
Abstract
Currently, mass spectrometry-based data-dependent acquisition protocols require several micrograms to milligram amounts of proteins to start with, and needs fractionation and enrichment or depletion protocols to identify low abundant proteins and their modifications. However, a data-independent acquisition (DIA) approach can help us to identify a large number of proteins irrespective of their abundance, from even a very low amount of protein. In the DIA protocol, mass spectrometry data are matched against a previously established tandem mass spectrometry (MS/MS) spectra for each peptide. Therefore, establishing a spectral library is a prerequisite for successful DIA protocol. However, the DIA protocol becomes extremely important to investigate biological systems, where there is a difficulty in gathering reasonable amounts of proteins. In this context, DIA can become a valuable tool to investigate proteome dynamics of slow growing pathogen such as Mycobacterium tuberculosis that causes tuberculosis. We report here a case study of the DIA approach that is ideal for M. tuberculosis, which cannot be scaled up easily as it requires specific BSL3 laboratory facilities to be grown. We generated a spectral library for M. tuberculosis proteome using six publicly available proteomic data sets. The in-house M. tuberculosis proteome spectral library contains MS/MS spectra for peptides corresponding to 88% of proteins when compared with the M. tuberculosis H37Rv proteome. We believe that the public availability of the M. tuberculosis spectral library is an important step forward to facilitate the research community to adopt DIA approaches, for example, to investigate M. tuberculosis proteome with greater depth and efficiency.
Introduction
Data-independent acquisition (DIA) is a powerful technique in which the mass spectrometer systematically acquires tandem mass spectrometry (MS/MS) spectra irrespective of precursor ion intensity and has been an alternative approach to data-dependent acquisition (DDA) (Gillet et al., 2012; Stahl et al., 1996; Venable et al., 2004). The selection of peptide ions in a DDA approach often depends on an abundance of precursor ions, before these are taken forward for fragmentation and further identification (Craft et al., 2013).
With this approach, efforts to increase whole proteome coverage have been focused on increasingly rigorous fractionation and enrichment of complex tryptic peptide mixtures before mass spectrometry analysis, which requires larger amounts of proteins from the samples to start with (Swaney et al., 2010). Such reliance on fractionation and enrichment can be jettisoned by adopting a DIA approach to identify low-abundant proteins and post-translational modifications (PTMs). With the DIA approach, one can select more peptide precursors than DDA and theoretically acquire MS/MS spectra in a sequential manner (Bruderer et al., 2017).
This might help in distinguishing peptide forms with identical parent mass, very similar chromatographic retention time, and only small differences in fragmentation pattern. The widely used analysis techniques for DIA data are based on spectral libraries to analyze extracted ion chromatograms to test for the presence and quantify the abundance of peptide ions in the reference library (Deutsch et al., 2018). Sequential window acquisition of all theoretical fragment ion spectra mass spectrometry (SWATH-MS) is a DIA technique that combines the multiplexing ability of shotgun proteomics with the high-precision data analysis of selected reaction monitoring and can quantify proteomes using single-shot MS/MS analysis (Kind et al., 2018; Zhu et al., 2020).
Mycobacterium tuberculosis, a pathogen causing tuberculosis, remains one of the biggest global bacterial causes of death (Zhang et al., 2017). M. tuberculosis is the leading cause of mortality among infectious diseases and claims 1.8 million lives annually (Calder et al., 2015). Whole-genome sequences of several clinical isolates and established type strains have been reported. As a result, a large number of novel single nucleotide variations are being reported in known drug-resistant genes in addition to previously reported drug-resistant mutations (Advani et al., 2019).
As the next frontier to genomics, mass spectrometry-based proteomic studies are uniquely poised to contribute to our understanding of this pathogen, which may ultimately lead to better management of the disease (Cohen et al., 2019). The M. tuberculosis proteome has been investigated using proteomics for over a decade, with increasingly sophisticated mass spectrometry technology and sensitive methods for comparative proteomic profiling.
The discovery of low-abundant proteins, altered expression, sequence variations, and multiple PTMs (multi-PTMs) in different strains may open up new avenues of diagnosis and therapy as well as contribute to deciphering pathogenesis. However, the challenge to meet a requirement of micrograms to milligram amounts of proteins for sample preparation in a typical in-depth proteomic and multi-PTMs experiments poses a challenge to streamline alternative methods, which do not require larger amounts of proteins to start with.
DIA-based proteomic data analysis depends on a spectral library with a high-proteome coverage that, in several cases, is built on MS/MS spectra from DDA-based analysis of the same sample. A spectral library is a collection of peptide fragment ions assigned to a precursor. Searching the MS/MS-derived proteomics data against the spectral library is the approach in peptide identifications (Lam, 2011; Schubert et al., 2015a). It is difficult to scale up culturing of M. tuberculosis as it is a slow-growing organism and needs specific biosafety requirements, the DIA-based approach can be a convenient method to study the dynamics of the M. tuberculosis proteome.
Therefore, as the first step toward this, we generated a spectral library of M. tuberculosis by integrating proteomic data from six previously derived raw MS/MS data sets on M. tuberculosis proteome. We subjected these raw MS/MS data sets to proteome search using the Proteome Discover platform to fetch MS/MS spectra for M. tuberculosis H37Rv and H37Ra strains for building a spectral library. We believe that such a ready availability of a spectral library will help biomedical researchers to carry out M. tuberculosis proteomic analysis in DIA mode without the need for building a spectral library as a prerequisite.
Materials and Methods
Data description and availability
The mass spectrometry raw data are publicly available at ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org). These data sets were downloaded from the PRIDE partner repository (https://www.ebi.ac.uk/pride/archive/). A total of six data sets were used for the study. The PRIDE accession IDs for respective data sets are PXD001188 (Jhingan et al., 2016), PXD004161 (Verma et al., 2017), PXD006039 (Cortes et al., 2017), PXD006117 (Yimer et al., 2017), PXD008555 (Pinto et al., 2018), and PXD009549 (Nieto et al., 2018). The list of data sets is given in Table 1. The respective data sets in the study were acquired in the DDA mode using Orbitrap series mass spectrometers and further used to search the proteomic data.
List of the Mycobacterium tuberculosis Proteomic Studies Chosen for Building a Spectral Library
Description of data acquisition
PXD001188
Jhingan et al. (2016) have acquired this data set using the EASY-nLC system (Thermo Fisher Scientific) coupled to LTQ Orbitrap-Velos mass spectrometer (Thermo Fisher Scientific). Data analysis was carried out using MaxQuant (MQ) software (version 1.4.1.2) using the M. tuberculosis UniProt reference proteome database.
PXD004161
Verma et al. (2017) have analyzed the samples on Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) interfaced with EASY-nLC II nanoflow liquid chromatography system (Thermo Scientific, Odense, Denmark). Raw data files were processed in Proteome Discoverer software version 2.0.1 using SequestHT and Mascot search algorithms (Thermo Fisher Scientific) and searched against the databases for H37Rv and H37Ra downloaded from Tuberculist (Release March 27, 2013) and NCBI (updated December 12, 2012).
PXD006039
Cortes et al. (2017) have analyzed the data sets on a nano-liquid chromatography (nano-LC) system (Eksigent Technologies) connected to an LTQ Orbitrap XL mass spectrometer equipped with a nanoelectrospray ion source (Thermo Fisher Scientific). The acquired MS2 spectra were searched with OMSSA47 and XTandem48 against an M. tuberculosis H37Rv protein database (TubercuList v2.6).
PXD006117
Yimer et al. (2017) have analyzed the samples using a Q Exactive hybrid quadrupole–orbitrap instrument interfaced with an EASY 1000-nano-LC electrospray ion source (Thermo-Fisher Scientific, Biberach, Germany). The MS/MS data analysis was performed using the MQ software package (version 1.4.0.5; Cox and Mann, 2008) with its integrated Andromeda search algorithms, searched against the M. tuberculosis H37Rv reference proteome UP000001584 (UniProt-proteome).
PXD008555
Pinto et al. (2018) have subjected the samples to LC-MS/MS analysis using Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific) coupled to EASY-nLC 1200 nanoflow ultra-high performance liquid chromatography (UHPLC) (Thermo Scientific). The data were searched using SequestHT and Mascot search algorithms against the H37Ra protein database (downloaded from NCBI, updated May 2016) using Proteome Discoverer software version 2.1 (Thermo Fisher Scientific).
PXD009549
Nieto et al. (2018) performed all the experiments using the OrbitrapVelos MS (Thermo Scientific, USA) coupled with the instrument EASY-nLC II nanoflow high performance liquid chromatography (HPLC) (Thermo Scientific). All MS/MS data were analyzed using Sorcerer2™ integrated data analysis platform (version 5.0.1; Sage-N Research, Milpitas, CA, USA) and SequestHT (version v. 3.5; Thermo Fisher Scientific, San Jose, CA, USA). The raw data were searched against the M. tuberculosis strain H37Rv Tuberculist database, version R25.
MS/MS data retrieval and processing using Proteome Discoverer software
The proteomic database search was performed in Proteome Discoverer v2.2 software using SequestHT and Mascot V.2.4 search algorithm. Mass spectrometry data were searched against a nonredundant proteome database comprising proteins from M. tuberculosis H37Rv and H37Ra (6526 proteins). The M. tuberculosis H37Rv was downloaded from Mycobrowser release 3 consisting of 4098 protein sequences, whereas proteins belonging to M. tuberculosis H37Ra were downloaded from NCBI that consisted of 4992 protein sequences. The search parameters included trypsin as a proteolytic enzyme with a maximum of two missed cleavages. Cysteine carbamidomethylation was specified as static modification and acetylation of protein N-terminus and oxidation of methionine were set as dynamic modifications, as mentioned in Table 2.
The Parameters Used for Searching Raw Data Sets Using Proteome Discoverer v2.2 Software
M. tuberculosis spectral library generation
The spectral library was created using the Bibliospec tool (version 1.0) with the .msf files generated from the total proteome search (Frewen and Maccoss, 2007). The output files (“.blib”) from Bibliospec were visualized using the Skyline (21.1.0.146) (MacLean et al., 2010) tool. The detailed workflow of the generation of the spectral library is shown in Figure 1.
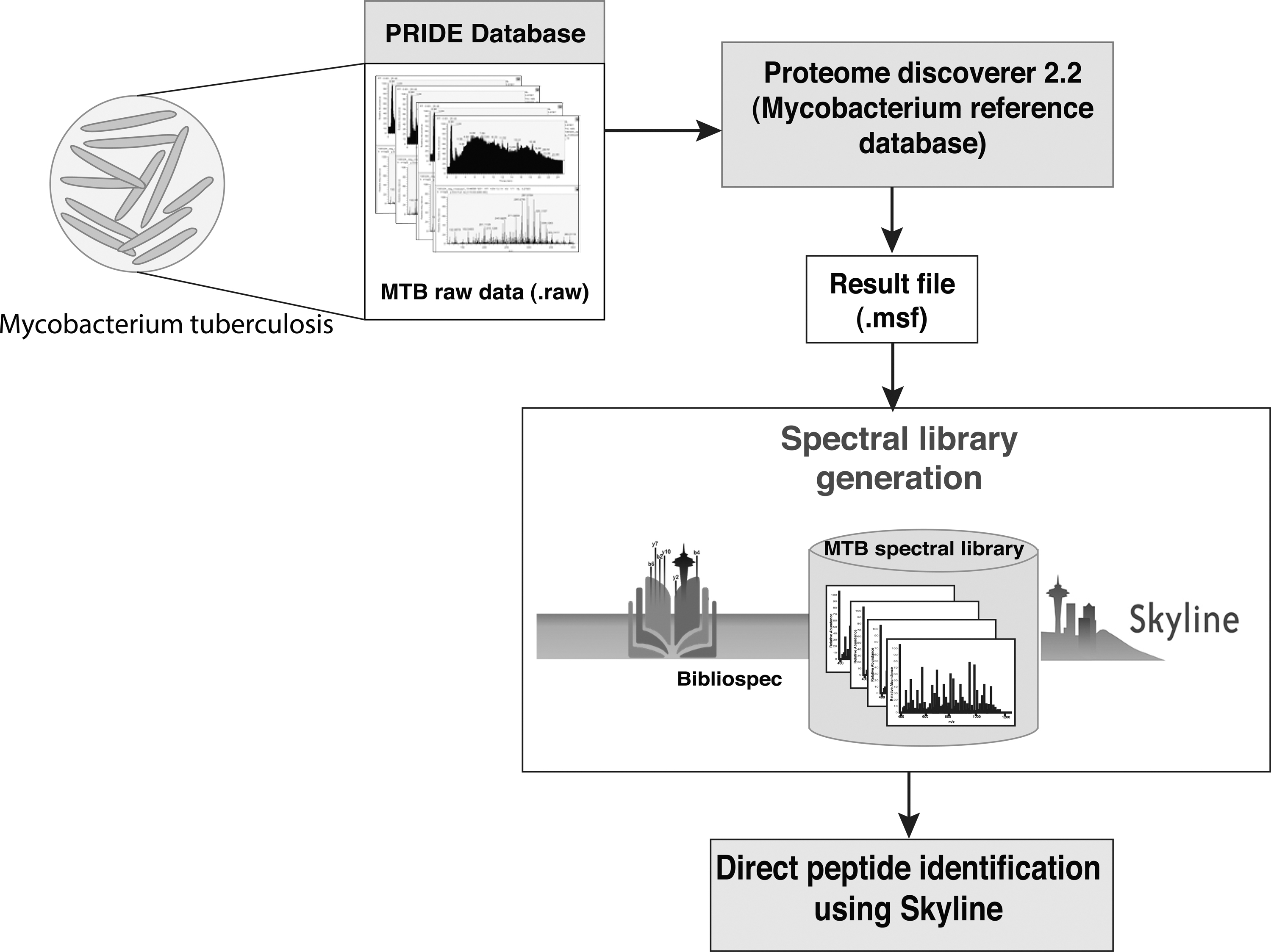
The workflow illustrating the development of spectral library in strains of Mycobacterium tuberculosis. The M. tuberculosis raw data obtained from PRIDE were searched for total proteome against the combined H37Rv and H37Ra reference database and the resulting .msf was used for building the spectral library using Bibliospec and Skyline.
Peptide precursors from the in-house generated M. tuberculosis spectral library were extracted with charge (z) details. This list was modified to a Prosit (https://www.proteomicsdb.org/prosit/) compatible comma-separated values (.csv) file format. This step was performed to remove the redundancy in spectral libraries since the spectral library was generated using publically available M. tuberculosis proteomic raw data acquired in different Orbitrap series instruments. The collision energy was fixed to 30 for all the peptide precursors (z = 2–6) with 7–30 amino acids and the .csv format file was subjected to in silico peptide spectral library generation using Prosit (https://www.proteomicsdb.org/prosit/). The prediction was performed using “Prosit_2020_intensity_hcd” and “Prosit_2019_iRT” intensity prediction and indexed Retention Time (iRT) prediction models, respectively.
Spectral library search of M. tuberculosis SWATH data
The publically available M. tuberculosis SWATH proteomic raw data files published by Schubert et al. with identifier PASS00655 (www.peptideatlas.org/PASS/PASS00655) were downloaded (Schubert et al., 2015b). The spectral library search of SWATH proteomic data of M. tuberculosis was performed using Skyline against the Prosit generated in silico peptide spectral library. First, the Prosit in silico peptide spectral library was indexed to Skyline document peptide setting parameters such as Enzyme: Trypsin [KR|P], maximum missed cleavages: 2, background proteome: M. tuberculosis H37Rv (mycobrowser V3). Biognosys-11 (iRT-C18) was selected as iRT standards for the retention time predictor calculator.
The minimum and maximum peptide lengths were set to 7 and 50, respectively. Carbamidomethylation of cysteine (C) was set as fixed modification while oxidation of methionine (M) and acetylation of N-termini as variable modifications. The spectral library peptides were associated with proteins with the “Add to only the first matching protein” option. An equal number of decoy peptides were generated corresponding to all the target peptides from a spectral library by reversing the peptide sequence.
Later, the raw SWATH data files (.wiff format) were imported into the Skyline document with transition setting parameters such as peptide precursor charges: 2–6; ion charges: 1–2; ion types: y, b, and p; product ion selection: “ion 3”; to “last ion”; and special ions: N-terminal to proline. DIA precursor window for exclusion was selected. Ion match tolerance was set to 0.05 m/z, from the filtered product ions of library spectrum, the most intense minimum three to maximum six product ions were selected. Min m/z and Max m/z of 100 and 2000 were also set.
Under the FullMS tab MS1 filtering options such as, isotope peaks included count, precursor mass analyzer: time of flight (TOF), peaks: 3, resolving power: 30,000 were set and MS/MS filtering parameters such as-acquisition method: DIA, product mass analyzer: TOF, isolation scheme: 25 m/z × 34 with 0.5 m/z margins for the range 400–1200, resolving power: 30,000 were set. The high selectivity extraction option was selected for scans within 5 min of predicted retention time (RT).
Once all the raw files were imported into the Skyline document, iRT values were calculated for all the peptides in the raw files. Peptide precursors with less than six transitions were discarded. Peptide precursors were reintegrated using decoy trained mProphet model. Finally, target peptide precursors with Q-value <0.01 and corresponding proteins were exported out from the Skyline document.
Data availability
The M. tuberculosis DIA spectral library is available at MassIVE repository.
Results and Discussion
Spectral library preparation using Bibliospec
The spectral libraries generated from the six data sets result in the identification of 11,308 peptides corresponding to 2252 proteins from PXD001188; 8849 peptides corresponding to 2582 proteins from PXD004161; 6579 peptides corresponding to 1445 proteins from PXD006039; 23,224 peptides corresponding to 3191 proteins from PXD006117; 23,670 peptides corresponding to 3234 proteins PXD008555; and 16,287 peptides corresponding to 2776 proteins from PXD009549, respectively. The identified precursor and transitions along with their corresponding proteins and peptides information are given in Table 3.
Number of Proteins, Peptides, Precursors, and Transitions from the Six Data Sets Using Skyline
Generation of a merged spectral library
The spectral libraries created from the six data sets were merged into one single spectral library using Bibliospec. Bibliospec chooses the spectrum that has the highest score to go into the merged library. The developed spectral library was visualized using Skyline to extract the information associated with proteins and peptides from these data sets with the protein database combined for M. tuberculosis H37Rv and H37Ra. The spectral library contained a total of 899,393 transitions belonging to 106,889 precursors of 81,474 peptides corresponding to 4705 proteins. This M. tuberculosis spectral library from six data sets was able to give coverage of 88% of M. tuberculosis H37Rv reported proteome. Merging of the spectral library from different data sets provided a larger coverage for peptide identifications.
Peptide precursor identifications
Peptide detectability is defined as the probability that a peptide is identified in an LC-MS/MS experiment and has been useful in protein identification. The fragmentation of the peptides can be used to improve peptide identification by combining multiple spectra for the same peptide. The spectral library generated consists of a total of nonredundant 106,889 precursor ions corresponding to 81,474 peptides obtained from six data sets (Supplementary Table S1).
There was a total of 5013 precursors lying within the m/z range of 200 to 400; 36,013 precursors with m/z in the 400–600 range. Similarly, 32,013 precursor ions fall in the range of 600 to 800 precursor m/z, whereas 19,280 precursor ions fall in the range 800 to 1000. The peptides covered the range from 200 to 2000 precursor m/z, respectively. A 52.8% of the peptides were falling within the 200 to 800 m/z range depicting higher confidence in the fragmentation of peptides (Fig. 2).
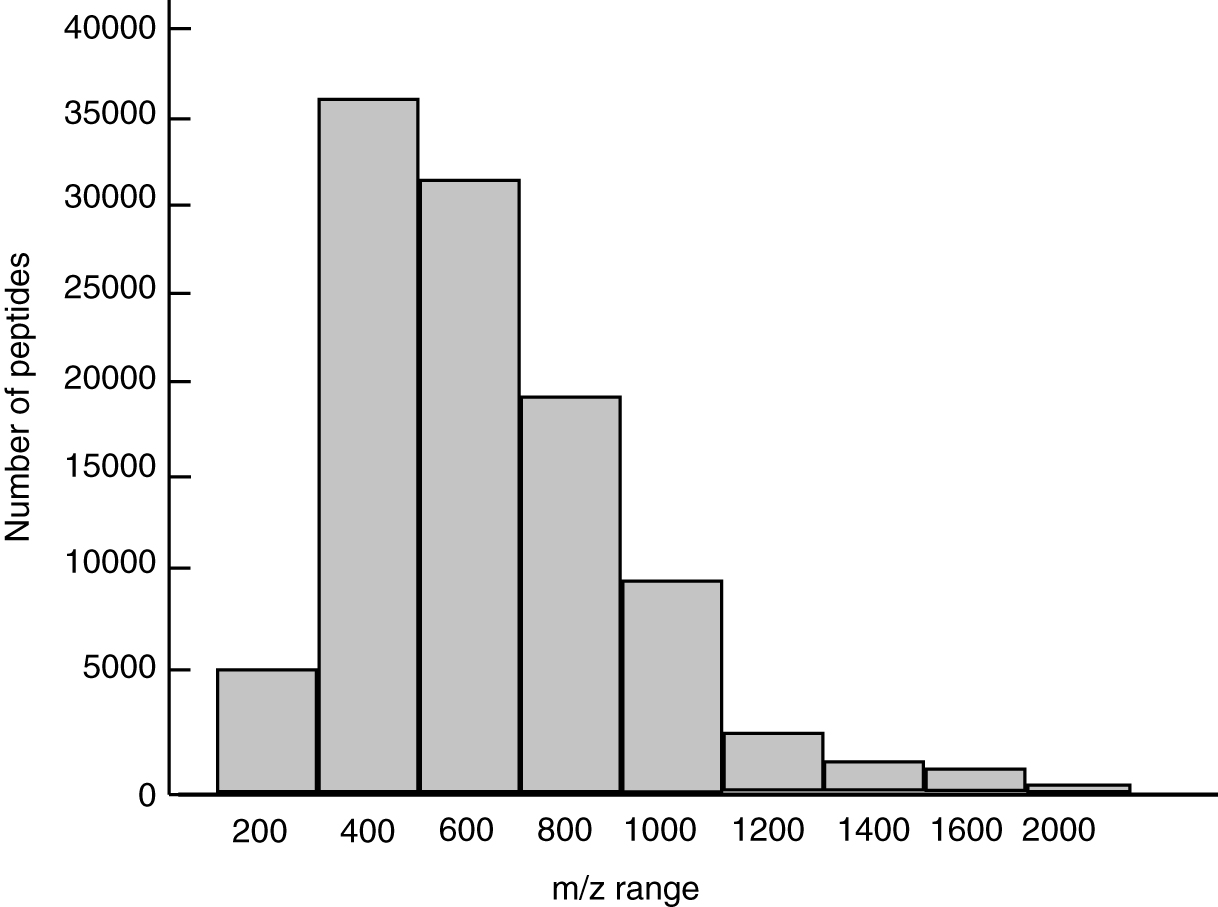
Histogram depicting the number of precursor peptide m/z in the merged spectral library.
Peptide precursor charge state
Precursor charge state is an important input to MS/MS sequence database search tools, the ability to accurately determine the precursor charge is helpful for the identification process (Sharma et al., 2010). There was a total of 47,093 peptides with precursor charge state 2, 46,085 peptides with the charge state 3, 11,645 peptides with the charge state 4, 1816 peptides with the precursor charge state 5, 233 peptides having precursor charge state 6, and 17 peptides having a precursor charge state 7, respectively. A total of 98.0% of the peptides are lying among charge states 2 to 4 (Fig. 3).
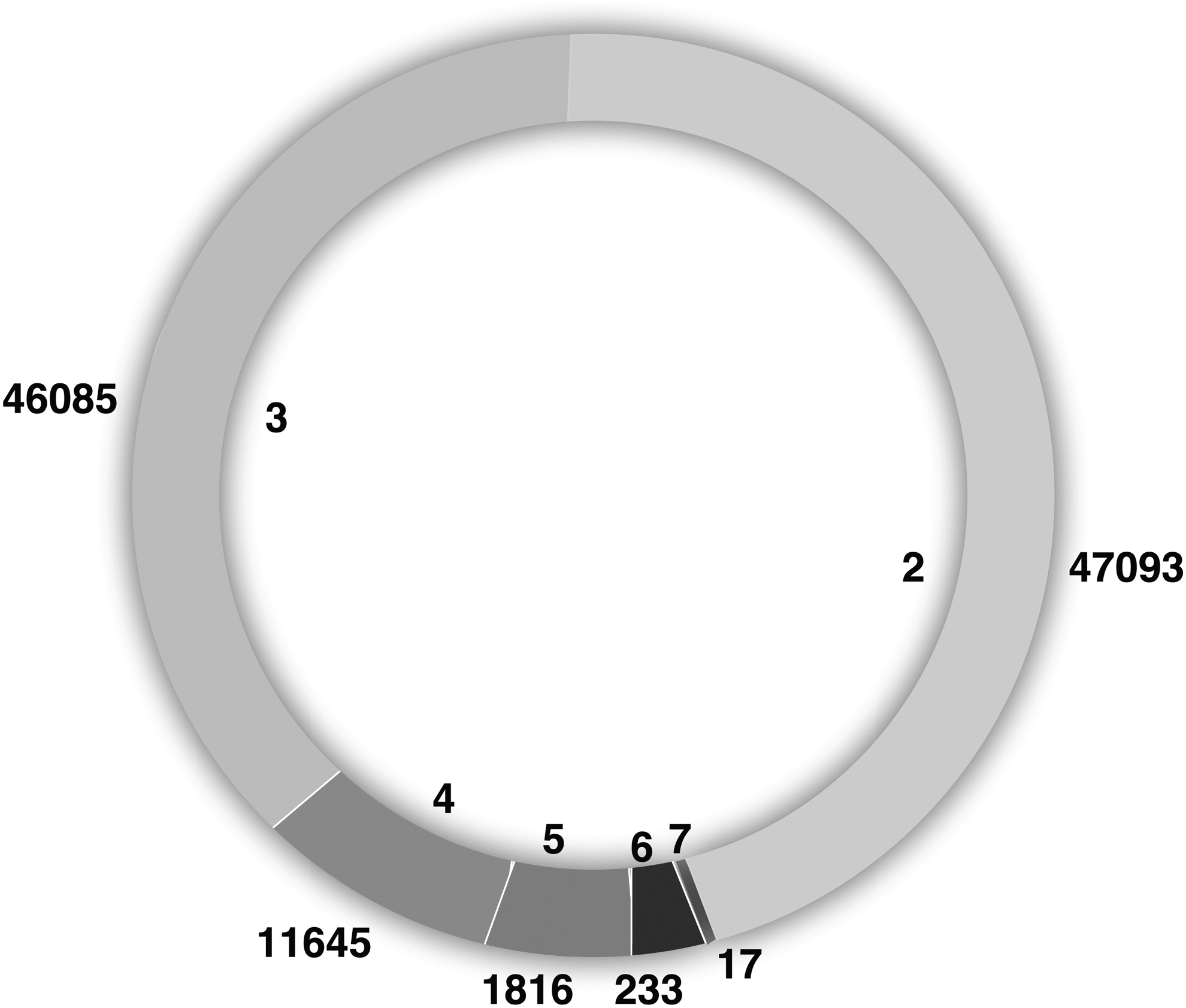
Pie chart showing number of peptides corresponding to precursor charge state.
Peptide sequence length
The merged M. tuberculosis spectral library generated had a peptide sequence length varying from 6 to 60 amino acids. The M. tuberculosis H37Rv spectral library consists of 93.7% peptides having at least one peptide of suitable length (7–35 residues) for mass spectrometry sequencing technology (Fig. 4).
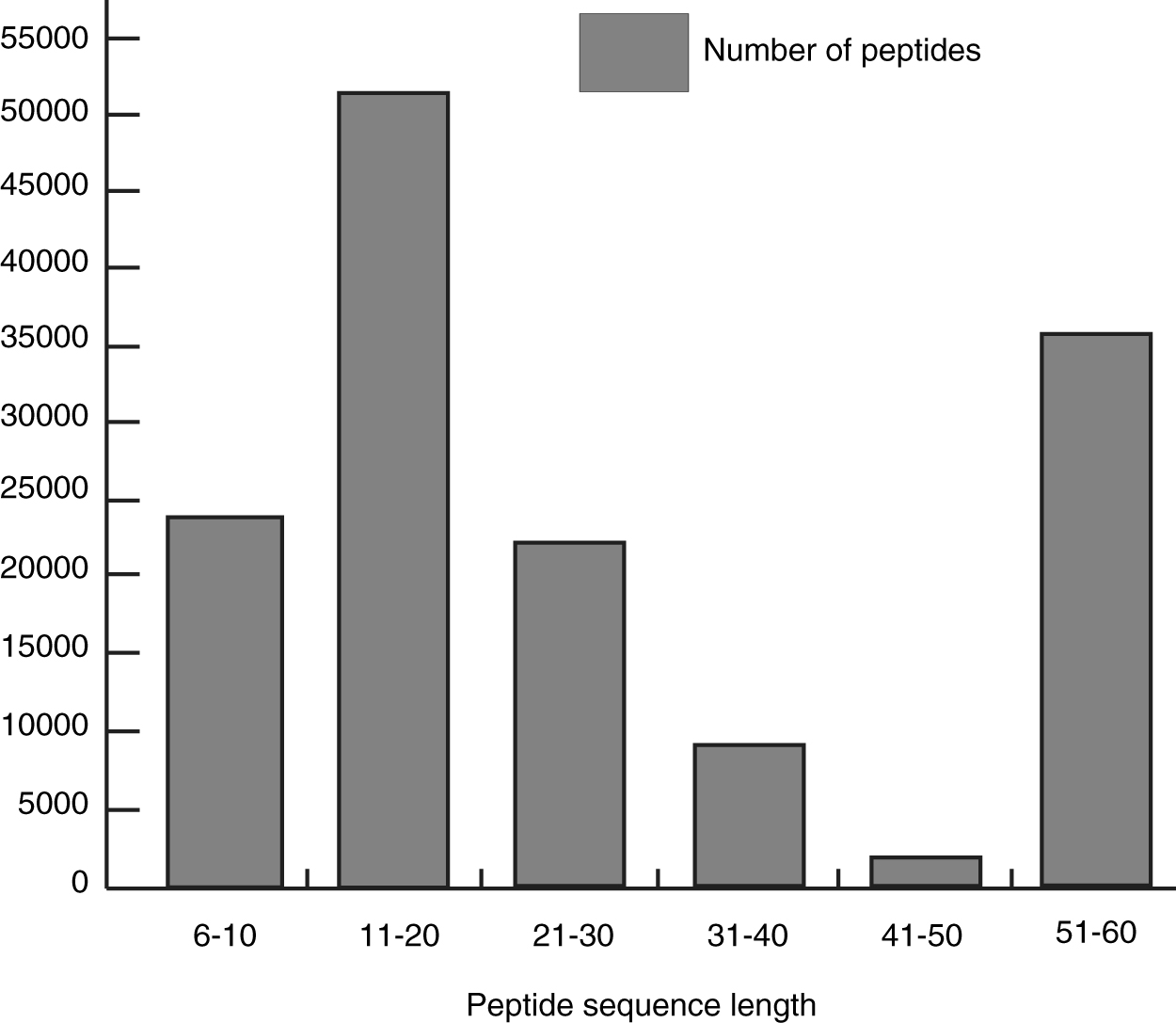
Histogram showing the number of peptides having a respective peptide sequence length.
Comparison of the spectral library generated in-house against the M. tuberculosis SWATH data
Schubert et al. (2015b) have identified 2458 proteins from M. tuberculosis using the SWATH approach using a triple TOF mass spectrometer. The same raw data were subjected to DIA analysis using our in-house spectral library to evaluate the effectiveness of DIA analysis using our spectral library. The in-house M. tuberculosis spectral library was converted into the Prosit spectral library to remove the heterogeneity. We identified 31,503 peptides corresponding to 3533 H37Rv proteins with 44,995 precursors. This demonstrates how the same data set results in better identification using a new merged M. tuberculosis spectral library prepared by us.
Building of spectral library provides identification of low abundant peptides and proteins. We compared M. tuberculosis H37Rv proteins from Schubert et al. group, in-house spectral library and proteins identified post-DIA analysis; we found a common of 3484 proteins (88.7%) in data sets depicting a larger coverage of the M. tuberculosis proteome (Fig. 5A, B). The peptide and precursor ion comparison is carried out across the respective M. tuberculosis spectral libraries (Fig. 5C). Peptide level comparison between in-house and Schubert et al. M. tuberculosis spectral library showed an overlap of 26,043 peptides (39.9%) (Fig. 5D). The DIA analysis could identify 87.6% of the total proteins when compared with the H37Rv protein database (version 3) (Fig. 5E).
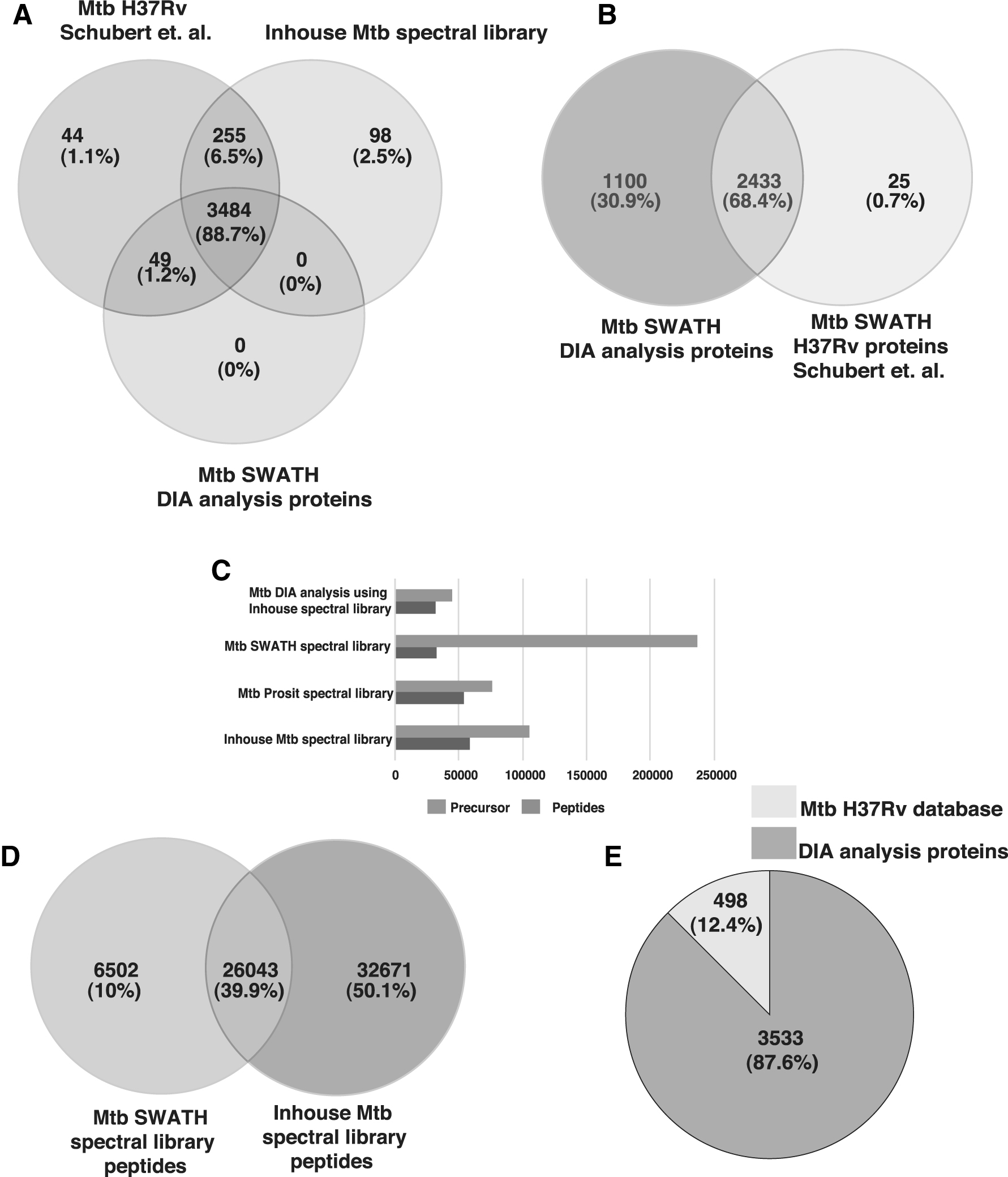
Comparison of the Mycobacterium tuberculosis spectral library.
Conclusions
A spectral library is used for peptide and protein identification in DIA-MS analysis. Spectral libraries remain a substantially underutilized resource in proteomics, with the potential to vastly increase the efficiency of mass spectrometry-based DIA analysis. The data provide information about the M. tuberculosis spectral library generated using high-resolution mass spectrometry. This study also provides composite information regarding precursor ion and peptide sequence length. An in-depth M. tuberculosis spectra can be built by using direct peptide sequences. This study deepens the coverage of the M. tuberculosis H37Rv proteome.
So far, the spectral library approach is limited toward the peptides that are not carrying modifications, except for carbamidomethylation of cysteine (C) was set as fixed modification, whereas oxidation of methionine (M) and acetylation of N-termini as variable modifications. In addition to envisage the M. tuberculosis phosphoproteome and multi-PTM spectral libraries that will aid in proteomics and multi-PTM studies. Recent proof-of-concept studies by Gessulat et al. (2019), and Tiwary et al. (2019) have demonstrated that in addition to DDA-MS-derived spectral libraries, one can also develop virtual spectral libraries without the need for DDA-MS experiments, which will further open up the development of new custom spectral libraries, which will simplify DIA-MS strategies in times to come.
Footnotes
Acknowledgments
We thank “Olav Thon Foundation” for funding toward salaries of K.A. and C.N.K. under a project—Discovering new therapeutic targets and drugs to combat AMR tuberculosis (project no.: 65l530/90305600). We thank Karnataka Biotechnology and Information Technology Services (KBITS), Government of Karnataka for the support to the Center for Systems Biology and Molecular Medicine at Yenepoya (Deemed to be University) under the Biotechnology Skill Enhancement Programme in Multiomics Technology (BiSEP GO ITD 02 MDA 2017). We thank Yenepoya (Deemed to be University), Mangalore, India for access to instrumentation. A.A. is a recipient of the junior research fellowship from the Council of Scientific and Industrial Research (CSIR), Government of India.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
No specific funding was received for this work.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.