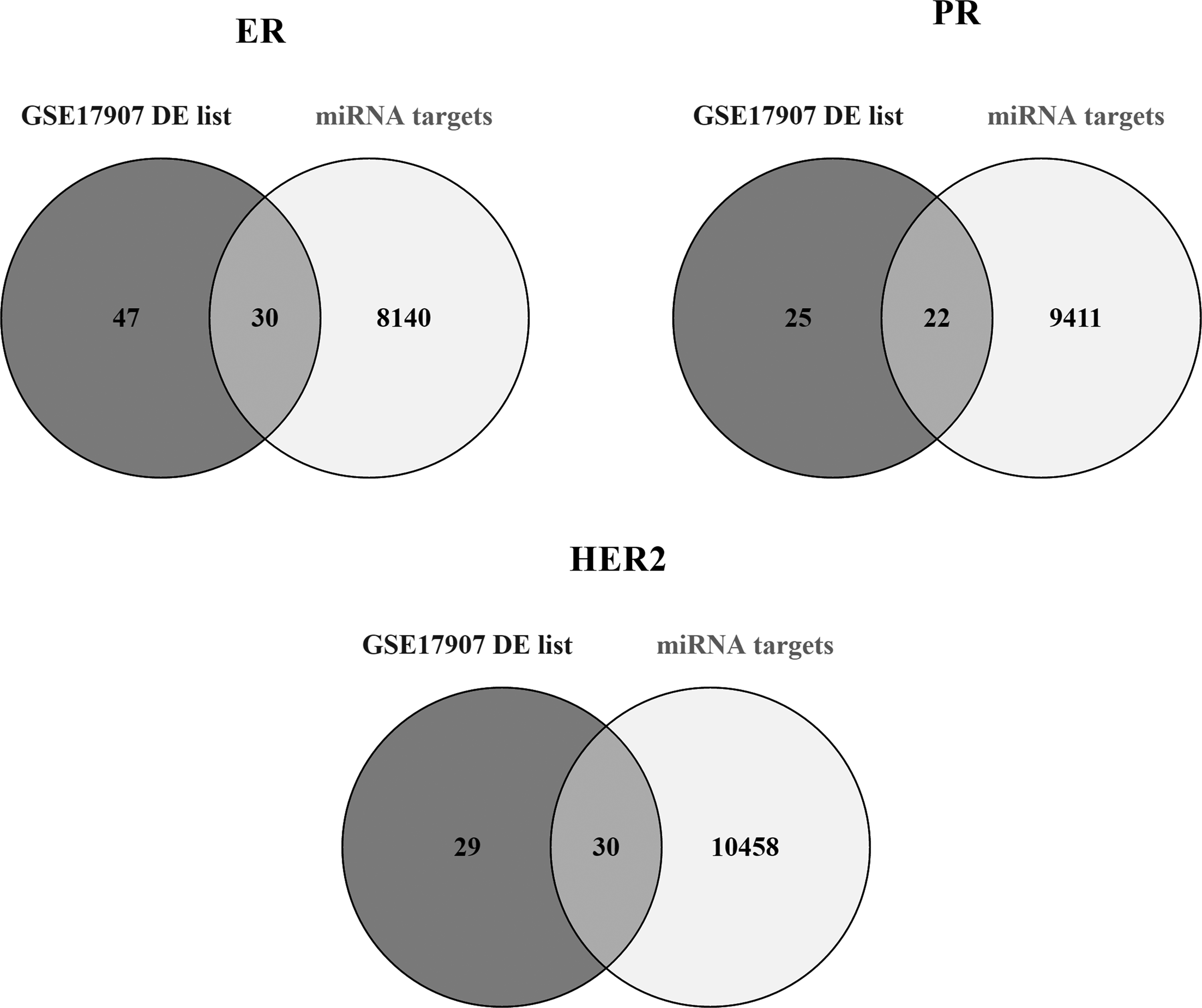
The intersection of meta-miRNA targets and DE genes that were obtained from GSE17907 data analysis. Thirty of the 77 genes for ER-status, 22 of 47 genes for PR-status, and 30 of 59 genes for HER2-status obtained based on the analysis of the data from GSE17907 are in common with the target genes of the meta-miRNAs. ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; miRNA, microRNA; PR, progesterone receptor.
Meta-miRNAs in diseases and pathways
For the functional characterization of the ER, PR, and HER2-status-specific miRNAs, two different tools miRCancer and PhenomiR were used.
The top 20 meta-miRNAs for three of the subtypes (ER, PR, and HER2) were compared with the dysregulated miRNA lists in miRCancer and the PhenomiR 2.0 databases. This comparison resulted with nine common miRNAs; let-7b-5p (MIMAT0000063), let-7c-5p (MIMAT0000064), let-7e-5p (MIMAT0000066), miR-130a-3p (MIMAT0000425), miR-30a-5p (MIMAT0000087), miR-92a-1-5p (MIMAT0004507), miR-211-5p (MIMAT0000268), miR-500a-3p (MIMAT0002871), and miR-516b-3p (MIMAT0002860).
The pathway enrichment analysis conducted with the targets of these nine miRNAs strengthens our confidence in the results of our meta-analysis method since we have identified a number of different pathways known to be important in cancer (Fig. 2).
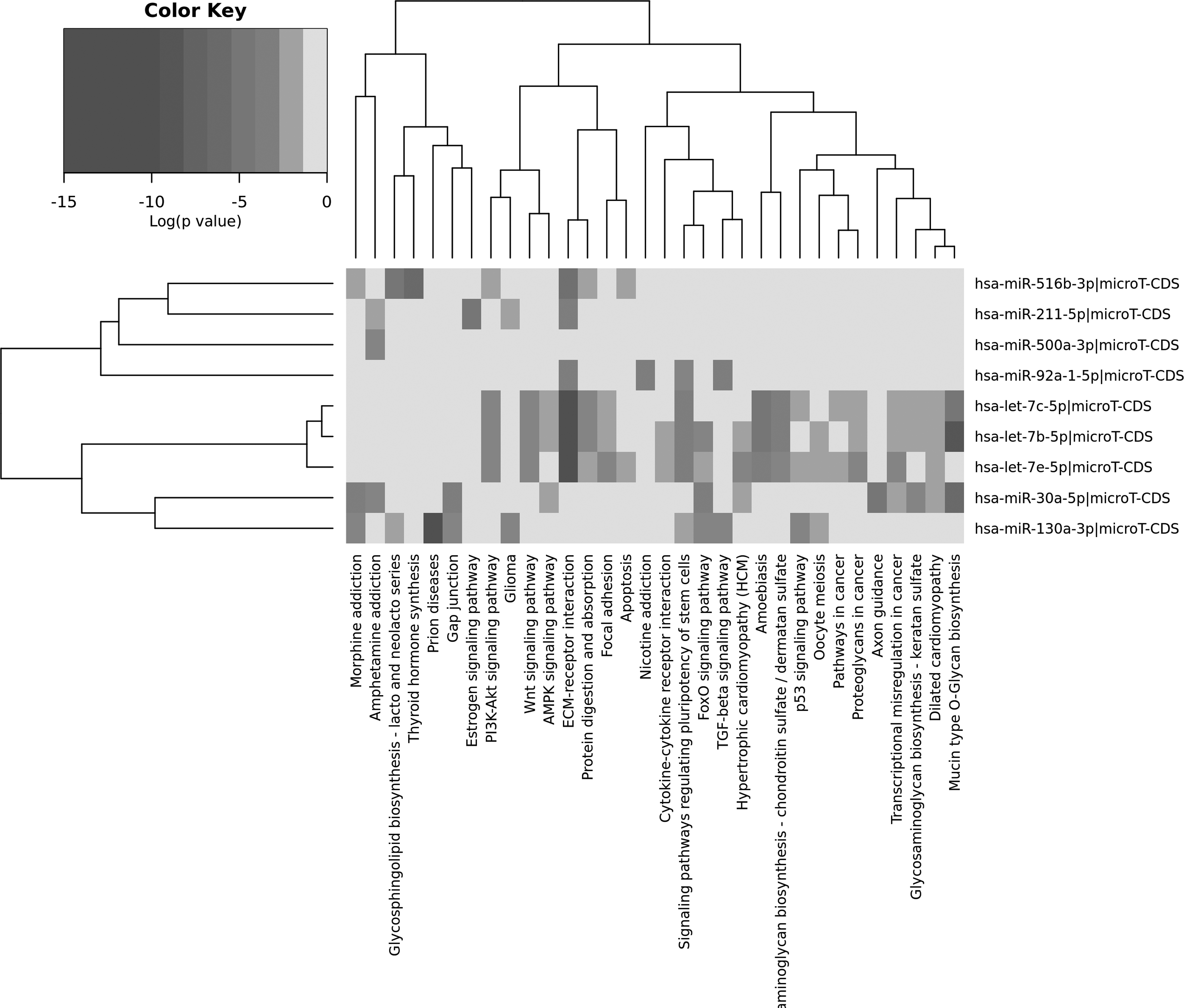
Pathway enrichment results of nine common meta-miRNA targets. Targets of the miRNAs and KEGG pathway results were used to construct the heatmap. Figure was obtained from DIANA-mirPath v.3. The intensity of color represents the logarithmic p-values.
Furthermore, we assessed the expression levels of these nine miRNAs in breast tumor tissues and examined their discriminative power in PAM50 subtypes by using TCGA Breast Cancer BRCA (n = 1247) data set through Xena Browser. Expression levels of let-7b-5p, let-7c-5p, miR-30a-5p, and miR-130a-3p were found to be significantly downregulated, whereas let-7e-5p was upregulated in tumor samples compared with normal samples (one-way ANOVA; p < 1.130e-8, 4.33 < f < 117) (Fig. 3). In contrast, according to PAM50 classification three of these miRNAs (let-7b-5p, let-7c-5p, and miR-30a-5p) consistently downregulated (Fig. 4), whereas miR-130a-3p, miR-92a-1-5p, miR-211-5p, and miR-500a-3p significantly upregulated in tumors from luminal A to basal-like subtypes (Fig. 4).
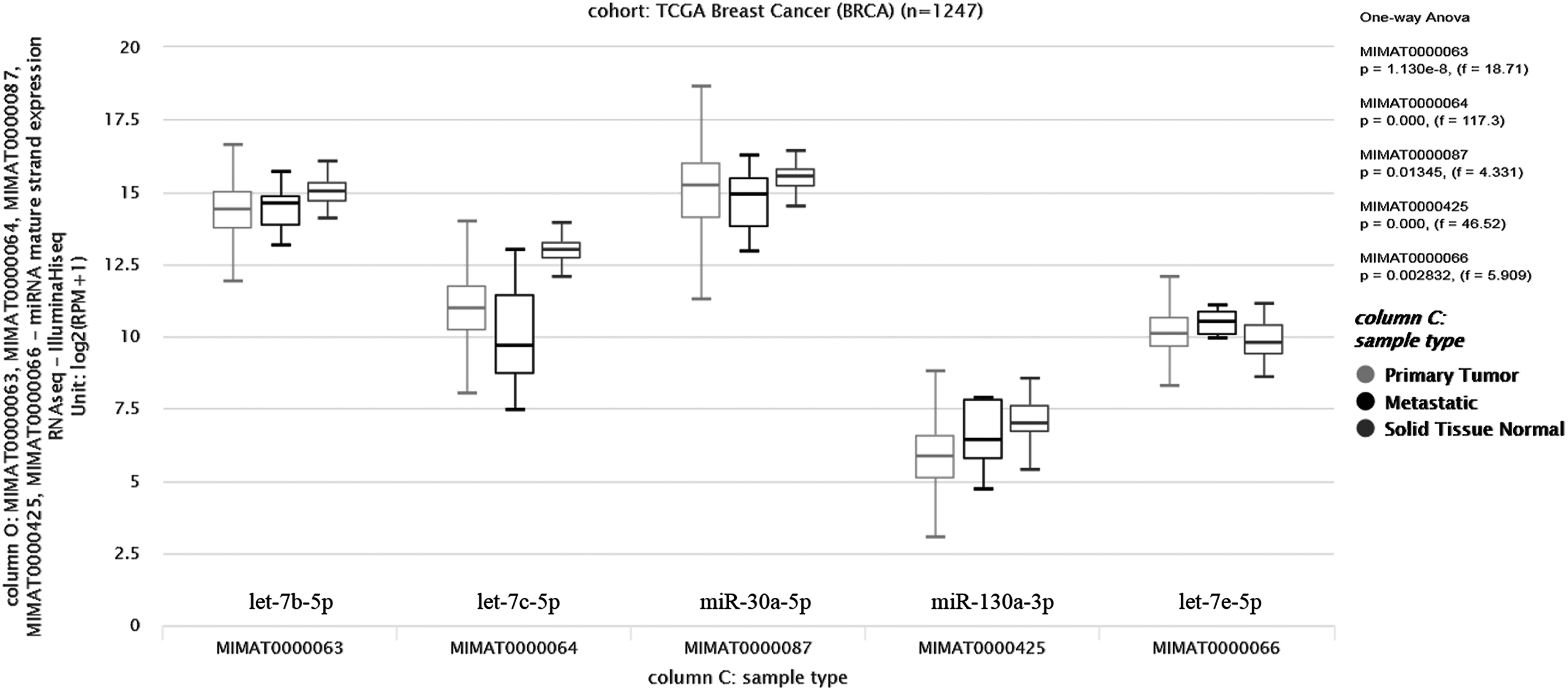
TGCA data analysis results of nine meta-miRNAs. Expression levels of let-7b-5p, let-7c-5p, miR-30a-5p, and miR-130a-3p, and let-7e-5p were differentially expressed in tumor samples compared with normal samples (one-way ANOVA; p < 1.130e-8, 4.33 < f < 117). ANOVA, analysis of variance; TGCA, The Cancer Genome Atlas.
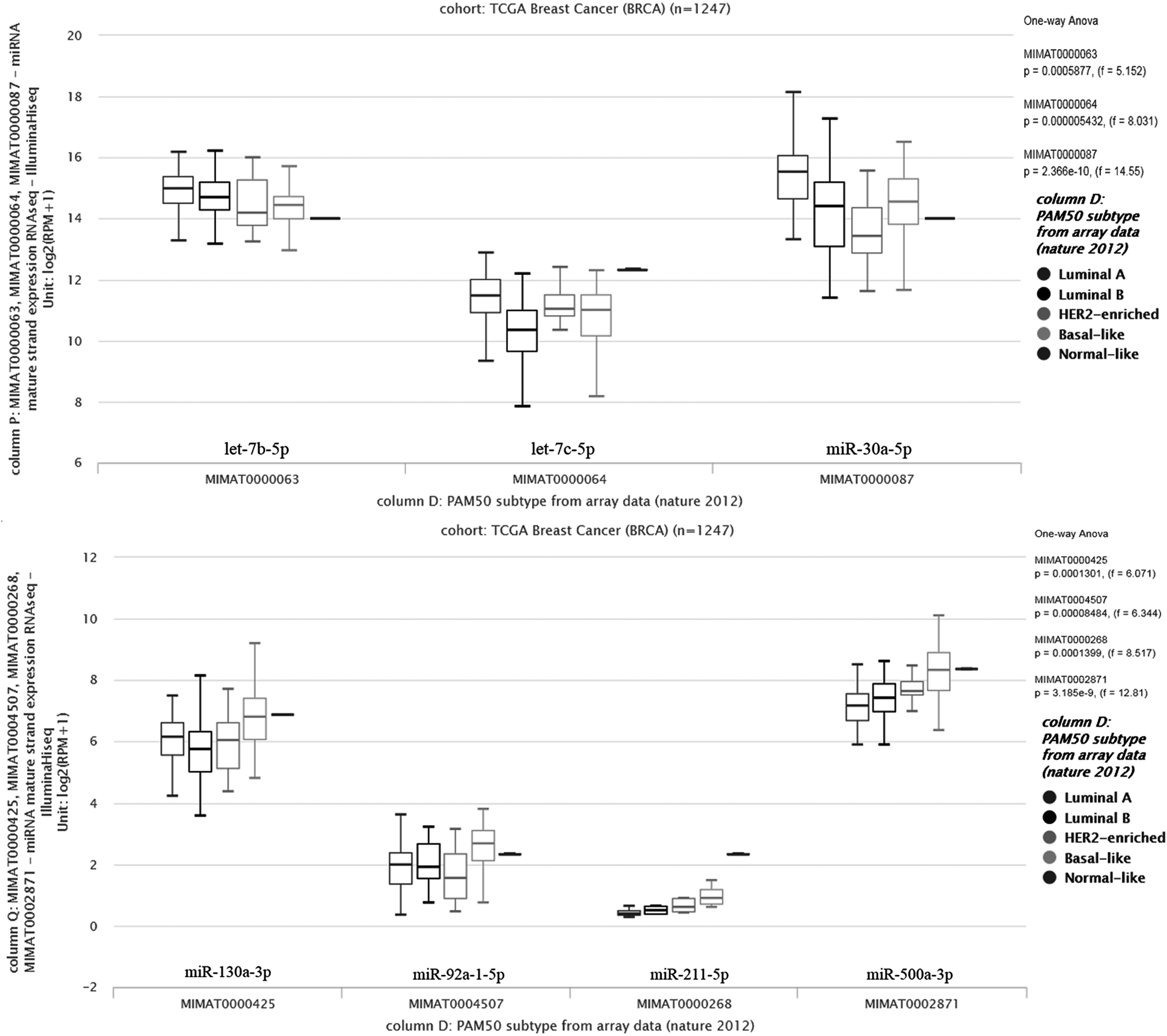
TGCA data analysis results of nine meta-miRNAs using PAM50 data set. let-7b-5p, let-7c-5p, and miR-30a-5p were downregulated in tumors from luminal A to basal-like subtypes according to PAM50 classification, whereas miR-130a-3p, miR-92a-1-5p, miR-211-5p, and miR-500a-3p were significantly upregulated in the same subtypes.
Meta-miRNAs targeting ESR1, PGR, and ERBB2
Since the main focus of this study is the regulation of miRNAs in the pathological subtypes of breast cancers related with three different receptors ER, PR, and HER2, we believe that it is important to identify miRNAs regulating the receptors themselves. Hence the miRNAs that were defined to be targeting the mRNAs, ESR1, PGR, and ERBB2, which are coding ER, PR, and HER2, respectively, were searched in our meta-miRNA lists. When ER-status-specific miRNAs were compared with ESR targeting miRNAs, let-7 family members were found to be dominated with four members of the family (let-7a-5p, let-7b-5p, let-7c-5p, and let-7e-5p).
The intersection of HER2-status-specific miRNAs with the ERBB2 targeting miRNAs resulted nine common miRNAs, which were miR-516b-3p, miR-211-5p, miR-138-5p, miR-940, miR-939-5p, miR-936, miR-93-5p, miR-92a-1-5p, and miR-135a-5p. Twenty miRNAs and these were found to be common when PGR targeting miRNAs and PR-status-specific miRNA list were intersected (miR-1197, miR-101-3p, let-7b-5p, let-7e-5p, miR-1290, miR-1237-3p, miR-125b-5p, miR-1248, miR-1275, miR-500a-3p, miR-1278, miR-1227-3p, miR-1267, miR-1184, miR-1202, miR-1244, miR-1224-3p, miR-519a-3p, miR-1181, and miR-1204).
Discussion
Precision medicine in breast cancer calls for rational classification of breast cancers using molecular biomarkers when possible. Thus far, ER, PR, and HER2 have been the biomarkers used for classification strategies in clinical decision making. These biomarkers help to determine which patients are likely to respond to targeted therapies such as tamoxifen or aromatase inhibitors for ER+/PR+ patients and trastuzumab or lapatinib for HER2/neu patients.
MiRNAs are important regulators of cell biology. Deregulation of miRNAs is noted in many types of cancer. In this study, miRNA lists, which could be specific to receptor status of the cells, were obtained by using ER, PR, and HER2 positive and negative breast tumor samples followed by a meta-analysis approach (Oztemur et al., 2015; Yu et al., 2018). The findings involved 927 tumor samples from 8 independent studies. As a result of the data analysis, miRNAs that are informative for ER+/ER−, PR+/PR−, and HER2+/HER2− tumors were identified in the metadata.
As a result of our ranking-based meta-analysis, we obtained 697 and 677 miRNAs for the classification of ER+/ER− and PR+/PR− tumors, respectively, and they were ranked according to their real ranking scores instead of the significance values. The studies conducted for the classification of pathological subtypes of breast cancer according to miRNA expression profiles showed that we had miRNAs in common, which have potentials to become biomarkers for the precise classifications of pathological subtypes.
Concordant with the study of Iorio et al., miR-30a-5p, miR-26a, miR-29b, and miR-30b were found to be discriminative miRNAs for ER- and PR-status and ranked in the top 30 list of our meta-list (Iorio et al., 2005). In addition, five miRNAs, miR-30a-3p, miR-30a-5p, let-7a, miR-93, and miR-199a*, which were listed in the top 20 list of ER-status-specific meta-miRNAs, were shown to be candidate biomarkers in the study of Blenkiron et al. (2007). MiR-491, let-7a, and miR-452, which were ranked at seventh, eighth, and ninth positions in the ER-status meta-list, were also presented as ER-status-specific biomarkers in an independent study (Rothe et al., 2011). We also obtained consistent results for HER2-status-related miRNAs and similar with the study of Lowery, miR-302c was found to be discriminative between HER2+ and HER2− tumors (Lowery et al., 2009).
It is well known that miRNAs show their effects on mRNAs (Guo et al., 2010; Oztemur et al., 2015). For this reason, the targets of pathological subtype status-specific 20 miRNAs were intersected with DE gene lists that were obtained from an independent breast cancer mRNA data (GSE17907), which could be regulated by our meta-miRNAs.
A limitation of the study is that the targets of the miRNAs were predicted targets and needed to be experimentally validated. These genes were significantly enriched in the breast cancer pathway, which we believe is an important confirmation that our miRNA and gene lists may contain biomarker candidates. The importance of many of the genes listed in our candidate list were shown in the literature before and among those GREB1, TFF1, and SLC16A6 were found to be the members of ER-specific meta-miRNA-related mRNA list in our study. GREB1 was shown to have important roles in hormone-responsive tissues as well as in tumor. Its expression profile was shown to be highly correlated with ER positivity in a study conducted with 39 breast cancer cell lines with known ER-status (Rae et al., 2005), and it is associated with changes in ER activity after breast cancer treatment (Dunbier et al., 2010).
In addition, GREB is essential for the proliferation of ER+ breast cancer cells and overexpression of it induces the colony formation (Liu et al., 2012). SLC16A6 was presented to be one of the 36 genes that were designated as estrogen-regulated genes that could predict disease-free survival in tamoxifen-treated patients (Lippman et al., 2008). In addition to these genes, ESR1, GATA3, TFF1, TFF3, CCND1, and XBP1, which were reported to be overexpressed in luminal cell lines (ER+), were also in our list and they were shown to be responsive to estrogen or related with estrogen signaling (Charafe-Jauffret et al., 2006). The significant presence of these three genes in our study may distinguish them to be suggested as miRNA-regulative luminal signature.
When our meta-miRNA lists were intersected with the disease-miRNA relation databases, miRCancer and PhenomiR 2.0, nine miRNAs were found to be common and when pathway analysis is performed with these miRNAs targets, they significantly enriched in important pathways in cancer (Fig. 2) such as PI3K-Akt signaling and p53 signaling pathways. The additional analysis performed with these miRNAs on TGCA data showed their potential roles in progression of breast cancer. The consistent downregulation of let-7b-5p, let-7c-5p, and miR-30a-5p and the upregulation of miR-130a-3p, miR-92a-1-5p, miR-211-5p, and miR-500a-3p in tumors with distinct clinical outcomes from luminal A to basal-like subtypes were observed. Pathway enrichment analysis together with TGCA data may indicate that the expression profile of these miRNAs could be suggested as prognostic parameters for breast cancer subtypes.
Since the focus of this study was to find out miRNA signatures that could be discriminative between pathological subtypes of breast cancer, we also compared the miRNAs that are targeting ESR1, PGR, and ERBB2 genes with our meta-miRNA lists.
let-7 family members were the prominent ones with four members of the family (let-7a-5p, let-7b-5p, let-7c-5p, and let-7e-5p) for ESR1 and ER-status comparison. In accordance with our findings, the regulative effects of let-7 family on ER signaling were emphasized previously in the literature. Barh et al. showed that let-7 family members could potentially regulate ER signaling and angiogenic pathways through targeting key proteins such as ESR1, MAPK4-6, and ITGB3. Furthermore, let-7 family members were shown to target CYP19A1, ESR1, and ESR2 and block ER signal in ER+ breast cancers as well as mitogenic signals through directly targeting TGFB, RAS, SKP2, MMP2, ILS, and MYC (Barh et al., 2008, 2010).
Moreover, miR-125b-5p and miR-500a-3p were among the miRNAs that were common between PGR targeting and PR-status-specific miRNAs. In humans, three copies of miR-125, miR-125a, miR-125b-1, and miR-125b-2, locate on three different chromosomes chr19, chr11, and chr21, respectively. It was defined that many of the proteins, which are taking roles in proliferation and apoptosis are targeted and regulated by miR-125a and miR-125b and downregulation of miR-125b-5p was reported in PR+ cancers (Lehmann et al., 2015).
miR-500a takes part in breast cancer-related pathways and related with the survival of the patients. The high activity of miR-500a was shown to be inversely correlated with survival in ER+ breast cancer patients (Aushev et al., 2016). In contrast, it is known that miR-500a-3p increases the sensitivity of the cells to tamoxifen by increasing apoptosis. Since endocrine therapy is still used as a treatment strategy for PR+ breast cancers, these two miRNAs could be suggested as biomarkers to define PR-status of the patients (Kim et al., 2016).
Conclusions
In this study, the heterogeneity of the pathological subtypes of breast tumors that are classified according to their ER, PR, and HER2 status were further classified in higher resolution according to their miRNA profiles. The meta-analysis approach we applied to the various microarray data sets enabled us to define robust and generalized miRNA classifiers for each subtype. These meta-miRNAs and the genes they are regulating offer new promise for future translational research and potential targets for precision medicine diagnostics.
Footnotes
Author Disclosure Statement
The authors declare that no competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.