
Abstract
Abstract
Metagenomics is not only one of the newest omics system science technologies but also one that has arguably the broadest set of applications and impacts globally. Metagenomics has found vast utility not only in environmental sciences, ecology, and public health but also in clinical medicine and looking into the future, in planetary health. In line with the One Health concept, metagenomics solicits collaboration between molecular biologists, geneticists, microbiologists, clinicians, computational biologists, plant biologists, veterinarians, and other health care professionals. Almost every ecological niche of our planet hosts an extremely diverse community of organisms that are still poorly characterized. Detailed characterization of the features of such communities is instrumental to our comprehension of ecological, biological, and clinical complexity. This expert review article evaluates how metagenomics is improving our knowledge of microbiota composition from environmental to human samples. Furthermore, we offer an analysis of the common technical and methodological challenges and potential pitfalls arising from metagenomics approaches, such as metagenomics study design, data processing, and interpretation. All in all, at this critical juncture of further growth of the metagenomics field, it is time to critically reflect on the lessons learned and the future prospects of next-generation metagenomics science, technology, and conceivable applications, particularly from the standpoint of a metagenomics methodology perspective.
Introduction
During evolution, microorganisms have adapted to an incredibly diverse set of environments. Almost every ecological niche of our planet hosts an extremely diverse community of unicellular organisms that are still poorly characterized. Detailed depiction of the features of such communities is instrumental to our comprehension of ecological, biological, and clinical issues. The study of the collective genome of microorganisms from an environmental sample is named metagenomics (Xia et al., 2018).
Metagenomic analyses of soil and marine ecosystems have proven that a hitherto unappreciated genetic diversity exists. Both soil (Daniel, 2005; Thompson et al., 2017) and oceanic (Bork et al., 2015; Rusch et al., 2007; Yooseph et al., 2007) metagenomes were investigated through various global sample collection efforts, unveiling unexpected diversities in prokaryotic, unicellular eukaryotic (Carradec et al., 2018) organisms and viruses (Schulz et al., 2018). These efforts yielded a large amount of genetic data that can be explored for ecological, biotechnology, and phylogenetic applications, among others.
Environmental metagenomics can be exploited to assess the effects of contamination by different pollutants on microbial communities (Ghosh and Das, 2018; Hemme et al., 2015; Jung et al., 2016), thus suggesting possible bioremediation strategies. Metagenomics has also been harnessed so as to investigate the microbiomes associated with agriculture and food (Liu et al., 2018; Orellana et al., 2018).
Mammals harbor complex communities of microorganisms that live on the surface and inside the host (skin, mucosal surfaces, gut, urogenital system, etc.). The mammalian microbiota includes all the domains of life (Archaea, Bacteria, and Eukaryota) and is fundamental to the health of the host (D'Argenio and Salvatore, 2015).
Studies in mammals have implicated the microbial communities present in the gastrointestinal tract (gut microbiota) in a range of physiologic processes with a significant impact on food digestion, metabolic processes, immunity, acquisition, and maintenance of overall wellness of the host. Accordingly, mounting evidence suggests a correlation between changes in composition of the gut microbiota and the pathophysiology of several disorders such as depression, autism, allergies, and chronic inflammatory diseases (Fazlollahi et al., 2018; Haberman et al., 2014; Hsiao et al., 2013; Imhann et al., 2018; Savage et al., 2018; Sekirov et al., 2010; Trompette et al., 2014).
Metagenomics is likely to have transformative and possibly game-changing impacts in clinical practice as well. On one hand, a microbial dysbiosis could be causally linked to a disease, suggesting targets of new therapeutic interventions aiming at dysbiosis correction and restoration of the so-called eubiosis. On the other hand, metagenomic profile could also represent a noninvasive, cost-effective, and rapid biomarker useful for diagnosis and/or prognosis. In infectious diseases, prediction of antimicrobial resistance adds a further layer of relevant information that can be retrieved from clinical specimens (Wilson et al., 2014). Furthermore, these approaches also impact public health with applications such as monitoring antimicrobial resistance in food supply by bacterial whole-genome sequencing (Chiu and Miller, 2019; Oniciuc et al., 2018).
Despite its relatively brief history, the study of microbial life through next-generation sequencing (NGS) technologies and computational biology is defining a new era in microbiology. Up to now, two main NGS-based strategies have been implemented for whole microbial genome analysis: 16S ribosomal DNA (rDNA) sequencing and shotgun metagenomics.
The former relies on PCR amplification of hypervariable regions of bacterial 16S rDNA through the use of degenerated primers followed by amplicon deep sequencing. Intrinsic limits of this approach are the exclusion of eukaryotic organisms and viruses from the analysis and the possible primer biases toward specific taxa (Tremblay et al., 2015). Since 16S amplicon sequencing focuses on the analysis of a tiny region of prokaryotic DNA, it is not formally defined as metagenomics (Xia et al., 2018). Differently, shotgun metagenomics (or simply metagenomics) relies instead on sequencing of randomly sampled DNA fragments isolated from a microorganism community.
Metagenomics allows the characterization of complex communities of microorganisms of specific environments, including human body sites, at unprecedented resolution, without the need of any a priori knowledge and without prior culturing (Escobar-Zepeda et al., 2015; Schloss and Handelsman, 2005; Tringe et al., 2005).
Several reports compared the performance of the two techniques. Comparison of 16S rDNA sequencing and metagenomics has been performed using a single sample on Solid (Mitra et al., 2013) and Illumina platforms (Ranjan et al., 2016). The use of synthetic samples has also been exploited to compare the two approaches (Jovel et al., 2016). Such comparisons showed that the 16S rDNA amplicon sequencing approach yields quantitatively and qualitatively different results compared to metagenomics. In a recent article, we compared the performance of metagenomics and 16S amplicon sequencing using Illumina platform by using DNA isolated from human fecal samples, showing that metagenomics outperforms 16S rDNA amplicon sequencing (Laudadio et al., 2018).
This expert review article evaluates how metagenomics is improving our knowledge of microbiota composition from environmental to human samples. Furthermore, we offer an analysis of the common technical and methodological challenges and potential pitfalls arising from metagenomics approaches, such as metagenomics study design, data processing, and interpretation.
Current Approaches and Future Challenges in Metagenomics
From experimental design to genome assembly
A major bottleneck of metagenomics NGS is the ability to translate the data into relevant information, to obtain clinically actionable results. A professional biostatistician needs to be consulted at the time of study design (before samples collection) to programmatically assess statistical power to be achieved and which metadata should be collected and included in the analysis (Fig. 1). Overlooking the relevance of this step will invariably lead to difficulties in the subsequent analysis of data and negatively affect the relevance of any finding. Furthermore, confounding factors such as diet, environment, and social behavior, which may affect human microbiota, have to be considered. To avoid biases due to cultural or social issues, it is recommended that broad cohorts of patients (and controls) are analyzed, possibly through large consortia, allowing to integrate in a single study cohort of patients from different continents.
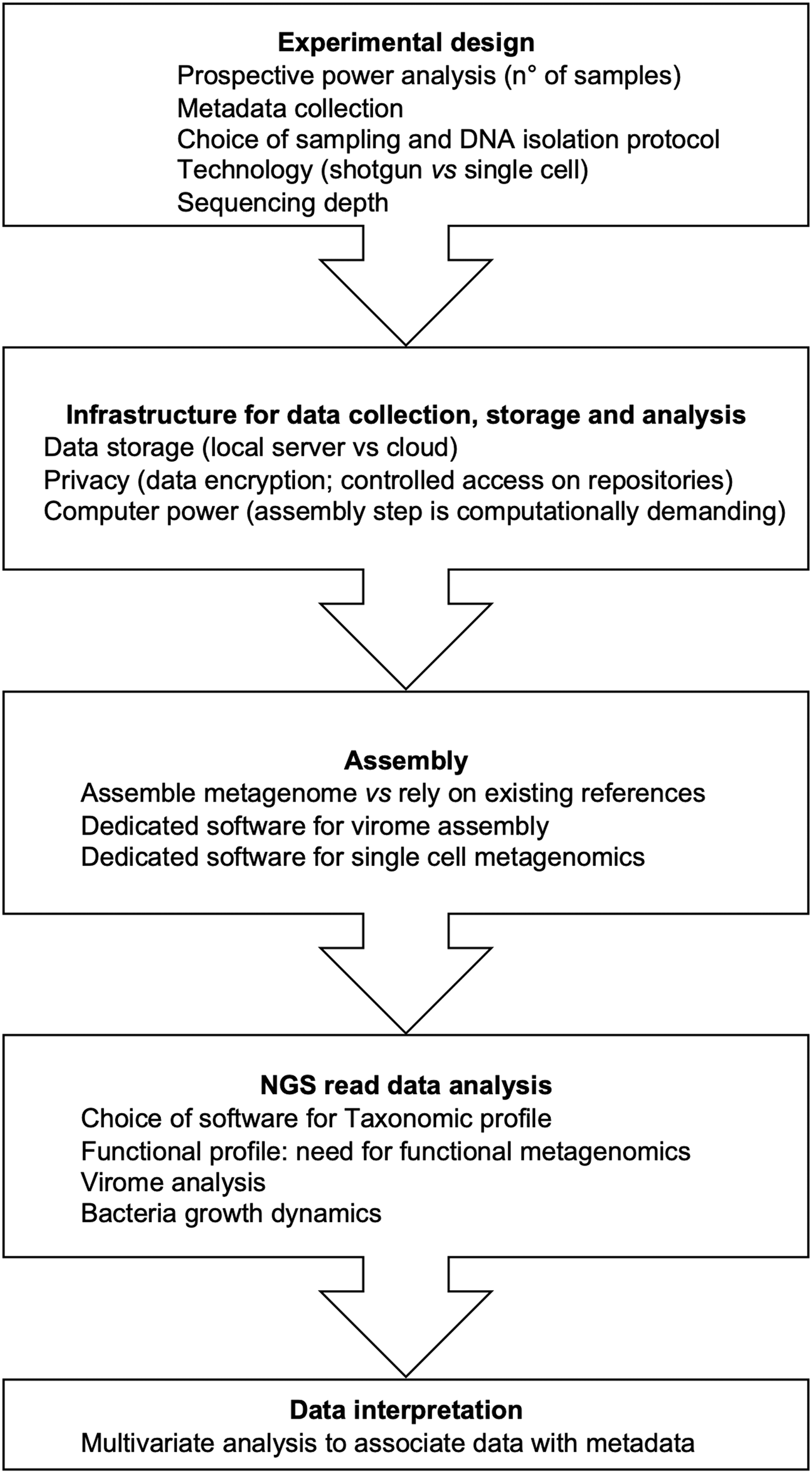
Typical metagenomic study workflow: key choices and critical parameters.
One of the most relevant constraints of metagenomics lies in the limited annotation of bacterial genomes. Human gut microbiome metagenomics took advantage of seminal studies, which allowed a very deep coverage on hundreds to thousands of samples and resulted in comprehensive coverage and extensive annotation of human gut bacterial genomes (Li et al., 2014; Pasolli et al., 2019; The Integrative Human Microbiome Project, 2014). On the other hand, metagenomes from poorly exploited environmental niche are still inadequately characterized (Uritskiy and DiRuggiero, 2019); hence, databases offer only limited coverage for alignment of reads. In these instances, the first challenging step is the assembly of a reference metagenome to use in the analysis.
Assembly is a key step of the analysis pipeline as all subsequent output will depend on its outcome. Some features of metagenomic data (i.e., uneven coverage across species, highly similar sequences in unrelated species due to horizontal gene transfer, and repeated sequences) require dedicated software. Binning co-abundant sequences before assembly has been proven to overcome limitations due to uneven coverage (Plaza Oñate et al., 2018).
Several different strategies can be exploited (Ghurye et al., 2016), and a wide array of software to assemble metagenomes have been developed and tested on both real samples (Olson et al., 2017; Wang et al., 2019) and synthetic bacterial communities (Greenwald et al., 2017). It has been shown that virome analysis (a branch of metagenomics focusing on bacteriophage communities) is particularly affected by the choice of the assembler software used (Sutton et al., 2019).
Metagenomics analysis: one raw reads dataset, several layers of information
Through the use of several available tools, bioinformatics analysis of metagenomics datasets can provide information at different levels. Even though, metagenomics datasets are most commonly investigated to achieve information on the taxonomic and functional profiles of a microbiome, tools to investigate the virome (Nooij et al., 2018; Ogilvie and Jones, 2015; Rampelli et al., 2016), the replication rates of bacteria (Korem et al., 2015), and the profile of antibiotic resistance (Rowe and Winn, 2018; Yang et al., 2016) have also been set up.
In particular, virome investigation through metagenomics has revolutionized the field of virology, disclosing a wealth of putative novel viruses (Schulz et al., 2018; Simmonds et al., 2017). Moreover, bacteriophages have recently emerged as key remodelers of microbial host communities, influencing the bacterial diversity, facilitating nutrient turnover, and conferring antibiotic resistance genes through horizontal transfer of genetic material (Modi et al., 2013; Ogilvie and Jones, 2015; Reyes et al., 2013).
Taxonomic profiles are obtained by mapping reads on a database of reference bacterial genomes. Most software rely on a small selection of genes (markers) to obtain a taxonomic profile (Nayfach et al., 2016; Segata et al., 2012; Truong et al., 2017). The choice of the selection of marker genes allows users to obtain taxonomic profiles with relatively low computing power.
Of note, contamination-free collection, homogenization, storage, and a subsequent efficient DNA extraction method have been shown to significantly affect the taxonomic profile yielded (Brooks et al., 2015; Wesolowska-Andersen et al., 2014). Several studies documented that contaminant DNA and cross-contamination can critically influence NGS-based microbiome analyses (Salter et al., 2014; Sinha et al., 2015). Contaminant DNA appears to originate from reagents, laboratory environments, human commensals on laboratory personnel, and sample processing.
Indeed, for the Human Microbiome Project, a rigorous study protocol and standardized instructions for body site sampling and specimen processing were set up (Aagaard et al., 2013). Moreover, Panek et al. (2018) described the influence of storage condition and sample extraction in detection and composition of the fecal bacterial community.
Functional metagenomics: from microbial community biodiversity to functional processes
Functional profiling aims at achieving a comprehensive view of the functions of proteins encoded by the metagenome. Such analysis is extremely relevant for both ecologic and clinic purposes, as it highlights the interplay between a specific microbial community and the environment. Metabolomic analyses are the natural complement of a functional profiling, allowing the researcher to correlate alteration in the abundance of specific metabolites and that of taxa and genes related to those metabolites (Frankel et al., 2017; Liu et al., 2017).
16S amplicon sequencing allowed functional profiling by assuming that it is the sum of the functional profiles of all the identified species (Aßhauer et al., 2015; Langille et al., 2013). Otherwise, metagenomics yields functional profiles through direct identification of the genes encoded by a metagenome rather than inferring them based on taxonomy. Furthermore, most pipelines can assign a function not only to reads mapping to annotated genes but also to a fraction of the reads that cannot be precisely assigned to an annotated genome. This is exerted through in silico translation of the DNA sequences, followed by homology search on protein sequence databases. This process, called “translated search,” allows the software to ascribe a putative function to the protein encoded by a DNA fragment (Buchfink et al., 2015; Franzosa et al., 2018; Huson et al., 2016).
While extensive annotation of the gut microbiome allows binning of most reads in gut microbiota, in environmental metagenomics, most reads cannot be assigned to any function, due to lack of annotation and poor knowledge of the function of most polypeptides encoded by microorganisms (Quince et al., 2017), reflecting the current bias of the annotation toward a small number of cultivable microorganisms. Functional metagenomics, aiming at isolation, cloning, and expressing genes into suitable model organisms to identify their function will be required to achieve a comprehensive annotation of microbial genes (Santana-Pereira and Liles, 2017), thus improving the resolution of functional profiles obtained through metagenomics.
An extremely relevant subfield of functional metagenomics is related to antibiotic resistance, aiming at the identification of genes that confer antibiotic resistance to microorganisms. By leveraging information collected in this field, it is possible to characterize the “resistome” in both environmental (D'Costa et al., 2006) and human samples (van Schaik Willem, 2015) with relevant applications in built environments such as hospitals (Mahnert et al., 2019).
In the past decade, transformation of biomedical big data into valuable knowledge has been a fundamental challenge in bioinformatics, and a significant role in the success of bioinformatics exploration from biological data came from machine learning and deep learning applications. The intrinsic characteristics of several steps of metagenomic analyses are suitable of being addressed through machine learning and neural networks. In fact, such methods have been exploited for metagenome assembly (Afiahayati et al., 2015; Ji et al., 2017; Wang et al., 2015), metagenomic reads binning to reference genomes (Vervier et al., 2018), gene prediction (Al-Ajlan and El Allali, 2018), gene function prediction (Li et al., 2018), and analyses of large datasets (LaPierre et al., 2019; Pasolli et al., 2016).
Big data storage and analysis considerations
The large amount of data typically generated by metagenomic opens two major challenges: data storage and computational power. While some tasks such as taxonomic analysis with marker genes can be achieved with limited resources (assuming that an appropriate reference metagenome is already available), more complex tasks (metagenome assembly, functional profiling, and gene discovery) are much more demanding in terms of computational resources. Cloud computing can certainly provide an affordable solution; indeed, several server implementations of metagenomics pipelines are available (Lee et al., 2018; Mitchell et al., 2018; Raknes and Bongo, 2018), while other packages are released as images installable on cloud computing servers (McIver et al., 2018).
Privacy concerns may arise when human microbiome data are being analyzed and stored. In fact, several seminal studies highlighted that precise identification of individuals by means of metagenomics is feasible (Franzosa et al., 2015; Leake et al., 2016; Schmedes et al., 2017). Therefore, this issue started to be addressed by implementation of metagenomics analysis using secure computation (Wagner et al., 2016).
Conclusions and Future Outlook
Shotgun metagenomics has rapidly expanded our understanding of environmental and clinical microbial communities, allowing to address previously unattainable biological questions as well as accelerating genome-based discovery of novel microbial genes (Santana-Pereira and Liles, 2017). One of the most intriguing fields propelled by metagenomics data will be functional metagenomics. The development of medium- to high-throughput tools to characterize the thousands of novel genes encoded by bacterial and viral genomes will likely result in a collection of exploitable enzymes, whose applications will range from biotechnology to material science and pharmacology. Investigation of the secondary metabolites produced by bacteria will also represent an invaluable resource (Table 1).
How Metagenomics Changed Our Approach to Microbiology and Future Directions
In the last decade, according to PubMed data (Mesh terms search), 4227 scientific reports have been published on “metagenomics,” 1163 of which attain the “microbiome, human.” Indeed, characterization of the human microbiome unveiled that microbial communities associated with human body sites are dynamic and subject to impressive modification in the course of host life and in response to many factors, including the interaction between human, animal, and environment. Changes in the microbiome have been associated with disease states, and in some instances, causal links were highlighted between microbiota and specific pathological conditions.
Accordingly, the “One Health” concept underlines the inseparable ecological relationships between human, animal, and environmental, recognizing that the health of people is connected to the health of animals and the environment. Indeed, the “One Health” Commission declared that nearly 75% of emerging human infectious diseases in the past three decades originated in animals (Murtaugh et al., 2017). The development of high-throughput tools to characterize entire microbial communities now offers new insights toward “One Health” concept. Actually, metagenomics approach will integrate the knowledge of the complex interactions from these three domains, open the potential for novel diagnostic tools, and pave the way to collaborative and integrated multidisciplinary approaches to treatment and intervention.
Most recent technological developments allowed single-cell metagenomics (Stepanauskas and Sieracki, 2007; Stepanauskas et al., 2017). This innovative approach overcomes some relevant limits intrinsic to the shotgun sequencing, as assembly of a genome from fragments arising from a single cell is computationally much less demanding. In fact, while metagenome assembly from shotgun sequencing requires multiple comparisons between many fragments, which in most cases arise from different genomes, single-cell metagenomics assembly takes advantage of smaller datasets of DNA reads, all arising from a single cell, allowing also a much easier parallelization of the assembly process.
Comparison of the two methods not only provided an implicit independent confirmation of the current metagenomics assembly methods but also highlighted a thus far overlooked variability between single cells belonging to the same taxon (Alneberg et al., 2018). Nevertheless, further improvements and widespread application of single-cell metagenomics are required to fully unleash the potential of this approach.
Many high-throughput techniques have been developed through step-wise improvements and have required, at some point, a standardization effort to allow proper comparison of data produced by different groups (Brazma et al., 2001; Taylor et al., 2007, 2008). The metagenomics community is currently setting up reliable and consistent standard procedures for sample collection, storage, and processing, data analysis, and metagenome reporting (Bowers et al., 2017; Roux et al., 2018). Further effort to standardize metagenomics database will be necessary to support consistent and productive development of the field.
Footnotes
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.