
Abstract
The rise of machine learning (ML) has recently buttressed the efforts for big data-driven precision oncology. This study used ensemble ML for precision oncology in breast cancer, which is one of the most common malignancies worldwide with marked heterogeneity of the underlying molecular mechanisms. We analyzed clinical and RNA-seq data from The Cancer Genome Atlas (TCGA) (844 patients with breast cancer and 113 healthy individuals) and the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) (1784 patients with breast cancer and 202 healthy individuals). We evaluated six algorithms in the context of ensemble modeling and identified a candidate mRNA diagnostic panel that can differentiate patients from healthy controls, and stratify breast cancer into molecular subtypes. The ensemble model included 50 mRNAs and displayed 82.55% accuracy, 79.22% specificity, and 84.55% sensitivity in stratifying patients into molecular subtypes in TCGA cohort. Its performance was markedly higher, however, in distinguishing the basal, LumB, and Her2+ breast cancer subtypes from healthy individuals. In overall survival analysis, the mRNA panel showed a hazard ratio of 2.25 (p = 5 × 10−7) for breast cancer and was significantly associated with molecular pathways related to carcinogenesis. In conclusion, an ensemble ML approach, including 50 mRNAs, was able to stratify patients with different breast cancer subtypes and differentiate them from healthy individuals. Future prospective studies in large samples with deep phenotyping can help advance the ensemble ML approaches in breast cancer. Advanced ML methods such as ensemble learning are timely additions to the precision oncology research toolbox.
Introduction
Human cancers are highly complex traits, as with anticancer treatment outcomes. The emerging field of precision oncology calls for individual tailoring of diagnoses and treatments for human cancers so as to acknowledge the vast molecular heterogeneity that underpins cancer biology and response to drugs, radiotherapy, and other health interventions.
Breast cancer is one of the most common malignancies worldwide and the second leading cause of cancer-related deaths in women. It is estimated that 287,850 new cases and 43,250 deaths from breast cancer are expected in the United States in 2022, and the probability of developing invasive cancer within the period from birth to death is estimated to be 12.9% (Siegel et al, 2022). In addition, the incidence of breast cancer in women has continued to increase by ∼0.5% per year since the mid-2000s (Pfeiffer et al, 2018).
At the molecular level, gene expression profiling has transformed our understanding of breast cancer by identifying four predominant molecular subtypes: luminal A (LumA), luminal B (LumB), human epidermal growth factor receptor 2 (HER2) enriched, and basal-like triple-negative breast cancer (TNBC) (Johnson et al, 2021). The substantial differences among these molecular subtypes are a consequence of dramatically altered genomic and proteomic profiles that manifest as changes in activated signaling networks (Gatza et al, 2014) and are reflected in differences in risk factors, incidence, age, disease progression, survival, and response to treatment.
Therefore, there is a clear need for development of innovative strategies to stratify breast cancer into molecular subtypes, not only to assess the prognosis of the disease but also to guide precision/personalized therapeutics.
The rise of machine learning (ML) has recently buttressed the efforts for big data-driven precision oncology. The conceivable and demonstrated applications of ML are wide-ranging in planetary health and clinical oncology. These include efforts for real-time, real-life patient-reported outcomes (PROs) and multiomics data analysis and integration, not to mention capturing of crucial metadata in clinical contexts that can help account for confounding in translational biomarker research.
Moreover, the promising role of ML approaches for the development and application of precision oncology in breast cancer has been suggested in several studies (Ozer et al, 2020). ML approaches have been applied to estimate breast cancer survival and help forecast outcomes with high accuracy (Montazeri et al, 2016; Turkki et al, 2019).
On the other hand, relatively little attention has been paid to molecular classification of breast cancer into molecular subtypes. Studies aimed at investigating the potential of ML for classifying breast cancer cases into molecular subtypes are limited and tend to focus on binary classifications, for example, considering TNBC and non-TNBC subtypes (Wu and Hicks, 2021).
Publicly available population-wide genomic and transcriptomic profiling projects such as the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) (Curtis et al, 2012) and The Cancer Genome Atlas (TCGA) (Ciriello et al, 2015) offer sufficiently comprehensive molecular datasets on breast cancer and thereby help open up new possibilities for discovery of multiomics biomarkers to enable precision oncology.
In this study, we hypothesized that changes at the level of gene expression could lead to new insights to differentiate patients with different breast cancer molecular subtypes as well as from healthy individuals. The present study reports original findings in breast cancer using an ensemble ML approach that harnessed comprehensive clinical and RNA-seq data from METABRIC and TCGA so as to develop a candidate mRNA diagnostic panel.
Materials and Methods
Gene expression datasets
The present study was conducted under the overall oversight of the university research ethics board. Data were obtained from public databases as described in detail below.
Clinical and RNA-seq data from breast cancer patients and healthy individuals were obtained from two population-wide genomic and transcriptomic profiling projects, TCGA (Weinstein et al, 2013) and METABRIC (Curtis et al, 2012).
METABRIC datasets containing reads per kilobase per million mapped (RPKM)-normalized expression data (EGAD00010000210, EGAD00010000211, and EGAD00010000212, n = 1992) for 25,186 probes were obtained from the European Genome–Phenome Archive (https://ega-archive.org) and employed under permission from the ICGC Data Access Committee. After filtering out the missing data, the METABRIC dataset consisted of gene expression profiles of 1784 breast cancer patients (331 TNBC, 240 Her2+, 721 LumA, and 492 LumB) and 202 healthy individuals.
TCGA dataset associated with breast invasive carcinoma (TCGA-BRCA, n = 1215) was downloaded from the National Cancer Institute Genomics Data Commons (GDC) data portal (https://portal.gdc.cancer.gov/projects/TCGA-BRCA) and included RPKM-normalized gene expression data for 56,908 probes. After filtering out the missing data, TCGA-BRCA dataset consisted of gene expression profiles of 844 breast cancer patients (139 TNBC, 68 Her2+, 469 LumA, and 168 LumB) and 113 healthy individuals.
The METABRIC dataset was used for model development and training and testing purposes, while the BRCA dataset was used for external validation of results and in biological interpretation of the results through survival analysis.
Data preprocessing
Missing data were filtered out from both datasets, and the remaining METABRIC cohort (n = 1986) was randomly partitioned in an 80/20 ratio into a training cohort (n = 1588) and an internal validation (testing) cohort (n = 398), whereas TCGA cohort (n = 957) was used for external validation (Table 1).
The Distribution of Samples into Subtypes
METABRIC, Molecular Taxonomy of Breast Cancer International Consortium; TCGA, The Cancer Genome Atlas; TNBC, basal-like triple-negative breast cancer.
Feature selection and scaling
Three efficient approaches to eliminate features from a training dataset for feature selection, that is, recursive feature elimination (RFE) (Guyon et al, 2002), SelectKBest (Pedregosa et al, 2011a), and SelectFromModel (Ferri et al, 1994), were used alternatively considering all probes defined in the METABRIC dataset. Feature sets of five different sizes were selected, including 20, 25, 30, 50, and 100.
Because the METABRIC datasets were RPKM normalized, no further scaling was applied. However, when scaling the BRCA dataset consisting of count data, the StandardScaler scaling method was used to minimize scaling variations associated with the different features and avoid the dominance of certain features in the model.
Model construction and tuning
Considering that the models with which each dataset works in harmony are different due to the nature of the dataset and disadvantages of the modeling algorithms, we preferred the combined use of algorithms and followed an ensemble learning approach to obtain the best combination of models by using five of the six algorithms in each simulation.
As a preliminary analysis for selection of modeling approaches, the python package, LazyPredict (Pandala, 2022), was applied to the RNA-seq datasets and the performance of 29 different ML algorithms was comparatively analyzed in terms of accuracy, balanced accuracy, and F1 score metrics. This preliminary analysis revealed six modeling methods, including the linear discriminant analysis (McLachlan, 2005), LightGBM (Ke et al, 2017), XGBoost (Chen and Guestrin, 2016), SVC (Platt, 1999), KNeighborsClassifier (KNN) (Guo et al, 2003), and CatBoost (Prokhorenkova et al, 2018), with considerable performance (accuracy >0.80, balanced accuracy >0.70, and F1 score >0.8).
We followed the ensemble learning approach, in which the six algorithms are used in combination to obtain the best combination of models. A two-level blending approach was followed: metamodels were generated by using five of the six algorithms through the VotingClassifier (Pedregosa et al, 2011b), and the RandomFrorestClassifier (RandomForest) (Breiman, 2001) was used to generate the ensemble models.
The python package, Optuna (Akiba et al, 2019), was used to tune the hyperparameters. The ensemble models were trained considering feature sets of five different sizes (i.e., 20, 25, 30, 50, and 100) and their performance was comparatively evaluated.
Performance evaluation
A confusion matrix was constructed based on the results of each simulation to determine true-positive, true-negative, false-positive, and false-negative metrics. The classification results of the models were evaluated by calculating overall accuracy, precision (positive predictive value), specificity, and sensitivity (recall) indicators, as well as the area under the curve (AUC) metric of receiver operating characteristic (ROC) curve analysis.
In addition, the principal component analysis (PCA) was performed to visualize the classification performance. Performance evaluations were conducted on the METABRIC test cohort as internal validation to select the best feature set in stratification of patients into molecular subtypes. For the selected best feature set, performance evaluation was repeated on TCGA-BRCA cohort as external validation.
Survival analysis
To determine the association of the selected feature set (mRNAs) with patients' overall survival, we performed a survival analysis using clinical data from TCGA cohort. The survival analysis was performed on 930 patients with available clinical information. Survival analysis was performed based on individual mRNA expression using the Kaplan–Meier (K–M) approach, through the survival package in R, by dividing the subjects into two groups (high- and low-risk groups) according to their prognostic index (PI), which is the linear component of the Cox proportional hazard model:
where
The signatures of survival in each group were estimated by K–M plots. A plot with log-rank p-value <0.05 was considered as statistically significant, and the hazard ratio (HR) was calculated to discover the significance of the survival plot based on the ratio between the relative death rates in each risk group.
Gene set overrepresentation analyses
Overrepresentation analyses were carried out using ConsensusPathDB (ver. 35) (Kamburov et al, 2013) to recognize functional annotations, such as metabolic, signaling, and disease pathways, and biological processes significantly related to the gene products.
The preferred data source for metabolic and signaling pathways was the Kyoto Encyclopedia of Genes and Genomes (KEGG, release 102.0) (Kanehisa et al, 2017), and Gene Ontology annotations (Ashburner et al, 2000) were employed to represent associated biological processes; p-values were obtained to determine significant biological processes and pathways through Fisher's exact test, followed by Benjamini–Hochberg correction as the multiple testing correction method. The enrichment results with adjusted p < 0.05 were considered statistically significant.
Results
Model construction
To identify a set of mRNAs that can distinguish patients with different breast cancer subtypes as well as from healthy individuals, three alternative feature selection algorithms (RFE, SelectKBest, and SelectFromModel), which are widely used to eliminate features with low importance from a training dataset, were employed on the METABRIC training cohort (Table 1).
The models with which each dataset works in harmony are different due to the nature of the dataset and disadvantages of modeling algorithms. Therefore, we considered the combined use of algorithms and followed an ensemble learning approach to obtain the best combination of models by using five of the six algorithms in each simulation. Considering feature sets of five different sizes (i.e., 20, 25, 30, 50, and 100), as a result, 15 different mRNA sets were created.
Performance evaluations
The performance of the ensemble models was evaluated on the METABRIC test cohort as an internal validation to select the best model (i.e., the most appropriate feature set) for stratification of patients into molecular subtypes. In addition to an overall analysis to stratify all subtypes and the healthy cohort, pairwise subtype analyses to distinguish each subtype from the healthy cohort were also considered when evaluating the performance of the models.
In addition to indicators of accuracy, precision, specificity, and sensitivity (recall) derived from confusion matrices, the AUC metric of the ROC curve analysis and PCA visualizations were also considered in the evaluation of model performance.
Performance evaluations on the METABRIC test cohort based on overall accuracy, precision, and recall in stratifying patients into molecular subtypes underscored several models. RFE as the feature elimination method and feature set sizes of 25, 50, and 100 came into prominence (Supplementary Table S1). Further analyses based on confusion matrices (Fig. 1), AUC metrics of ROC curve analyses (Fig. 2), and PCA visualization (Supplementary Fig. S1) indicated a high performance of the feature set, the so-called RFE-50, with a set size of 50 mRNAs selected by RFE.
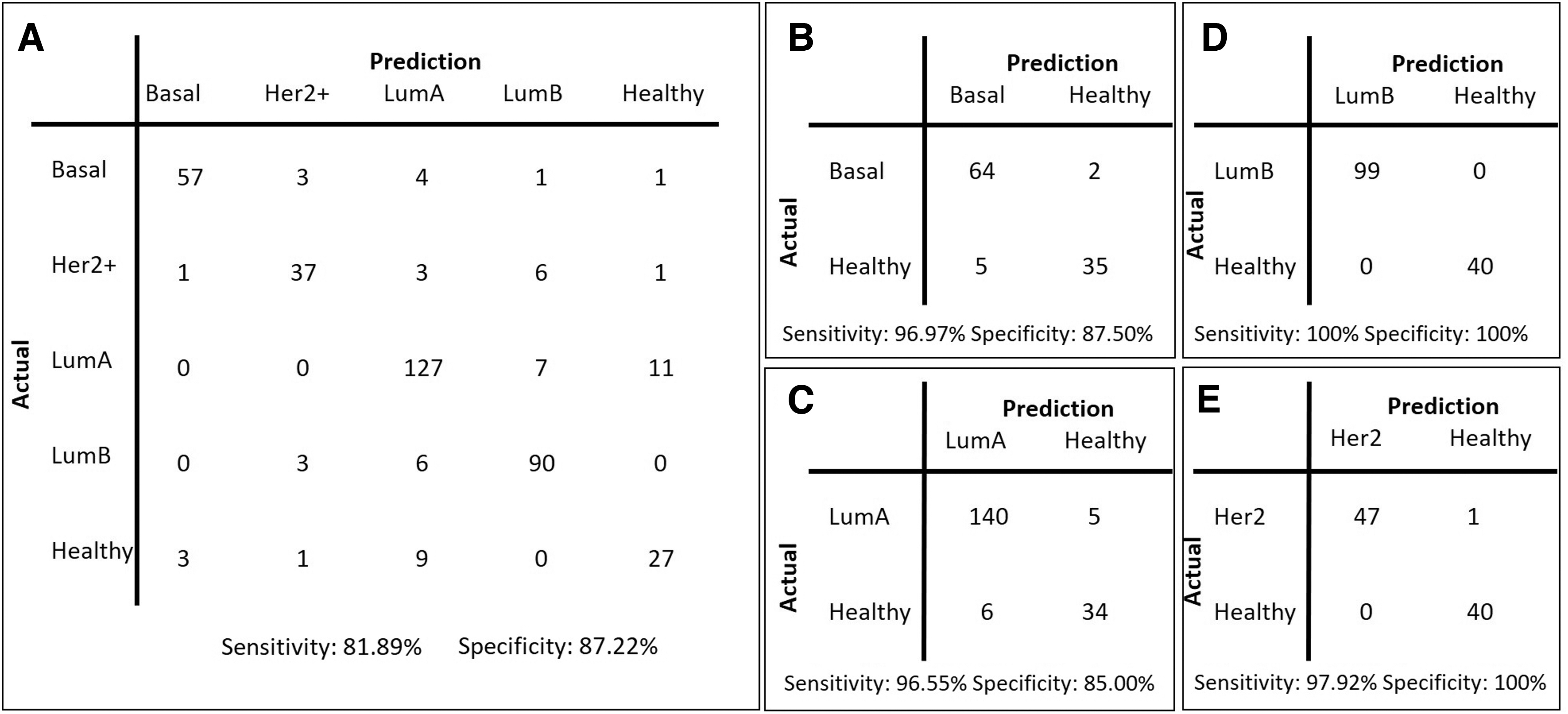
Confusion matrices for the METABRIC test cohort.
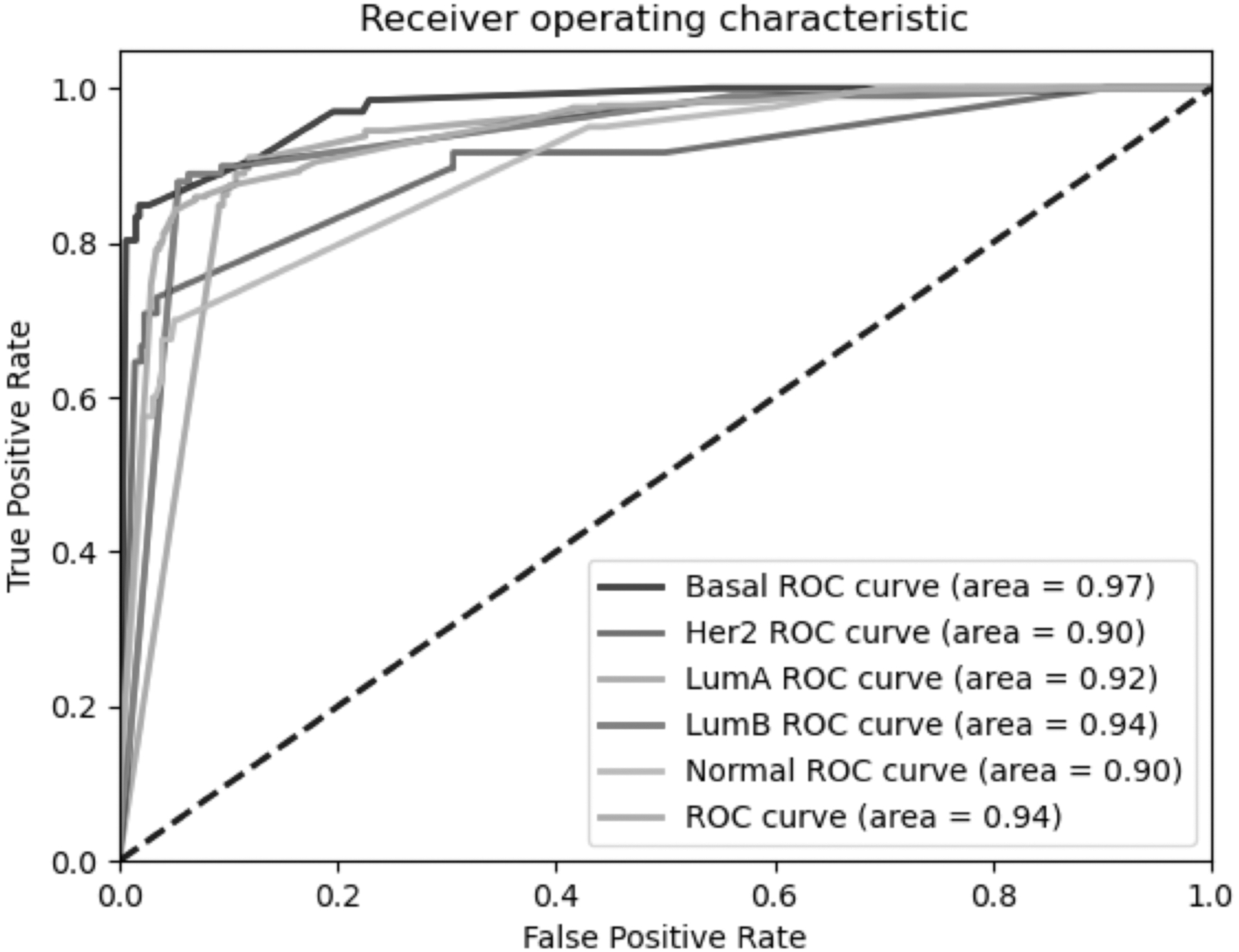
The ROC curve analysis results on the METABRIC test cohort. ROC, receiver operating characteristic.
The RFE-50 model achieved 84.92% accuracy, 83.36% precision, 87.22% specificity, 81.89% sensitivity, and an AUC value of 0.94 in stratifying patients into molecular subtypes (Figs. 1 and 2). However, in pairwise comparisons to distinguish each subtype from the healthy cohort, performance was much higher, with 100% accuracy, 100% sensitivity, and 100% specificity in distinguishing patients with LumB from healthy individuals and 98.86% accuracy, 97.92% sensitivity, and 100% specificity in distinguishing patients with Her2+ from healthy individuals (Fig. 1).
External validation
For the RFE-50 model, further performance evaluations were performed on TCGA-BRCA cohort for external validation purposes. Our model represented 82.55% accuracy, 84.09% precision, 79.22% specificity, and 84.55% sensitivity in stratifying patients into molecular subtypes; however, its discrimination performance displayed 100% accuracy, 100% sensitivity, and 100% specificity in distinguishing patients with basal, LumB, and Her2+ subtypes from healthy individuals and 95.65% accuracy, 94.74% sensitivity, and 100% specificity in distinguishing patients with LumA from healthy individuals (Fig. 3 and Supplementary Fig. S2).
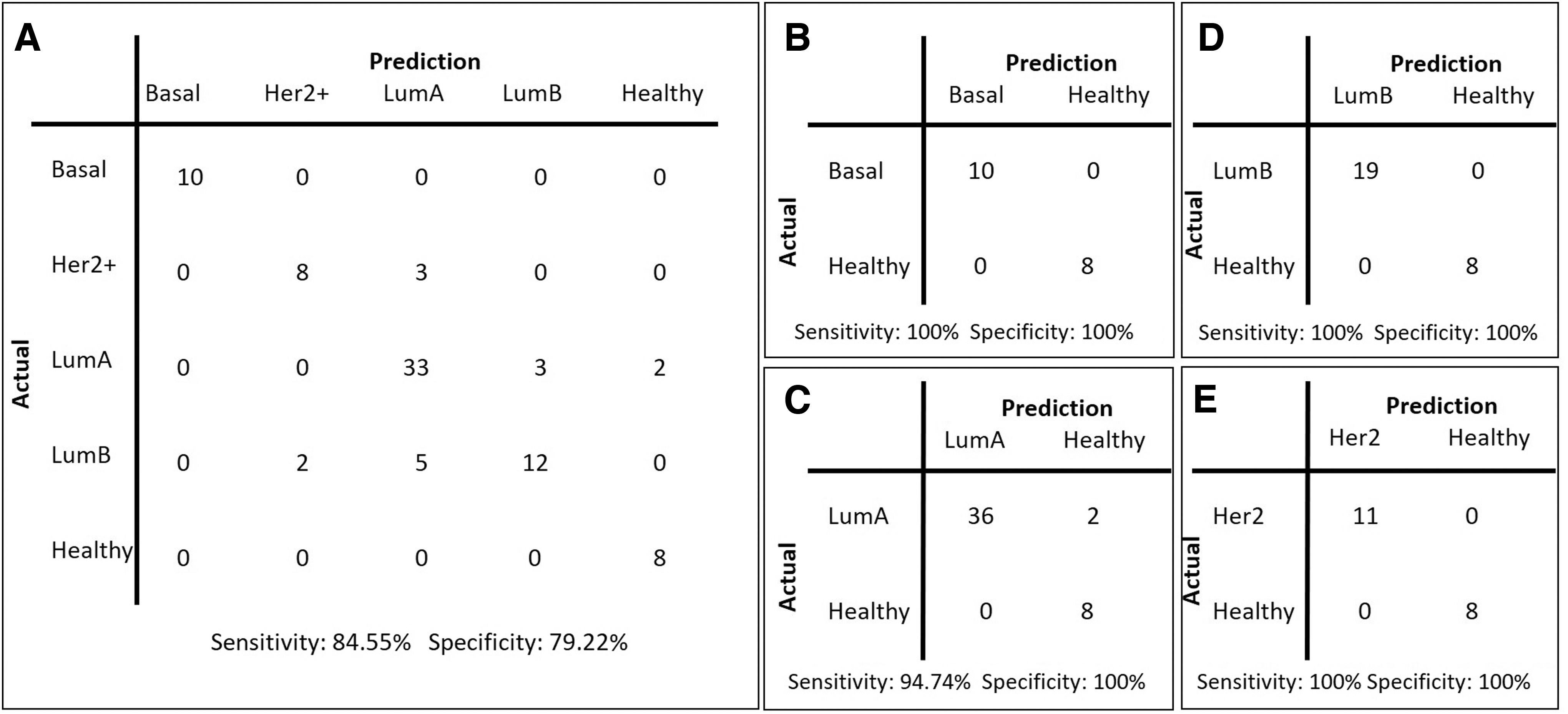
Confusion matrices for TCGA test cohort.
Biological interpretation
The genes encoding mRNAs present in the RFE-50 model (Fig. 4) were further investigated through functional enrichment and overall survival analyses. Gene set overrepresentation analyses revealed that these genes encode signaling and regulatory proteins rather than metabolic enzymes.
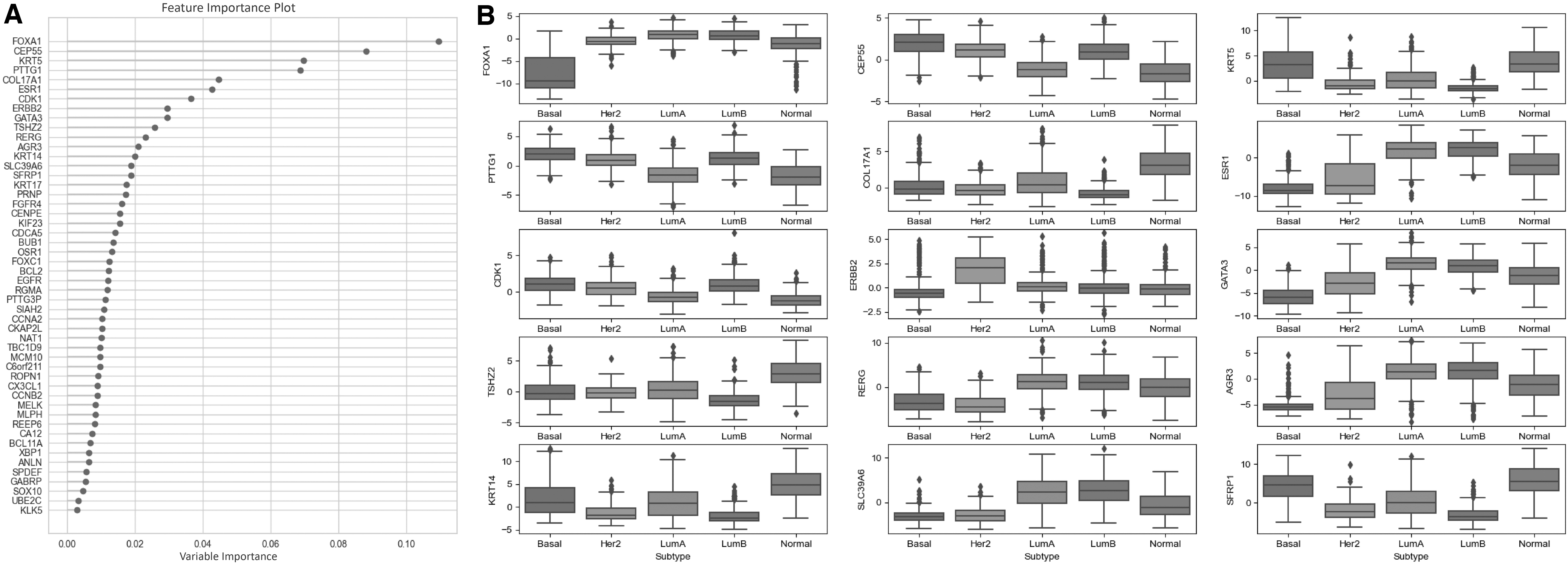
Genes encoding the mRNAs present in the RFE-50 model.
In addition, pathways and processes that enable the acquisition or maintenance of cancer hallmarks were highlighted. These include cancer pathways such as pathways in cancer (p = 0.018), miRNAs in cancer (p = 0.015), breast cancer (p = 0.032), and proteoglycans in cancer (p = 0.045); signaling pathways such as estrogen signaling (p = 0.001), p53 signaling (p = 0.011), HIF-1 signaling (p = 0.018), PI3K-Akt signaling (p = 0.045), and calcium signaling (p = 0.050); and biological processes such as cell cycle (p = 2.04 × 10−8), cell death (p = 1.66 × 10−7), chromosome segregation (p = 4.05 × 10−5), cell proliferation (p = 4.64 × 10−4), cell motility (p = 1.43 × 10−3), keratinization (p = 9.05 × 10−3), immune system development (p = 0.011), cellular homeostasis (p = 0.019), cellular localization (p = 0.0297), cellular senescence (p = 0.034), and focal adhesion (p = 0.0447) (Supplementary Table S2).
To assess the prognostic significance of model genes as potential system biomarkers, the patient cohort was divided into low- and high-risk groups based on the expression profiles of genes encoding mRNAs in the RFE-50 model, and multivariate survival analysis and risk assessments were performed. We found that the genes in the RFE-50 model had a high impact on overall survival (p = 5 × 10−7 and HR = 2.25) of patients with breast cancer (Supplementary Fig. S3).
Discussion
Precision oncology is built on the concept of individually tailoring each patient's clinical care from diagnosis, risk prevention, and assessment to personalized therapeutics using multiple strands of information such as molecular biomarkers, clinical variables, and social determinants of health (Yilmaz and Arga, 2022). Precision oncology has made significant strides in the last few decades.
Yet, developing accurate and computationally intensive algorithms to classify patients to support diagnostic and therapeutic decisions at the point of care is still one of the greatest challenges. Artificial intelligence techniques, particularly ML and deep learning, offer tremendous potential to advance precision oncology (Gulfidan et al, 2021).
Breast cancer is one of the most common malignancies worldwide, with multiple underlying molecular etiologies and subtypes that differ in risk factors, incidence, disease progression, survival, and response to treatment. Thus, there is a clear and urgent need for development of precise strategies to stratify breast cancer into molecular subtypes, not only to assess the prognosis of the disease but also to enable effective, safe, and individualized therapy. Previous studies have shown that ML approaches offer new solutions and avenues for biomedical, bioengineering, and clinical applications (Ozer et al, 2020).
In this study, we applied a data mining approach to comprehensive RNA-seq data from TCGA (n = 1215) and METABRIC (n = 1992) projects and hypothesized that changes at the level of gene expression could lead to differentiation of patients with different breast cancer subtypes as well as from healthy individuals. Using alternative feature elimination methods, considering different feature sizes, and following an ensemble learning approach to find the best combination of various ML models, we report here a candidate panel and ensemble model containing 50 mRNAs as features to stratify patients with different breast cancer subtypes as well as to distinguish them from healthy individuals.
The performance of the model reached up to 100% accuracy in pairwise comparisons to distinguish each subtype from the healthy cohort. Moreover, the mRNA panel showed high prognostic performance with an HR of 2.25 (p = 5 × 10−7) in breast cancer overall survival.
The mRNA panel consists of several genes, including BCL2, CCNA2, CCNB2, CDCA5, CDK1, EGFR, ERBB2, ESR1, FGFR4, KIF23, KRT14, and KRT17, which have previously been linked to cancer pathways in the KEGG database and encode proteins that function in signaling pathways, such as estrogen signaling, p53 signaling, HIF-1 signaling, PI3K-Akt signaling, and calcium signaling, whose association with cancer development and progression is well established in the literature.
In addition, the other genes in the candidate panel are significantly associated with biological processes such as cell cycle, cell death, cell proliferation, chromosome segregation, and cell motility that enable the acquisition or maintenance of hallmarks of cancer. Collectively, these offer mechanistic plausibility and support for the proposed panel for future evaluation and validation in breast cancer cohorts.
Chief among the novel aspects of the present investigation is that it is the first study to classify patients with different breast cancer subtypes as well as distinguish them from healthy individuals using an ensemble ML approach and utilizing both comprehensive RNA-seq datasets from two large consortia, METABRIC and TCGA, for internal and external validation purposes.
Recognizing that each modeling algorithm has advantages and disadvantages, we followed the ensemble learning approach, in which six algorithms were used in combination to obtain the best combination of models, and followed a two-level blending approach. We reported here an ensemble model, including 50 mRNAs as features, to stratify patients with different breast cancer subtypes as well as healthy individuals.
With up to 100% accuracy in pairwise comparisons for distinguishing each cancer subtype from the healthy cohort, the ensemble learning approach offers promise over other ML algorithms (Ozer et al, 2020). Hence, the ensemble learning approach presented here offers new pathways to innovation for precision/personalized medicine in oncology.
Several limitations of the study are noteworthy. First, comparisons of the ensemble learning approach herein with other methods for patient stratification, and using prospective data that account for clinical and other confounders, are necessary.
Second, although we used the two most comprehensive RNA-seq datasets for breast cancer, the study design was unbalanced with respect to size of patient groups. This has the practical consequence of reducing the statistical power of models and introducing sampling errors in feature selection.
Third, clinical data were limited. Future studies could include in the modeling process larger sample sizes with more detailed/deep phenotyping.
Fourth, although we evaluated six algorithms in the context of ensemble modeling, there are many algorithms that we did not consider here.
Finally, the precision oncology models need validation in independent clinical cohorts and world populations while also evaluating in tandem the preclinical and clinical biological and mechanistic plausibility of the forecasts made to best inform precision oncology.
Conclusions
There is a real-life need for precision oncology worldwide especially in breast cancer so as to stratify cancer into molecular subtypes. We proposed here an ensemble learning approach to find the best combination of different ML models and presented an ensemble model, including 50 mRNAs, to stratify patients with different breast cancer subtypes and differentiate them from healthy individuals.
The findings offer marked potential to inform future precision oncology efforts in the field of breast cancer. Clinical forecasting for precision medicine is an arduous and complex task particularly in the context of cancers. Future studies in large samples with deep phenotyping, which prospectively account for numerous clinical and other confounders, will help advance the proposed approach and the field of precision oncology.
Furthermore, this study has suggested that accounting for gene expression is useful for precision oncology in breast cancer. We recommend further research to strengthen the performance of our strategy by integrating deep phenotyping and prospective study designs. Applications of advanced ML methods such as ensemble learning to develop gene panels for stratification of cancer patients are timely as powerful tools to improve diagnosis and targeted therapy.
Footnotes
Authors' Contributions
F.K. was involved in data curation (lead); formal analysis (lead); software (lead); and writing—original draft (equal). M.A was involved in conceptualization (lead); methodology (lead); supervision (lead); and writing—review and editing (equal). K.Y.A. was involved in conceptualization (supporting); data curation (supporting); supervision (supporting); writing—original draft (equal); and writing—review and editing (lead).
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.