
Abstract
Cattle breed identification is crucial for livestock research and sustainable food systems, and advances in genomics and artificial intelligence present new opportunities to address these challenges. This study investigates the identification of the Tharparkar cattle breed using genomics tools combined with machine learning (ML) techniques. By leveraging data from the Bovine SNP 50K chip, we developed a breed-specific panel of single nucleotide polymorphisms (SNPs) for Tharparkar cattle and integrated data from seven other Indian cattle populations to enhance panel robustness. Genome-wide association studies (GWAS) and principal component analysis were employed to identify 500 SNPs, which were then refined using ML models—AdaBoost, bagging tree, gradient boosting machines, and random forest—to determine the minimal number of SNPs needed for accurate breed identification. Panels of 23 and 48 SNPs achieved accuracy rates of 95.2–98.4%. Importantly, the identified SNPs were associated with key productive and adaptive traits, thus attesting to the value and potentials of digital transformation in livestock genomics. The ML-aided ultra-low-density SNP panel approach reported here not only facilitates breed identification but also contributes to preserving genetic diversity and guiding future breeding programs.
Introduction
Developing a combination of genetic markers that can accurately classify a particular cattle breed population could have multipronged positive impacts on planetary food sustainability and benefits in the livestock industry (Cegelski et al., 2003; Castric and Bernatchez, 2004; Peter et al., 2006). This, in turn, could help preserve native genetic resources in each country with benefits for population health and the local and global economies. Advances in livestock genomics and digital transformation by artificial intelligence (AI) and machine learning (ML) offer veritable prospects to address these overarching challenges and promises in planetary health and ecology.
Over the past two decades, allocation methods have gained popularity in population genetic studies, including determining the extent of genetic exchange between populations, identifying immigrants, and detecting hidden population structures in livestock genomics (Dalvit et al., 2008; Kumar et al., 2021; Shackell et al., 2001; Trenkel et al., 2020). These methods have practical applications, such as tracing animals and animal products back to their breed of origin, which is particularly relevant as comprehensive and integrated food safety policies become increasingly important. The underlying principle behind the allocation of animals and products is the genetic diversity present among populations and breeds, which may exceed that present within a group of animals (Bertolini et al., 2015).
DNA markers can identify differences between livestock breeds, which can be used for breeding and verifying monobreed-labeled products. Studies have investigated the potential use of high polymorphic microsatellites, specific genes affecting breed characteristics (Fontanesi et al., 2011; Russo et al., 2007), and a small number of randomly selected single nucleotide polymorphisms (SNPs) (e.g., Pant et al., 2012; Ramos et al., 2011). SNP chip data have been increasingly used for breed identification with the development of SNP Bead Chips and public databases (Kim et al., 2015; Singh et al., 2019; Trenkel et al., 2020). However, selecting SNP markers that can differentiate between breeds is still challenging because SNPs are typically identical across all breeds.
Attempts have been made to use a combination of SNP markers with automated platforms for target breed identification (Brooks et al., 2016; Suekawa et al., 2010), and a Tharparkar-specific SNP panel has been established for cattle breed identification using a combination of minor allele frequency (MAF)–linkage disequilibrium (LD) and preselection statistics (Kumar et al., 2021). This approach can correctly assign an unknown sample to its breed, and efforts are underway to develop technologies for assessing the purity of Tharparkar cattle for conservation purposes using SNP genotyping data.
An example of current digital transformation in livestock genomics is the use of AI and ML algorithms to identify SNP panels for breed purity in livestock and poultry species. ML is a form of AI that involves developing algorithms to enable computers to learn from training data and make predictions (Kumar et al., 2021a). Genomic data and a range of classification models can be used to differentiate between animal populations using pattern analysis. Previous research on genetic diversity has hitherto infrequently employed ML algorithms. However, some recent studies have used high-density SNPs to compare the classification abilities of support vector machine and random forest (RF) models (Bertolini et al., 2017; Bertolini et al., 2015; Pasupa et al., 2020; Schiavo et al., 2020).
In the past, the classification models derived from ML algorithms have tended to make comparisons based on the F statistic, delta statistic, and eigenvalue of principal component analysis (PCA) to evaluate populations based on genomic information. However, the SNP marker combinations used by different ML algorithms often vary greatly, even though some common SNPs’ usefulness has been confirmed by the resulting SNP marker combinations (Schiavo et al., 2020). In a previous study, it was found that using a small subset of preselected SNPs instead of all genomic SNPs improved the classification performance of the ML model (Judge et al., 2017). More recently, ML algorithms have been successfully used to test high-purity buffalo meat, and using breed tag SNPs increased identification accuracy when compared with random SNPs (Xu et al., 2021). In an effort to identify chicken-target populations, another research team used three key ML algorithms, namely RF, decision trees, and AdaBoost (AB) (Seo et al., 2021). Liu et al. (2023) also used six ML classification algorithms in pig breed identification using SNP’s array data.
The aim of the present study was to create a low-density SNP panel for identifying the indigenous Tharparkar cattle breed and to utilize these findings for genetic conservation, diversity analysis, and product traceability certification as well as to develop comparable technologies for other livestock species and products. By utilizing a 50k SNP genotyping array for cattle, this research was set up to determine the minimum number of SNP markers needed to differentiate and authenticate a target cattle population (Tharparkar) from other cattle populations. In all, this investigation presents a framework and methodology for future studies on genetic purity and breed identification, utilizing the combination of genome-wide association studies (GWAS) and ML algorithms, and the ways in which such a combination offers synergies for digital transformation in livestock genomics and ecology research broadly.
Materials and Methods
Animals
A total of 72 Tharparkar individuals were randomly selected from a cattle farm at the Indian Council of Agricultural Research (ICAR)–Indian Veterinary Research Institute (IVRI), Bareilly, India. The Tharparkar cattle herd at Indian Veterinary Research Institute was established in the year 2007 by procuring animals of different age groups from Jaisalmer and adjacent areas of Rajasthan, India, that is, breeding the home tract of the Tharparkar cattle breed. The elite breeding bulls were from Central Cattle Breeding Farm, Suratgarh (Rajasthan, India). Tharparkar cattle herd maintained at IVRI is proven to be genomically diverse through genetic diversity studies (Saravanan et al., 2022). The present study was approved by the Institutional Animal Ethical Committee of ICAR-IVRI, Bareilly, India.
Genotyping
Blood samples were collected in vacutainers under aseptic conditions after due approval and following standard guidelines issued by the Institutional Animal Ethical Committee of the ICAR-IVRI, Bareilly, India. DNA isolation was performed using the QIAampR DNA kit (Qiagen, Germany). The concentration and quality assessment of DNA samples will be done using NanoDrop_1000_Spectrophotometer (Thermo Fisher Scientific, MA, USA) and agarose gel electrophoresis, respectively. After the quality check, DNA samples will be genotyped using the Illumina BovineSNP50 v3 BeadChip (Illumina, Inc., San Diego, CA, USA).
Genotype data from other breeds
In this study, data on three indicative cattle breeds (Sahiwal, Ongole, and Hariana) were obtained from online genotype data sources. The data were then merged with similar data from four exotic breeds (Holstein Friesian, Jersey, Guernsey, and Ayrshire) that were extensively used in India’s cross-breeding programs. The details of the number of animals used in this study for each breed are provided in Table S1.
Description of the Details of the Breed-Wise Number of Animals Used in the Construction of Tharparkar-Specific SNP Panel
Description of the Details of the Breed-Wise Number of Animals Used in the Construction of Tharparkar-Specific SNP Panel
SNP, single nucleotide polymorphism.
The genotypic data were accessed from the dryad repository and assembled online at the Web-Interfaced Next-Generation Database dedicated to Genetic Diversity Exploration (WIDDE).
Quality control of the datasets
The SNP datasets used in the study were aligned with the ARS UCD 1.2 bovine reference genome assembly, which is available at the National Center for Biotechnology Information. Unique positions on autosomal chromosomes were selected for SNP analysis. SNPs that were monomorphic in all breeds were removed from further analysis. Basic statistics were calculated for all animals using PLINK v1.9, as described by Purcell et al. (2007) and Chang et al. (2015): SNPs with a genotyping call rate >99% and an MAF of <0.05. The quality control (QC) parameters were applied to the merged dataset of 216 animals, including 72 Tharparkar cattle. Only common SNPs found in all eight cattle breeds were retained for downstream analysis.
Population clustering and stratification
To further verify the presence of eight breeds in our reference population, PCA was performed using R programming.
Selection of candidate SNP markers for identification of the target (Tharparkar) population
Figure 1 illustrates the step-by-step procedure used to identify candidate SNP markers that could differentiate the Tharparkar cattle population from other breeds. To select a marker combination that could distinguish Tharparkar cattle individuals, we utilized two primary strategies. First, we used PLINK 1.9’s case–control association analysis tool, where the Tharparkar cattle breed served as the case population and the remaining seven cattle breeds served as the control population, and SNPs were chosen based on their p- values (Chang et al., 2015). Using a two-step approach, we identified two sets of significant SNP marker combinations, consisting of 23 and 48 SNPs. The first step involved selecting SNPs with significantly lower p-values from a GWAS association test. Second, ML algorithms were employed.
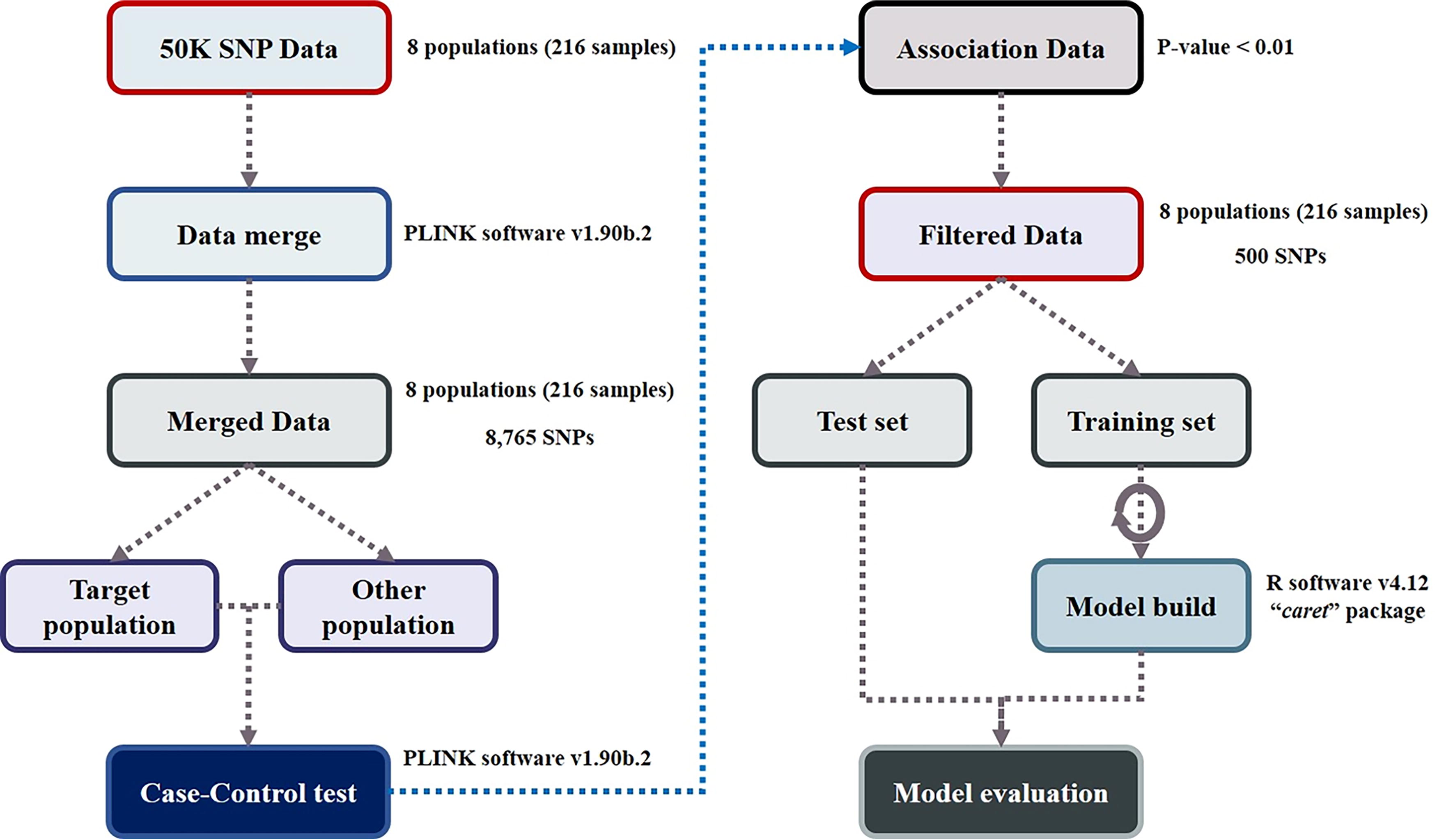
Pipeline for selection of Tharparkar-specific SNP markers for identification of the target population using machine learning and GWAS. GWAS, genome-wide association study; SNP, single nucleotide polymorphism.
ML to determine the minimum number of markers needed for breed identification
The selected SNPs, which were identified with the 50k SNP genotyping array, were used as the training and test dataset in the proportion of 70% and 30%. The training dataset had 153 individuals, and the test dataset had 63 individuals from all eight cattle breeds. The training dataset was further divided into a validation set (42%) and a training set (28%). Hence, a balanced split of data was obtained. To increase the accuracy of the results, 25-fold cross-validation was applied. The target population was identified by using classification algorithms of ML algorithms. Four models were applied to classify populations/breeds: RF, AB, bagging tree (BT), and gradient boosting machines (GBMs). The “caret” ML package in R software was used to build a classification model (Kuhn et al., 2022). The four ML models shared a common taxonomy. The resampling method used to fit each model was the “cross-validation” method. In all algorithms, the default settings apply to every parameter.
RF is an ensemble learning algorithm that employs decision trees as its primary classifier, and it randomly selects labels and features for classifier modeling before voting to determine the prediction outcome. The “carret” R package is used to construct the RF classifier. In contrast, adaptive boosting or AB is a meta-learning ensemble ML algorithm that improves weak classifiers by minimizing residual errors using an iterative approach. AB employs sequence ensembling and combines several weak classifiers to produce a strong classifier with high accuracy. BT or bootstrap aggregating is an ensemble learning approach that enhances the predictive accuracy of decision trees, reducing bias-variance trade-offs and preventing overfitting in regression and classification models (Breiman, 1996). GBM is an ensemble learning technique that creates a final model by combining individual models in sequence, often decision trees, using the gradient to minimize the loss function, similar to neural networks’ gradient descent approach (Breiman, 2001).
To evaluate the reliability of classifying the target population, each ML model has used its own set of criteria as proposed by Breiman (1996, 2001) and Kégl (2013).
The true positive rate (TPR), also known as the sensitivity, is the percentage of positive values that were correctly determined:
The true negative rate (TNR), or the percentage of negative values that were accurately determined, is referred to as specificity:
Tharparkar-specific SNP annotation
Genes annotated in the bovine ARS UCD 1.2 genome version were retrieved using the Ensembl-Variant Effect Predictor web tool (https://asia.ensembl.org). The closest genes to these SNPs were then obtained using the Variant Effect Predictor (McLaren et al., 2016). Functional annotation of SNPs was done. The proportion of SNPs showing important biological pathways was identified using Panther database release 17.0. We also attempted to check the genes carrying breed-specific SNPs overlapping with previously reported quantitative trait loci (QTLs) (https://www.animalgenome.org/cgi-bin/QTLdb) available in the cattle QTLdb v49.
Results
The entire research pipeline used in this study is illustrated in Figure 1.
Characteristics of the genomic datasets
We genotyped 72 Tharparkar animals using the Illumina BovineSNP50 v3 BeadChip (version 3), which resulted in 53,212 SNPs with a genotyping rate of 99.23%. We filtered out SNPs belonging to mitochondrial DNA and sex chromosomes, leaving a final dataset of 8765 common markers with a genotyping rate of over 99%. The final dataset consisted of 216 individuals from 8 cattle breeds, and the test and training datasets included 63 and 153 individuals, respectively.
Population clustering and stratification
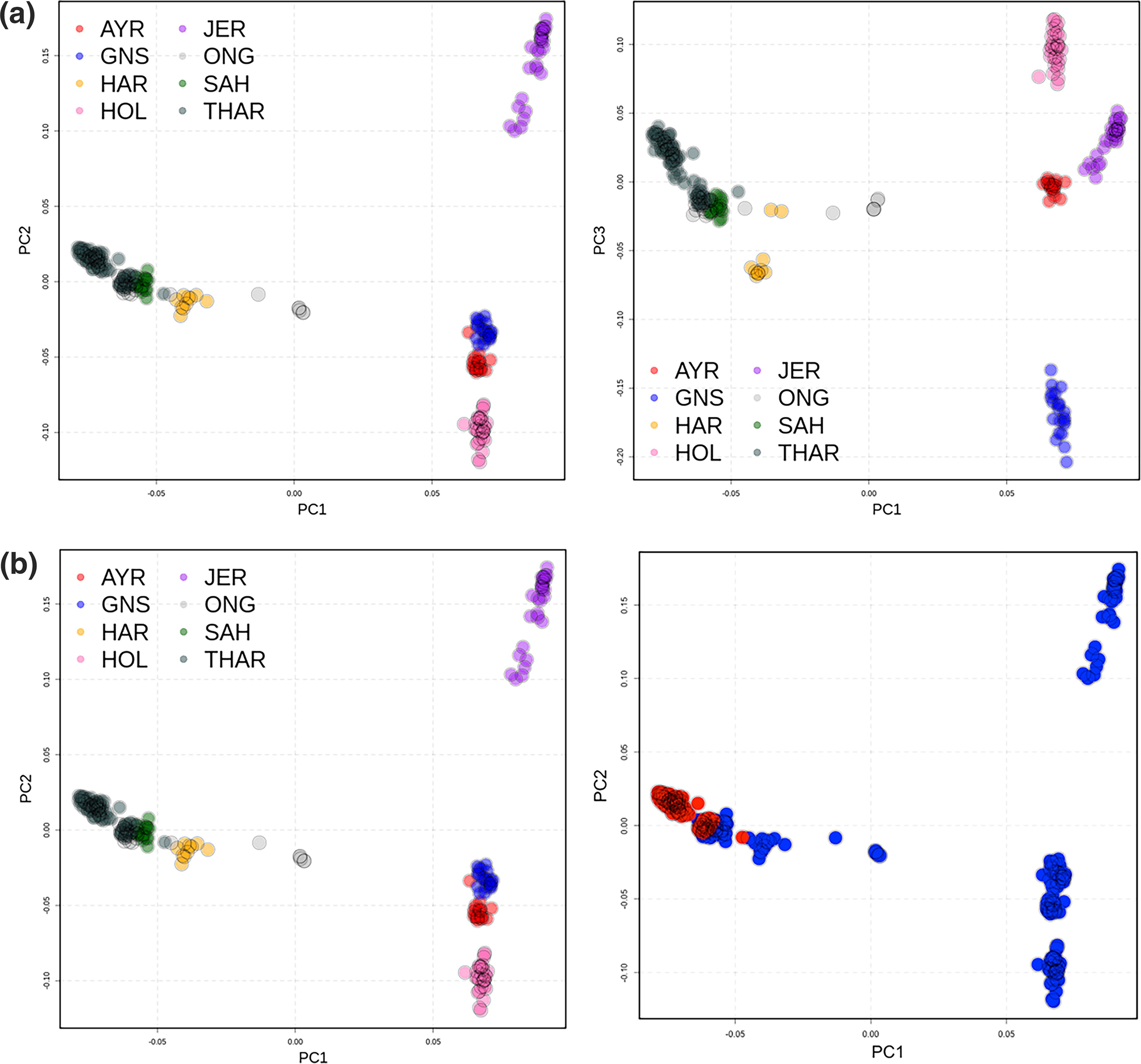
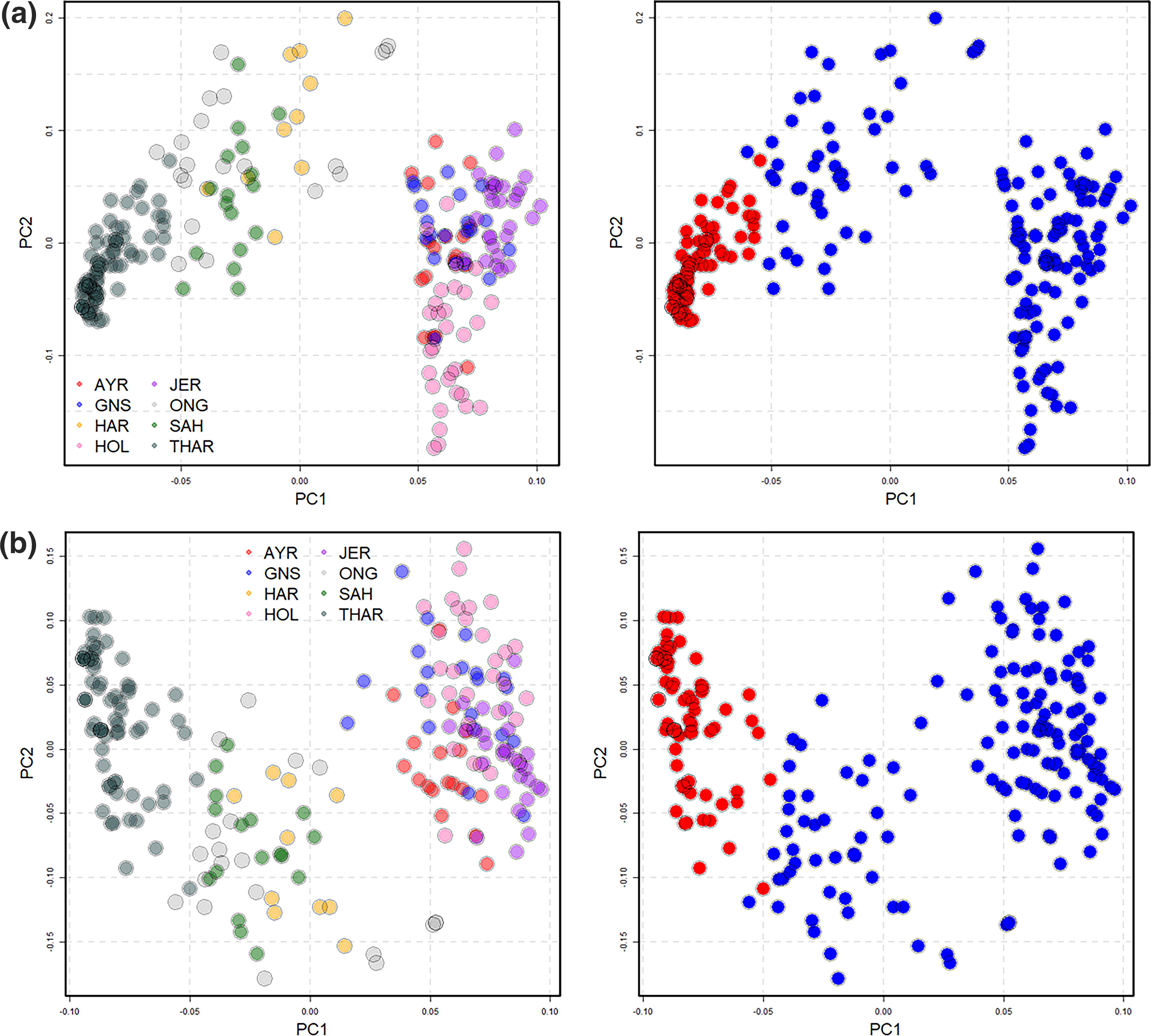
After QC analyses, a total of 8765 markers were left in the 8 breeds merged dataset. PCA analysis using 216 animals in the base population was done. This confirmed the presence of eight distinct clusters in the dataset following their different breeds of origin, validating the presence of a total of eight breeds. Figure 2 shows the distinct clustering of all eight breeds in the form of PCA plot.
GWAS and SNP selection
The study used 8765 SNPs to differentiate between the target and control cattle populations, which represented approximately 15% of the 50k SNP genotyping array data. The selection of SNPs was done by implementing a Bonferroni correction to GWAS analysis results with a p-value of 0.01, resulting in 8765 SNPs.
The number of SNPs was further reduced to 500 case-related SNPs with the lowest p-value. The discriminatory power of these 500 SNPs was evaluated by comparing the results of the PCA analysis. The findings indicated that these 500 SNPs could distinguish between the target and control cattle populations as the case and control groups were separated (Fig. 3).
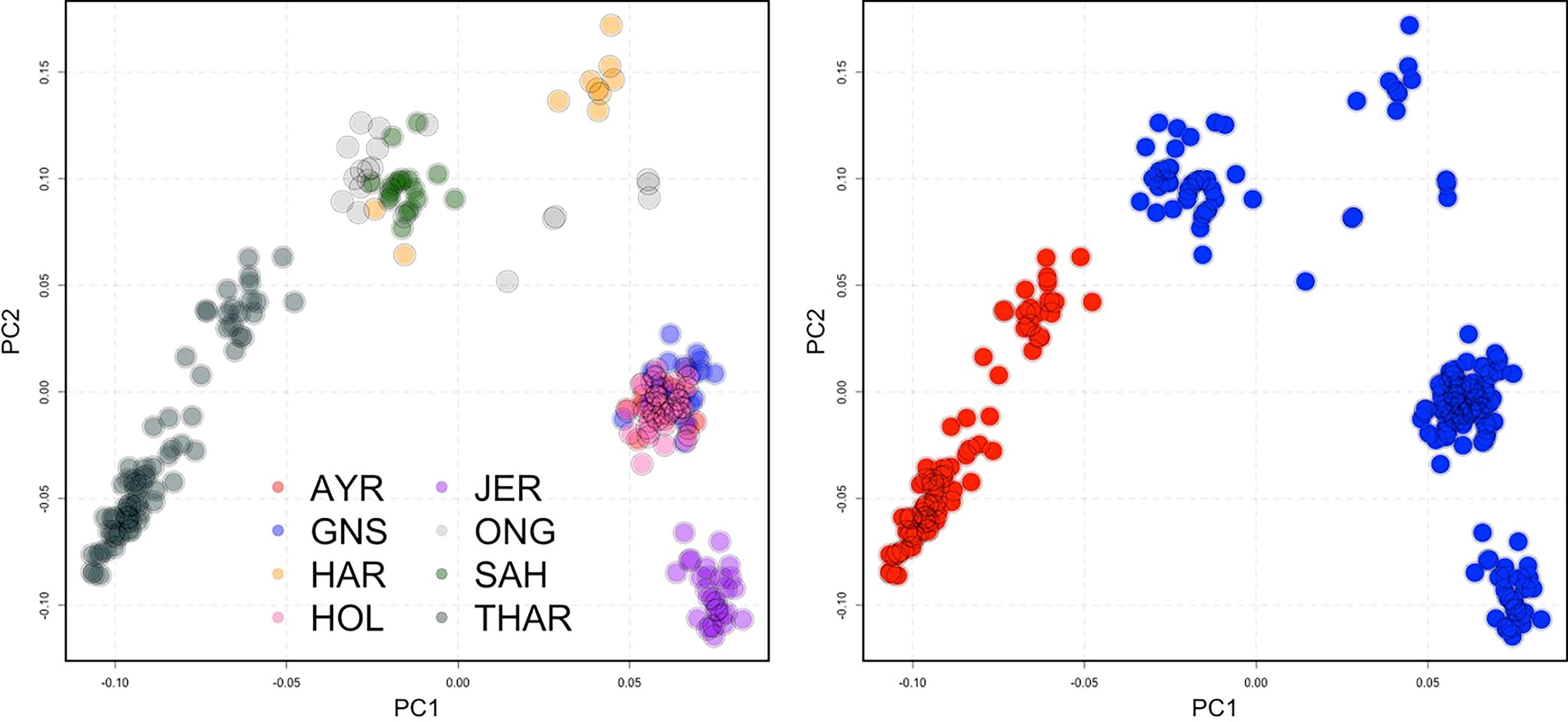
A PCA plot was constructed to show the degree of separation of the target group from the other populations (filtered 500 SNPs PCA results).
Breed identification by ML algorithms using the minimum number of SNPs
We used the four ML classification models to classify the target Tharparkar group based on the 500 selected markers. We assessed two panels for classification accuracy after feature selection, that is., 23 and 48 SNPs. We obtained a feature selection plot for each ML model (Fig. 4). As shown in the figure, the score of feature importance (FI) according to each model is confirmed differently, and in our analyses, the average value of the scaled FI of all models was used.
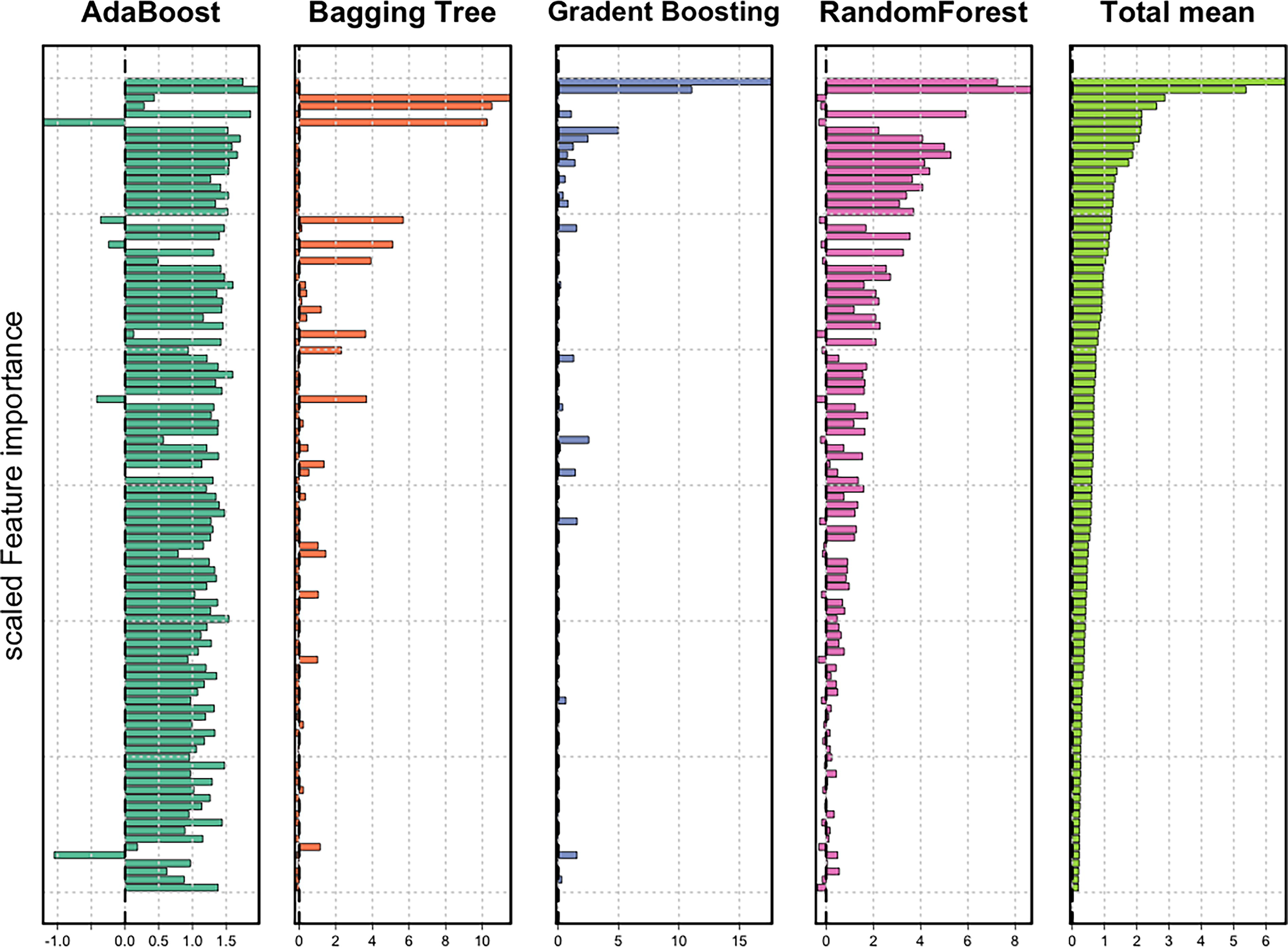
Plots for selection of candidate SNP markers for identification of the target population (SNPs with feature importance for all the models and their mean).
With the use of feature selection, 23 and 48 SNP markers were selected by the four ML models, that is, AB, which had good classification accuracies (Table S2 and Table S3).
Feature Selected 23 SNPs Classification Accuracy
Feature Selected 48 SNPs Classification Accuracy
Thus, the target group Tharparkar could be classified by using a small number of markers. More than 98% of the case and control samples were distinguished in all models in both the cases of 23 and 48 SNP panels (Fig. 5).
All models except the GBM had 100% identification power, in terms of the sensitivity to confirm TPs, while all models had >97% identification power in terms of specificity to confirm FPs, except for BT. RF and AB had the highest balanced accuracy of >98%.
SNP panel
All models except the AB had 100% identification power, in terms of the sensitivity to confirm TPs, while all models had >97% identification power in terms of specificity to confirm FPs, except for BT. RF and GBM had the highest balanced accuracy of >98%.
Since there was not any significant loss in accuracy when reducing the SNP markers to 23, it could be concluded that these 23 SNP markers can distinguish Tharparkar cattle individuals from the rest of the control breeds. For a complete list and details of SNPs, see Table 4.
Enlisting of 23 SNP Panel Markers
Annotation of 23 Tharparkar-specific SNPs
A total of 23 Tharparkar-specific SNPs were annotated in detail. Seventeen genes and 32 transcripts were found to be overlapped with the Bovine Genome Reference Assembly. Seventy-three percent of total variants were reported as intron variants, and intergenic variants constituted 15%.
Discussion
The process of crossbreeding has resulted in the dilution of the germplasm of indigenous cattle breeds. To prevent further dilution and conserve pure germplasm, there is a need for technologies that can accurately assess the genetic purity of animals. In this study that builds on emerging digital transformation in livestock genomics and ML in particular, we aimed to determine the minimum number of marker combinations required to classify Tharparkar individuals accurately.
The Tharparkar indigenous cattle breed is noted for its exceptional heat tolerance, resistance to diseases, and adaptability to harsh climates, distinguishing it from exotic and crossbred counterparts (Bharati et al., 2017). However, rampant cross-breeding practices in India have diluted the unique and pure germplasm of Tharparkar cattle, resulting in only a handful of purebred animals remaining in their native region. Consequently, the identification of distinguishing SNPs for the Tharparkar breed serves not only conservation purposes but also offers a technical means to certify products or derivatives from such distinctive breeds. This holds true not only for cattle but also for other species (Bharati et al., 2017).
Significance of SNPs in breed identification
SNPs have emerged as a valuable tool for breed assignment, product authentication, and pedigree identification in domestic animals, driven by advancements in animal gene chips and high-throughput sequencing technologies. Notably, Ramos et al. (2011) identified 193 breed-specific SNPs in five pig breeds, showcasing their effectiveness in breed assignment and traceability by accurately classifying over 99% of additional 490 individuals from the same breeds. Various methods, including MAF, LD, delta statistics, the fixation index, PCA, and ML algorithms, have been proposed to identify informative markers in livestock and poultry populations (Bertolini et al., 2015; Hulsegge et al., 2013; Kumar et al., 2021; Lewis et al., 2011; Seo et al., 2021; Wilkinson et al., 2011). These techniques have consistently demonstrated high accuracy in breed assignment. For instance, Seo et al. (2021) employed the RF model to classify 283 chicken individuals from 20 populations, achieving an accuracy of up to 98% using just 44 SNPs as minimal markers. However, in cases where numerous breeds exist or there are close genetic relationships among local breeds, a slightly larger set of highly informative SNPs may be necessary for accurate breed identification, even though this could increase genotyping costs.
Challenges of high-density SNP arrays
The use of high-density SNP arrays for genotyping often results in a multitude of LD associations between SNPs, potentially affecting estimation accuracy. Thus, our initial step in this study involved using the plink software’s GWAS to filter out such SNPs. Subsequently, we conducted PCA to identify significant variances among the Tharparkar cattle breed and seven control cattle breeds. While local cattle breeds may have undergone extensive natural and artificial selection, they might share common ancestors or possess close genetic relationships, making their distinction challenging in a PCA plot. To address this, a low-density SNP panel, refined through ML approaches, was employed, enabling the accurate identification of the Tharparkar cattle breed.
GWAS analysis for target population identification
In this study, classification analysis was employed to address the limitations of population-specific markers. The Tharparkar cattle population was designated as the case group, with the remaining seven cattle populations serving as the control group. GWAS analysis utilizing 500 SNP markers revealed a close association with the case group, although the case and control groups tended to form separate clusters without clear differentiation. Previous studies have identified numerous breed-specific SNPs for discriminating between different cattle breeds. For instance, Judge et al. (2017) and Kumar et al. (2019) identified over 300 and 591 breed-specific SNPs, respectively, for distinguishing between different cattle breeds. In a different approach, Bertolini et al. (2017) employed the delta statistic, PCA, and an RF algorithm to identify marker combinations that can discriminate between different cattle breeds using fewer markers. However, our study utilized GWAS analysis to isolate the target population, making direct comparisons with previous research challenging. Nevertheless, the results indicated that the genetics of the Tharparkar cattle breed could be effectively explained by 500 SNP markers, although genotyping this many markers may be relatively expensive when compared with ultra-low-density SNP panels.
ML algorithms for classification
ML methods have gained popularity in breed identification due to their capacity to minimize loci, reduce costs, and maintain high accuracy. By combining ML algorithms with SNP chip data, breed identification has become an attractive prospect, particularly with the availability of shared reference breed data. Previous research has demonstrated that using artificial neural networks to reduce loci can achieve up to 96.6% accuracy in goat breed identification (Iquebal et al., 2014).
In our study, we employed four ML classification models to efficiently identify the Tharparkar cattle population. To assess whether the case and control groups could be differentiated, PCA was conducted using a ML algorithm based on 23 SNP markers. All classification algorithms for the 23 SNP panel demonstrated 100% breed classification sensitivity, except for the AB model, which achieved 90.5% accuracy. Typically, ML models employ a feature-selection process to simplify the model, shorten training time, avoid overfitting, and reduce the curse of dimensionality. In our study, the AB, RF, BT, and GBM models selected 23 SNP markers, each achieving classification accuracy of 95.2%, 98.4%, 95.2%, and 98.4%, respectively. Therefore, the Tharparkar cattle population can be accurately classified using a small number of markers.
Genes associated with important pathways
The study identified 23 SNPs linked to essential genes associated with multiple biological pathways. Notably, SNP rs43710710, associated with the PLCB1 gene, was linked to 12 distinct pathways, highlighting its potential significance. Other SNPs, including rs41849820, rs42687393, and rs41706473, were also associated with crucial genes and pathways.
Genes associated with adaptive/productive/disease traits
Several important candidate genes were linked to physiological roles and adaptive traits. For instance, the association of SNP rs109897885 with the CLASP1 gene suggests a role in stress response in cattle (Iung LH de et al., 2018). Another SNP, rs110391265, was associated with the EDN2 gene, which plays a role in paratuberculosis susceptibility in multibreed cattle populations (Çınar et al., 2020). The rs110941657 SNP was found to be associated with the BDKRB1 gene, which has potential implications as a biological indicator of heat stress tolerance in dairy cows (Garner et al., 2020).
Genes associated with important QTL traits
Further analysis using QTLdb-SNP query search revealed that six Tharparkar-specific SNPs were closely linked to candidate genes associated with production, reproduction, and disease-related traits. The potential applications of this research are substantial, especially in protecting intellectual property rights, instilling consumer confidence, and minimizing the time, cost, and expertise required for conventional mtDNA and microsatellite markers.
Future applications and lessons for digital transformation in livestock genomics
There has been a growing interest in harnessing AI and ML in genomics applications in diverse contexts such as precision/personalized medicine (Lin and Wu, 2022) and precision livestock farming (Klingström et al. 2024). In this overarching context, the present study’s findings usefully illuminate the transformative potential of combining genomics with ML for livestock breed identification, offering significant insights into broader applications and future directions at the intersections of digital transformation, ecology, and livestock genomics (Biscarini et al., 2015). Such integrative approaches and digital transformation hold promising implications for fields such as forensic science, conservation genetics, and livestock traceability systems. Accurate breed identification methods, such as those demonstrated here, can enhance forensic investigations by tracing livestock origins and verifying breed authenticity. In conservation genetics, these techniques could play a crucial role in monitoring and managing endangered breeds, providing insights into their evolutionary history and conservation status.
In the context of livestock traceability systems, the integration of genetic identification could address the growing consumer demand for transparency in food sourcing. By enabling precise breed identification throughout the supply chain, this approach supports claims about the authenticity and quality of meat and dairy products, aligning with regulatory requirements and consumer expectations. Looking ahead, there are several key research directions. One promising avenue is the application of these methods to predict binary traits, such as disease resistance or production efficiency, using minimal genetic markers. Adapting approaches similar to those used by Biscarini et al. (2015) for predicting root vigor in sugar beets could streamline marker selection for critical traits in livestock, optimizing genetic evaluations and breeding programs.
Moreover, there is potential to extend these methods to explore genetic changes within populations and classify complex multiclass groups using multiomic data. Developing advanced ML and deep learning models to handle such intricate datasets will allow for more detailed analyses of genetic diversity and breed distinctions, enhancing our understanding of livestock genetics. The ability to monitor genetic diversity and maintain breed sovereignty is another critical area where these technologies can make a significant impact. Identifying and tracking genetic markers associated with key phenotypic traits will help preserve the unique characteristics of various breeds and ensure their adaptability to changing environmental conditions. Future research should focus on how best to utilize these tools for conservation and sustainable livestock management.
The integration of genomic data with ML algorithms, as demonstrated in this study, offers valuable lessons for future research. This approach underscores the importance of using comprehensive genomic datasets and advanced analytical techniques to derive actionable insights and improve accuracy in breed identification. Customizing SNP panels to specific breeds, as shown for Tharparkar cattle herein, is crucial for achieving precise genetic analyses. Future work should continue to refine these panels and adapt them to different breeds and traits. Additionally, ensuring that methods developed are scalable and applicable across various livestock populations and environmental conditions is essential for maximizing the impact of digital transformation in genomics. Research should aim to create adaptable models that can be generalized beyond specific cases.
Finally, as we advance in using genomic and ML technologies, addressing ethical and practical considerations is vital. Issues such as data privacy, genetic data ownership, and the implications for small-scale farmers and indigenous breeds must be carefully managed to ensure responsible and equitable use of these technologies. Engaging with critical social sciences and humanities research has been shown to help broaden the technical discourses in medicine and planetary health to a larger societal realm (Reinhart, 2023; Yetiskin, 2022). Ecology and livestock genomics, too, can be brought to bear on pressing needs in the field by engaging with the societal dimensions of emerging technologies. Overall, the integration of genomics and digital transformation tools presents significant opportunities to advance livestock genomics, drive innovation in livestock management, and contribute to sustainable agricultural practices in the coming decades.
Conclusions
This study underscores the effectiveness of combining ML classification algorithms and SNP chip data to efficiently identify and classify the Tharparkar cattle population. By employing a feature-selection process, we identified a minimal set of 23 informative SNPs, which proved sufficient for accurate differentiation of the target breed from other cattle populations. The utilization of such a minimal marker set has the potential to reduce genotyping costs and facilitate the development of SNP marker combinations for use in various fields, including forensic science, conservation genetics, and livestock traceability systems. Furthermore, this innovative approach can be extended to explore markers associated with other phenotypic traits and for distinguishing among multiple groups simultaneously. The executive summary of this investigation is presented in Box 1.
Executive summary points from the present study
Footnotes
Acknowledgments
This study was conducted at the IVRI, Izatnagar, an institute under the ICAR, Department of Agricultural Research and Education, Government of India, and funded by ICAR-National Agricultural Science Foundation (NASF). The authors would like to express their gratitude to the Director, IVRI, for providing all requisite facilities as well.
Authors’ Contributions
H.K.: Sample Collection, Data curation, Analysis, Writing—Original Draft M.P.: Conceptualization, Methods, Writing—Review & editing D.S.: Data curation, Writing—Review & editing S.C.: Coding, Figures, Data curation, Writing—Review & editing B.B.: Resources & Supervision T.D.: Resources & Supervision.
Data Availability Statement
Author Disclosure Statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Funding Information
No funding was received for this article.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.